
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pentaenolate activation in the organocatalytic allylic alkylation of indene-2-carbaldehydes†
Adam
Cieśliński
,
Sebastian
Frankowski
and
Łukasz
Albrecht
 *
*
Institute of Organic Chemistry, Faculty of Chemistry Lodz University of Technology Żeromskiego 116, Łódź 90-924, Poland. E-mail: lukasz.albrecht@p.lodz.pl
First published on 1st June 2023
Abstract
In this manuscript, the application of pentaenolate intermediates in the allylic alkylation of indene-2-carbaldehydes with Morita–Baylis–Hillman (MBH) carbonates is described. The reaction has been carried out in a highly enantio- and diastereoselective manner due to the use of a chiral tertiary amine as a nucleophilic catalyst. The developed reactivity constitutes the first application of organocatalytic pentaenolate activation in asymmetric synthesis, expanding the arsenal of catalytic methods.
One of the primary goals of contemporary organic chemistry is to develop novel synthetic tools and reactivities to broaden the range of chemical transformations.1 Interestingly, one of the simplest modifications of an already established reactivity can be achieved by applying the principle of vinylogy, which is defined as the transmission of electronic properties through a conjugated double bond system. This concept was first introduced in 1926 by Ludwig Claisen and, in the nearly 100 years since then, has brought about the development of an enormous number of new reaction pathways. It is also worth noting that the concept of vinylogy has been readily employed in asymmetric organocatalysis.2 Within the organocatalytic vinylogous reactivities, a great deal of attention has been devoted to the development of various polyenolate species3 and polyenamine chemistry.4 Generation of polyenolate intermediates under Brønsted base activation primarily uses pronucleophiles bearing two electron-withdrawing groups installed at a position enabling stabilization of negatively charged carbanions, thus providing an easily-affordable carbon vinylogous nucleophile (Scheme 1, top).5 This approach has been mainly exploited in the dienolate reactivity, where electron-poor olefin is functionalized at the γ-position, giving access to different alkylation and formal cycloaddition products. Interestingly, only a few examples of trienolate reactivity6 which use doubly unsaturated dicyanocompounds as substrates are known (Scheme 1, middle). Moreover, this idea has led to the use of dicyano-indole-based olefins in such reactions, which proved to be a convenient method for the dearomatization of heterocyclic indole rings via the formation of trienolate.7 Further development of the potential of polyenolates by adding more conjugated double bonds to the system was carried out using multi-unsaturated silyl enol ethers under chiral metal complex catalysis (Scheme 1, bottom).8 On the other hand, great advances in vinylogous reactions have been possible due to the development of polyenamine reactivity (Scheme 2, top).4 This involves the formation of enamine from diversely designed, both linear and cyclic, unsaturated aldehydes or ketones under aminocatalytic conditions. Recently, the aminocatalytic activation of indene-2-carbaldehydes9 providing formal pentaenamine alkylation products has been developed in our laboratories (Scheme 2, middle).10
 | ||
| Scheme 1 Polyenolates in stereocontrolled organic synthesis. | ||
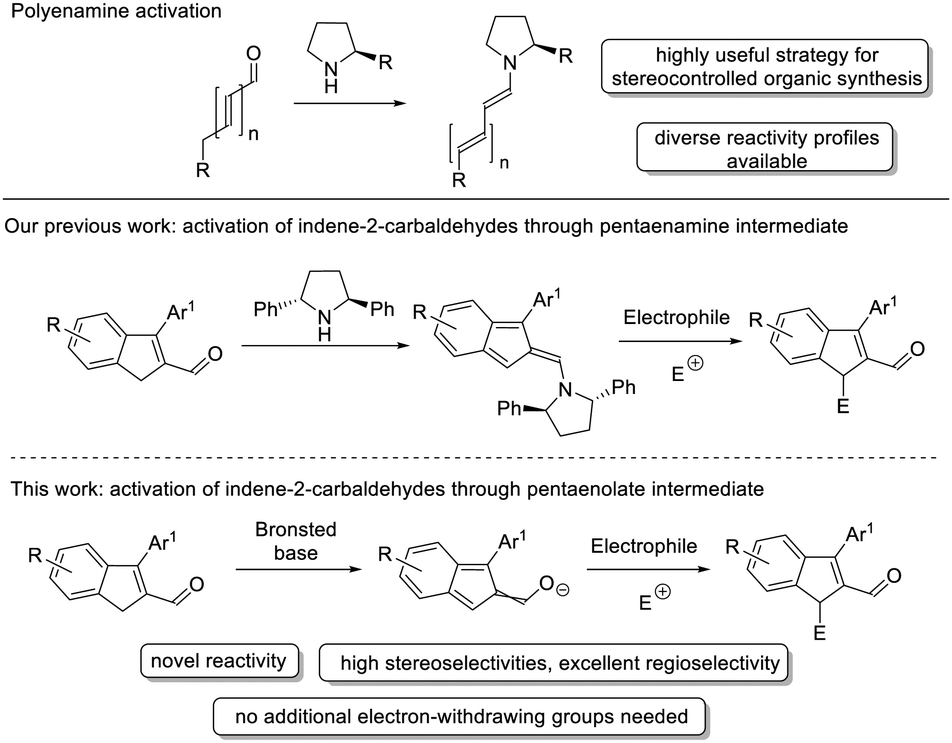 | ||
| Scheme 2 Polyenamines in organic synthesis and the objectives of our study. | ||
Inspired by our previous results, we decided to attempt organocatalytic pentaenolate alkylation using indene-2-carbaldehydes under basic conditions (Scheme 2, bottom). Morita–Baylis–Hillman (MBH) carbonates were selected as model electrophiles for the devised reactivity due to the fact that their ability to participate in the asymmetric allylic alkylations is well-recognized.11 It was anticipated that the application of chiral tertiary amines as nucleophilic catalysts would ensure high stereocontrol of the process.
Optimization studies were performed using 3-phenylindene-2-carbaldehyde 1a and MBH carbonate 2a as model reactants (Table 1). To our delight, commercially available quinine 4a promoted the reaction with moderate conversion (Table 1, entry 1). However, product 3a was racemic. Therefore, catalyst screening was carried out. The application of β-isocupreidine 4b or cupreine derivative 4c as a catalyst improved the conversion of 1a but the product 3a was either racemic (Table 1, entry 2) or the reaction stereoselectivity was low (Table 1, entry 3). It was found that when a catalyst 4d bearing H-bond donor moieties was employed, the reactivity was not observed (Table 1, entry 4). Next, the ability of the three dimeric cinchona alkaloid catalysts 4e–g to promote the desired reaction was investigated (Table 1, entries 5–7). We were pleased to observe that the use of 4g led to an increase in the enantioselectivity of the developed reaction. However, the diastereoselectivity was still unsatisfactory. Consequently, the influence of the bulkiness of the ester group in 2 on the stereochemical reaction outcome was evaluated. To our delight, the use of an MBH-carbonate 2b bearing a sterically demanding tert-butyl group led to an increase in the diastereoselectivity of the process (Table 1, entries 8 and 9). Next in the optimization part of the study, the influence of the concentration (Table 1, entry 10) and relative ratio of substrates (Table 1, entry 11) on the reaction outcome was evaluated. The modification of the concentration (from 0.25 M to 0.5 M) led to a significant improvement in the conversion while also ensuring high stereoselectivity of the process. The use of a 2-fold excess of MBH carbonate 2b had a similar effect on the reaction outcome (Table 1, entry 11). Subsequently, solvent screening was performed. Among the tested solvents (Table 1, compare entries 12–17), dichloromethane proved the most suitable reaction medium for the developed allylic alkylation as it provided the best stereoselectivity and efficiency of the process (Table 1, entry 12). It is worth noticing that the developed reaction was readily carried out on a 30-fold higher scale generating 3b with similar results (Table 1, entry 18).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| 2 | Cat. | Solvent | Time | Conv. (yield)b [%] | drc | erd | |
| a Reactions performed on a 0.05 mmol scale using 1a (1 equiv.) and 2 (1.2 equiv.) in 0.2 mL of the solvent. b Conversion as determined by 1H NMR for 3 of a crude reaction mixture. In parentheses yield of isolated 3 is given. c Determined for 3 by 1H NMR of a crude reaction mixture. d Determined by a chiral stationary phase HPLC for 3. e Reaction performed in 0.1 mL of the solvent. f Reaction performed using 1a (1.0 equiv.) and 2 (2.0 equiv.). g Reaction performed on a 1.5 mmol scale. | |||||||
| 1 | 2a | 4a | CH2Cl2 | 24 | 60 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
50:50 |
| 2 | 2a | 4b | CH2Cl2 | 24 | >95 | 2:1 |
50:50 |
| 3 | 2a | 4c | CH2Cl2 | 24 | 80 | 2.3:1 |
55:45 |
| 4 | 2a | 4d | CH2Cl2 | 24 | <5 | n. d. | n. d. |
| 5 | 2a | 4e | CH2Cl2 | 24 | 50 | 2.5:1 |
85:15 |
| 6 | 2a | 4f | CH2Cl2 | 24 | 20 | 2:1 |
n. d. |
| 7 | 2a | 4g | CH2Cl2 | 24 | 77 | 2.8:1 |
95:5 |
| 8 | 2b | 4g | CH2Cl2 | 24 | 26 | 5:1 |
n.d |
| 9 | 2b | 4g | CH2Cl2 | 48 | 40 | 5:1 |
95:5 |
| 10e | 2b | 4g | CH2Cl2 | 48 | 85 | 5:1 |
95:5 |
| 11f | 2b | 4g | CH2Cl2 | 48 | 80 | 5:1 |
95:5 |
| 12ef | 2b | 4g | CH2Cl2 | 48 | >95(79) | 5:1 |
95:5 |
| 13ef | 2b | 4g | Et2O | 48 | 40 | 5.2:1 |
n.d |
| 14ef | 2b | 4g | Toluene | 48 | 34 | 5:1 |
n.d |
| 15ef | 2b | 4g | MTBE | 48 | 50 | 5:1 |
n.d |
| 16ef | 2b | 4g | THF | 48 | 92 | 4.7:1 |
96:4 |
| 17ef | 2b | 4g | DCE | 48 | >95 | 4:1 |
93:7 |
| 18efg | 2b | 4g | CH2Cl2 | 48 | >95(63) | 4.7:1 |
95:5 |

Once the optimal reaction conditions were established, the scope and limitations of the method were examined. Initially, a variety of indene-2-carbaldehydes 1a–j were reacted with 2b under optimized conditions (Scheme 3). Surprisingly, 5-methyl- and 5-methoxy-indene-2-carbaldehydes 1b–c provided products 3c–d with good diastereoselectivity but with lower yield and diminished enantioselectivity, presumably for steric reasons. The use of 6-bromo or 6-methoxy-substituted aldehydes 1d–e led to products 3e–f with good diastereo- and enantioselectivities. Transformation proved unbiased toward the presence of both electron-withdrawing and electron-donating substituents at the 3-position of 1 (Scheme 3, compounds 3g–h). Replacing the phenyl ring at the 3 position of 1 by more sterically demanding 2-naphthyl or 1,1′-biphenyl substituents afforded access to the desired products 3i–j in lower yields but with good stereoselectivities. Interestingly, the use of unsubstituted indene-2-carbaldehyde 1j led to the isomeric product 3k′ with the double bond at a different position in the indene framework. A plausible explanation is that, under the reaction conditions, the initially generated product 3k underwent isomerization (via deprotonation/protonation mechanism) to give more substituted olefin 3k′.
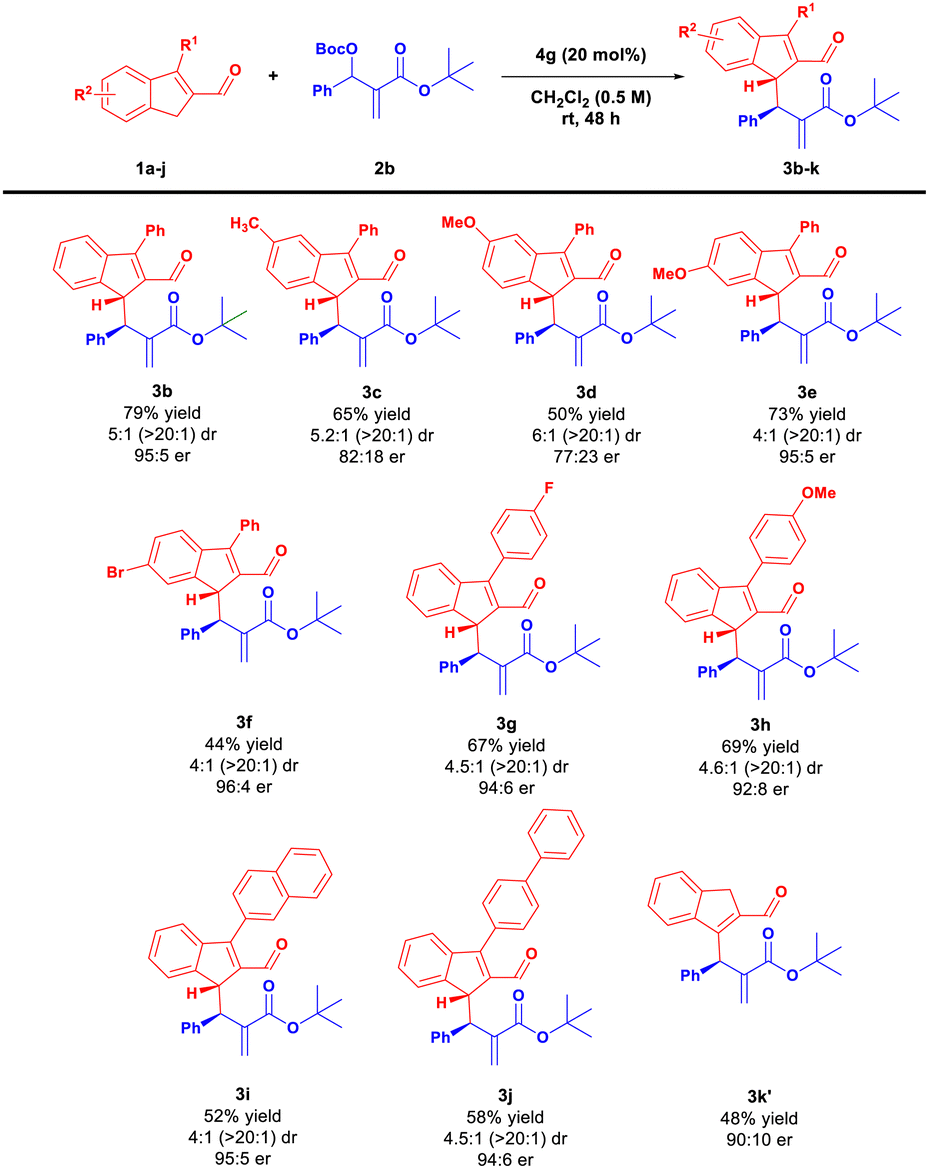 | ||
| Scheme 3 Pentaenolate activation in the allylic alkylation of indene-2-carbaldehydes 1 – indene-2-carbaldehydes 1 scope. Yields of isolated products 3 are given. Diastereomeric ratios as determined by 1H NMR of a crude reaction mixture. In parentheses dr of the isolated product is given. | ||
The second part of the study focused on the use of structurally diversified MBH carbonates 2a–l. To our delight, carbonate 2c bearing a bromine substituent at the para position gave access to the desired product 3l with good yield and stereoselectivity. The electron-donating groups appear to have an effect on the reactivity since substrate 2d and 2f gave access to 3m,o with lower yield but satisfying stereoselectivity. It was found that the presence of electron-withdrawing groups on the aromatic ring in 2e did not significantly influence the reaction outcome. The position of substituents on the aromatic ring in 2 had a significant influence on the diastereoselectivity of the developed transformation (Scheme 4, compare 3pvs.3q). The ortho-substituted carbonate 2h provided product 3q with moderate yield, good enantioselectivity, and low diastereoselectivity. The use of the double substitution pattern on the aryl ring in 2 was also possible but with diminished diastereoselectivity (Scheme 4, product 3r). Substrate 2j bearing a heteroaromatic ring reacted to give 3s with low yield and stereoselectivity. In the last part of the study, the possibility to modify the electron-withdrawing-group in the MBH carbonate 2 was investigated. The desired products 3t–u were produced in moderate yields and with low diastereoselectivity. As for the cyano-group-substituted MBH carbonate 2k, slightly lower enantioselectivity was observed after longer reaction time. Attempts to expand the scope of the reaction by including MBH-carbonates 2 derived from aliphatic aldehydes were unsuccessful. Under the optimized conditions, the formation of complex reaction mixtures was observed.
 | ||
| Scheme 4 Pentaenolate activation in the allylic alkylation of indene-2-carbaldehydes 1 – MBH carbonates 2 scope. Yields of isolated products 3 are given. Diastereomeric ratios as determined by 1H NMR of a crude reaction mixture. In parentheses dr of the isolated product is given. | ||
In order to demonstrate the synthetic utility of the allylic alkylation product 3, the selected transformations of 3 were attempted. Initially, the dipolar (3+2)-cycloaddition involving the alkene moiety in 3b with the dipole generated from imidoyl chloride 5 under basic conditions was performed to give 6b in a very good yield. Subsequently, the reaction sequence involving the reduction of 3b,o with NaBH4 to alcohols 7b,o followed by acid-induced intramolecular Friedel–Crafts alkylation was performed. Products 8b,o containing an additional six-membered ring were efficiently obtained (Scheme 5). Both reaction sequences occurred with the preservation of the optical purity of 3b,o as all products were obtained as single diastereoisomers (Scheme 5). The absolute configuration of product 3b was unambiguously determined by single crystal X-ray diffraction analysis of 3b.12 The stereochemistry of products 3 and as well as 6–8 was assigned by analogy.
 | ||
| Scheme 5 Pentaenolate activation in the allylic alkylation of indene-2-carbaldehydes 1 – transformations of 3b. | ||
In conclusion, we have demonstrated the possibility to generate highly extended pentaenolate species using asymmetric organocatalysis. In contrast to classical activation of indene-2-carbaldehydes 1 (via polyenamine formation),9,10 their use as pentaenolate precursors generated under basic conditions is unprecedented in the literature. Allylic alkylation of 1 with MBH-carbonates 2 was efficiently carried out under nucleophilic catalysis conditions using dimeric cinchona alkaloid derivative 4g as a reaction promotor. The advantages of this methodology are its broad scope, high efficiency, and excellent stereoselectivity. Further studies leading to the expansion of pentaenolate activation to more-demanding linear substrates are on-going in our laboratories.
This project was realized within the Opus programme (UMO-2021/41/B/ST4/03385) from the National Science Centre, Poland. Thanks are expressed to Dr Lesław Sieroń (Lodz University of Technology) for the X-ray analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed; (b) K. Mikami and M. Lautens, New Frontiers in Asymmetric Catalysis, Wiley–Interscience, Hoboken, NJ, 2007 CrossRef.
- For selected reviews, see: (a) R. C. Fuson, Chem. Rev., 1935, 16, 1 CrossRef CAS; (b) C. Schneider and F. Abels, Org. Biomol. Chem., 2014, 12, 3531 RSC; (c) H. B. Hepburn, L. Dell’Amico and P. Melchiorre, Chem. Rec., 2016, 16, 1787 CrossRef CAS PubMed; (d) L. Battistini, C. Curti, G. Rassu, A. Sartori and F. Zanardi, Synthesis, 2017, 2297 CAS; (e) P. Chauhan, U. Kaya and D. Enders, Adv. Synth. Catal., 2017, 359, 888 CrossRef CAS; (f) C. Curti, L. Battistini, A. Sartori and F. Zanardi, Chem. Rev., 2020, 120, 2448 CrossRef CAS PubMed.
- For selected examples, see: (a) C. Curti, G. Rassu, V. Zambrano, L. Pinna, G. Pelosi, A. Sartori, L. Battistini, F. Zanardi and G. Casiraghi, Angew. Chem., Int. Ed., 2012, 51, 6200 CrossRef CAS; (b) G. Rassu, V. Zambrano, L. Pinna, C. Curti, L. Battistini, A. Sartori, G. Pelosi, F. Zanardi and G. Casiraghi, Adv. Synth. Catal., 2013, 355, 1881 CrossRef CAS; (c) L. Dell’Amico, Ł. Albrecht, T. Naicker, P. H. Poulsen and K. A. Jørgensen, J. Am. Chem. Soc., 2013, 135, 8063 CrossRef; (d) Q. Chen, G. Wang, X. Jiang, Z. Xu, L. Lin and R. Wang, Org. Lett., 2014, 16, 1394 CrossRef CAS PubMed; (e) T.-P. Gao, J.-B. Lin, X.-Q. Hu and P.-F. Xu, Chem. Commun., 2014, 50, 8934 RSC; (f) Y. Zhong, S. Ma, Z. Xu, M. Chang and R. Wang, RSC Adv., 2014, 4, 49930 RSC; (g) G. Rassu, V. Zambrano, L. Pinna, C. Curti, L. Battistini, A. Sartori, G. Pelosi, G. Casiraghi and F. Zanardi, Adv. Synth. Catal., 2014, 356, 2330 CrossRef CAS; (h) N. Di Iorio, P. Righi, S. Ranieri, A. Mazzanti, R. G. Margutta and G. Bencivenni, J. Org. Chem., 2015, 80, 7158 CrossRef CAS PubMed; (i) X. Xiao, H. Mei, Q. Chen, X. Zhao, L. Lin, X. Liu and X. Feng, Chem. Commun., 2015, 51, 580 RSC; (j) J.-L. Han and C.-H. Chang, Chem. Commun., 2016, 52, 2322 RSC.
- For selected reviews, see: (a) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (b) S. Reboredo, A. Parra and J. Alemán, Asymmetric Catal., 2013, 1, 24 Search PubMed; (c) V. Marcos and J. Alemán, Chem. Soc. Rev., 2016, 45, 6812 RSC; (d) A. Przydacz, A. Skrzyńska and Ł. Albrecht, Angew. Chem., Int. Ed., 2019, 58, 63 CrossRef CAS PubMed; (e) T. J. Pawar, S. B. Mitkari, E. Peña-Cabrera, C. V. Gómez and D. C. Cruz, Eur. J. Org. Chem., 2020, 6044 CrossRef CAS.
- (a) T. B. Poulsen, C. Alemparte and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 11614 CrossRef CAS PubMed; (b) T. B. Poulsen, M. Bell and K. A. Jørgensen, Org. Biomol. Chem., 2005, 4, 63 RSC; (c) T.-Y. Liu, H.-L. Cui, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, J. Am. Chem. Soc., 2007, 129, 1878 CrossRef CAS PubMed; (d) J.-W. Xie, L. Yue, D. Xue, X.-L. Ma, Y.-C. Chen, Y. Wu, J. Zhu and J.-G. Deng, Chem. Commun., 2006, 1563 RSC; (e) J. Lu, F. Liu and T.-P. Loh, Adv. Synth. Catal., 2008, 350, 1781 CrossRef CAS; (f) S. Rout, S. K. Ray, R. A. Unhale and V. K. Singh, Org. Lett., 2014, 16, 5568 CrossRef CAS PubMed; (g) J. Alemán, C. B. Jacobsen, K. Frisch, J. Overgaard and K. A. Jørgensen, Chem. Commun., 2008, 632 RSC; (h) X. Li, X. Xu, W. Wei, A. Lin and H. Yao, Org. Lett., 2016, 18, 428 CrossRef CAS PubMed; (i) R. Zhou, J. Wang, J. Tian and Z. He, Org. Biomol. Chem., 2012, 10, 773 RSC.
- (a) Q.-Z. Li, J. Gua and Y.-C. Chen, RSC Adv., 2014, 4, 37522 RSC; (b) L. Dell’Amico, G. Rassu, V. Zambrano, A. Sartori, C. Curti, L. Battistini, G. Pelosi, G. Casiraghi and F. Zanardi, J. Am. Chem. Soc., 2014, 136, 11107 CrossRef PubMed; (c) C. Mou, L. Zhou, R. Song, H. Chai, L. Hao and Y. R. Chi, Org. Lett., 2020, 22, 2542 CrossRef CAS PubMed; (d) N. Brindani, G. Rassu, L. Dell'Amico, V. Zambrano, L. Pinna, C. Curti, A. Sartori, L. Battistini, G. Casiraghi, G. Pelosi, D. Greco and F. Zanardi, Angew. Chem., Int. Ed., 2015, 54, 7386 CrossRef CAS PubMed.
- (a) G. Rassu, C. Curti, V. Zambrano, L. Pinna, N. Brindani, G. Pelosi and F. Zanardi, Chem. – Eur. J., 2016, 22, 12637 CrossRef CAS PubMed; (b) B.-Q. Gu, H. Zhang, R.-H. Su and W.-P. Deng, Tetrahedron, 2016, 72, 6595 CrossRef CAS.
- (a) L. Ratjen, P. Garcia-Garcia, F. Lay, M. E. Beck and B. List, Angew. Chem., Int. Ed., 2011, 50, 754 CrossRef CAS PubMed; (b) C. Curti, L. Battistini, A. Sartori, A. Lodola, M. Mor, G. Rassu, G. Pelosi, F. Zanardi and G. Casiraghi, Org. Lett., 2011, 13, 4738 CrossRef CAS PubMed; (c) C. Curti, A. Sartori, L. Battistini, N. Brindani, G. Rassu, G. Pelosi, A. Lodola, M. Mor, G. Casiraghi and F. Zanardi, Chem. – Eur. J., 2015, 21, 6433 CrossRef CAS PubMed.
- For the application of indene-2-carbaldehydes in higher-order cycloadditions, see: (a) B. S. Donslund, A. Monleon, T. A. Palazzo, M. L. Christensen, A. Dahlgaard, J. D. Erickson and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 1246 CrossRef CAS PubMed; (b) B. S. Donslund, N. I. Jessen, G. Bertuzzi, M. Giardinetti, T. A. Palazzo, M. L. Christensen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2018, 57, 13182 CrossRef CAS PubMed; (c) M. Giardinetti, N. I. Jessen, M. L. Christensen and K. A. Jørgensen, Chem. Commun., 2019, 55, 202 RSC.
- A. Przydacz, A. Topolska, A. Skrzyńska, S. Frankowski and Ł. Albrecht, Adv. Synth. Catal., 2022, 364, 974 CrossRef CAS.
- For a review, see: (a) T.-Y. Liu, M. Xie and Y.-C. Chen, Chem. Soc. Rev., 2012, 41, 4101 RSC ; for selected examples, see: ; (b) H.-L. Cui, J.-R. Huang, J. Lei, Z.-F. Wang, S. Chen, L. Wu and Y.-C. Chen, Org. Lett., 2010, 12, 720 CrossRef CAS PubMed; (c) J. Feng, X. Li and J.-P. Cheng, Chem. Commun., 2015, 51, 14342 RSC; (d) D. Kowalczyk and Ł. Albrecht, Adv. Synth. Catal., 2018, 360, 406 CrossRef CAS; (e) Z. Li, M. Frings, H. Yu, G. Raabe and C. Bolm, Org. Lett., 2018, 20, 7367 CrossRef CAS PubMed; (f) D. Kowalczyk-Dworak, M. Kwit and Ł. Albrecht, J. Org. Chem., 2020, 85, 2938 CrossRef CAS PubMed; (g) J.-X. Xu, K.-T. Chu, M.-H. Chiang and J.-L. Han, Org. Biomol. Chem., 2021, 19, 1503 RSC.
- CCDC 2192825 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/structures.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 2192825. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc00900a |
| This journal is © The Royal Society of Chemistry 2023 |