
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Automated glycan assembly of highly branched heptadecasaccharide repeating unit of arabinogalactan polysaccharide HH1-1 from Carthamus tinctorius†
Narayana Murthy
Sabbavarapu
a and
Peter H.
Seeberger
 *ab
*ab
aDepartment of Biomolecular Systems, Max-Planck-Institute of Colloids and Interfaces, Potsdam 14476, Germany. E-mail: peter.seeberger@mpikg.mpg.de
bFreieUniversität Berlin, Institute of Chemistry and Biochemistry, Berlin 14195, Germany
First published on 27th March 2023
Abstract
Polysaccharides that are part of the human diet of fruits and vegetables influence the immune system via multiple signaling pathways. Given the immense complexity and diversity of naturally occurring polysaccharides and the difficulties associated isolating pure samples, few structure-activity relationships have been established. Rapid access to well-defined polysaccharides of biological relevance by automated glycan assembly (AGA) is important to create chemical tools to determine the link between nutritional oligo- and polysaccharides and the immune response. Here, we describe AGA of a hyper branched heptadecasaccharide repeating unit of arabinogalactan polysaccharide HH1-1 from Carthamus tinctorius.
Polysaccharides isolated from Carthamus tinctorius L. possess anti-pancreatic cancer activity by selectively targeting galectin-3 and modulate the human immune system as they influence macrophage phagocytosis as well as lymphokine secretion such as IL-2.1,2 The isolation of sufficient quantities of pure oligosaccharides from natural sourcesis difficult and often impossible to establish structure-function correlations. The highly branched heptadecasaccharide repeating unit (Fig. 1) comprises a linear backbone of β-(1 → 6)-linked-D-Galp branched at C-3 with two highly crowded side chains [L-Araf-α-(1 → 5)]-[D-Galp-β-(1 → 3)]-L-Araf-α-(1 → 3)-D-Galp and[L-Araf-α-(1 → 5)]-[L-Araf-α-(1 → 3)]-L-Araf-α-(1 → 3)-D-Galp as well as a side chain of D-Galp-β-(1 → 3)-D-Galp. The structural complexity of this polysaccharide repeating unit is an intriguing synthetic challenge for automated glycan assembly (AGA) that has been used to prepare a host of oligo- and polysaccharides of increasing complexity. A recent synthesis of a heptadecasaccharide relied on the convergent coupling strategy, where independently prepared cassettes assembled by a one-pot glycosylation strategy from monosaccharides. The photo-assisted convergent (6+4+7) one-pot coupling strategy incorporated in situ removal of a O-nitrobenzyl protecting groups to generate the acceptor for the concomitant glycosylation.3
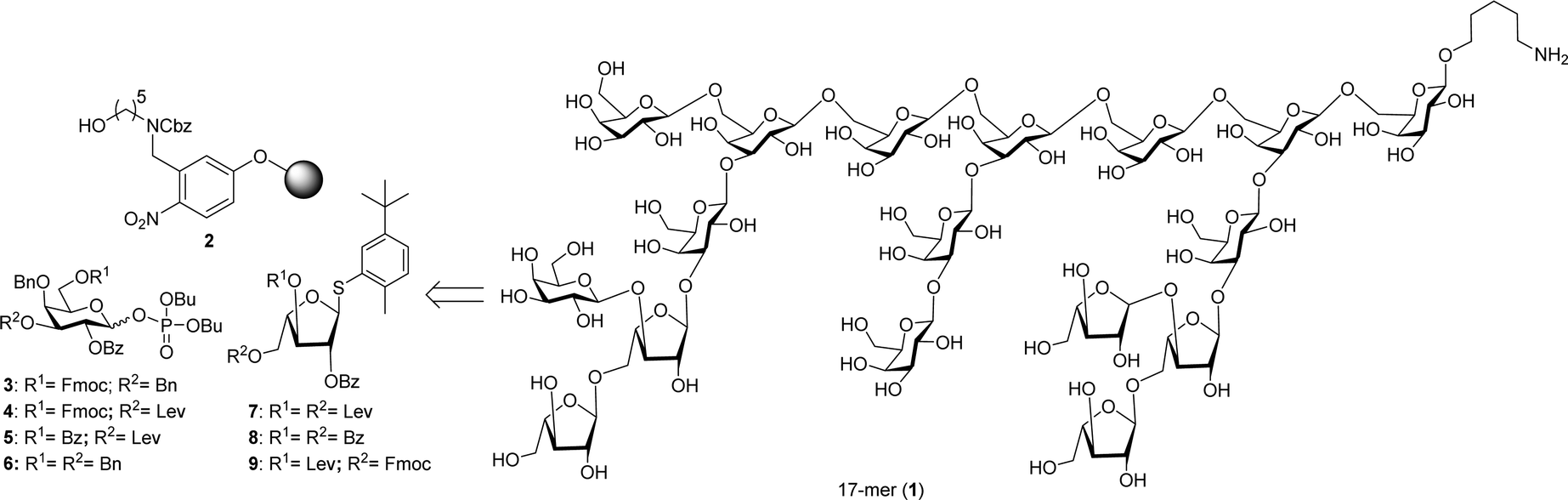 | ||
| Fig. 1 Retrosynthetic analysis of heptadecasaccharide repeating unit of arabinogalactan polysaccharide HH1-1 (1). | ||
Solution phase oligosaccharide synthesis4–6 involving isolation of intermediates is still most common.7,8 To simplify and accelerate oligosaccharide synthesis, automated glycan assembly (AGA),9 has proven a fast and robust technology to procure well-defined polysaccharides.10,11 Herein, we disclose the stereoselective AGA synthesis of a highly branched heptadecasaccharide repeating domain (Fig. 1) of polysaccharides isolated from Carthamus tinctorius L.
Highly branched heptadecasaccharide backbone 1 can be synthesized by AGA using orthogonally protected building blocks 3–9 (Fig. 1). The monosaccharides 3–9, are either commercially available or were synthesized in multi-gram scale starting from commercial intermediates (see ESI† for details). Initial efforts synthesizing target molecule 1 employing galactopyranosyl thioglycosides resulted in significant amounts of (n–1) oligomers in addition to the desired sequence due to the low reactivity of thioglycosides. To increase building block reactivity and to ensure complete conversion upon activation with TMSOTf in dichloromethane, galactopyranosyl phosphate building blocks were prepared. The synthesis of building blocks 4 and 5 started from commercially available thioglycoside 12.
Dibutyltin oxide catalyzed selective naphthylation of 12, followed by benzoylation and subsequent regioselective ring opening of benzylideneacetal furnished thioglycoside 14. Fmoc carbonylation of the hydroxyl in 14, followed DDQ-mediated oxidative cleavage of 2-naphthylmethyl (NAP) ether afforded 16. 3-O-Levulinoylation of 16, followed by coupling dibutyl phosphate under NIS/TfOH promotion afforded the desired dibutyl phosphate building block 4 in excellent yield. Benzoylation of the primary hydroxyl in 14, oxidative removal 2-naphthylmethyl (NAP) ether and 3-O-levulinoylation furnished thiogalactose 18. NIS/TfOH-promoted coupling of 18 with dibutyl phosphate resulted in building block 5. Similarly, coupling of commercially available 10 and 11 with dibutyl phosphate by treatment with NIS/TfOH provided the corresponding dibutyl phosphate building blocks 3 and 6 (Scheme 1).
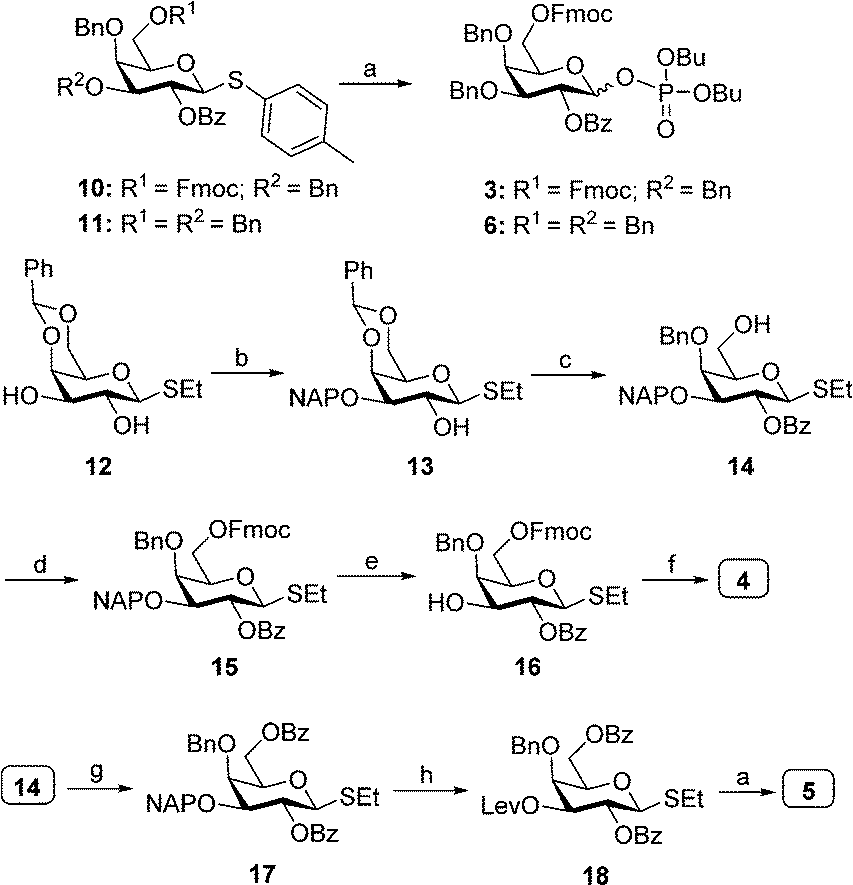 | ||
| Scheme 1 Synthesis of building blocks 3, 4, 5 and 6. Reagents and conditions: (a) (BuO)2P(O)OH, NIS, TfOH, 4 Å MS, CH2Cl2, (yields: 3 = 78%; 6 = 80%; 5 = 72%); (b) Bu2SnO, 2-NAPBr, CSF, DMF, 69%; (c) (i) PhCOCl, 4-DMAP, Py.; (ii) BH3·THF, TMSOTf, CH2Cl2, 75% (2 steps); (d) FmocCl, Py., 88%; (e) DDQ, CH2Cl2/H2O,81%; (f) (i) LevOH, DCC, 4-DMAP, CH2Cl2, 62%; (ii) (BuO)2P(O)OH, NIS, TfOH, 4Å MS, CH2Cl2, 79%; (g) PhCOCl, 4-DMAP, pyridine, 86%; (h) (i) DDQ, CH2Cl2/H2O; (ii) LevOH, EDC.HCl, 4-DMAP, CH2Cl2, 53% (two steps). | ||
L-Arabinothiofuranoside donors 7–9 were prepared from L-arabinose that was transformed into corresponding perbenzoylthiofuranoside 8 following literature precedent.12 Methanolysis of 8 under Zemplen's conditions followed by 3,5-O-cyclic protection of thiofuranoside 8 using 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane generated the corresponding 3,5-O-tetraisopropyldisiloxanylidene, followed by benzoylation and subsequent HF/pyridine-mediated removal of 3,5-O-tetraisopropyldisiloxanylidene protection afforded 20. Levulinoylation of the diol in 20 delivered building block 7. Selective protection of the primary hydroxyl as the corresponding trityl ether, followed by levulinoylation furnished the fully protected thioglycoside. Acid-mediated removal of the trityl group and subsequent Fmoc carbonylation of the resultant hydroxyl afforded the desired thioglycoside building block 9 in excellent yield (Scheme 2).
 | ||
| Scheme 2 Synthesis of building blocks 7, 8 and 9. Reagents and conditions: (a) (i) NaOMe, MeOH, 89%; (ii) TIPDSCl, Py., then PhCOCl, 4-DMAP; (iii) HF/pyridine, THF, 81% (two steps); (b) LevOH, DCC, 4-DMAP, CH2Cl2, 80%; (c) (i) TrCl, Py.; (ii) LevOH, EDC.HCl, 4-DMAP, CH2Cl2; (iii) CSA, MeOH/H2O; (iv) FmocCl, pyridine, CH2Cl2, 80% (four steps). | ||
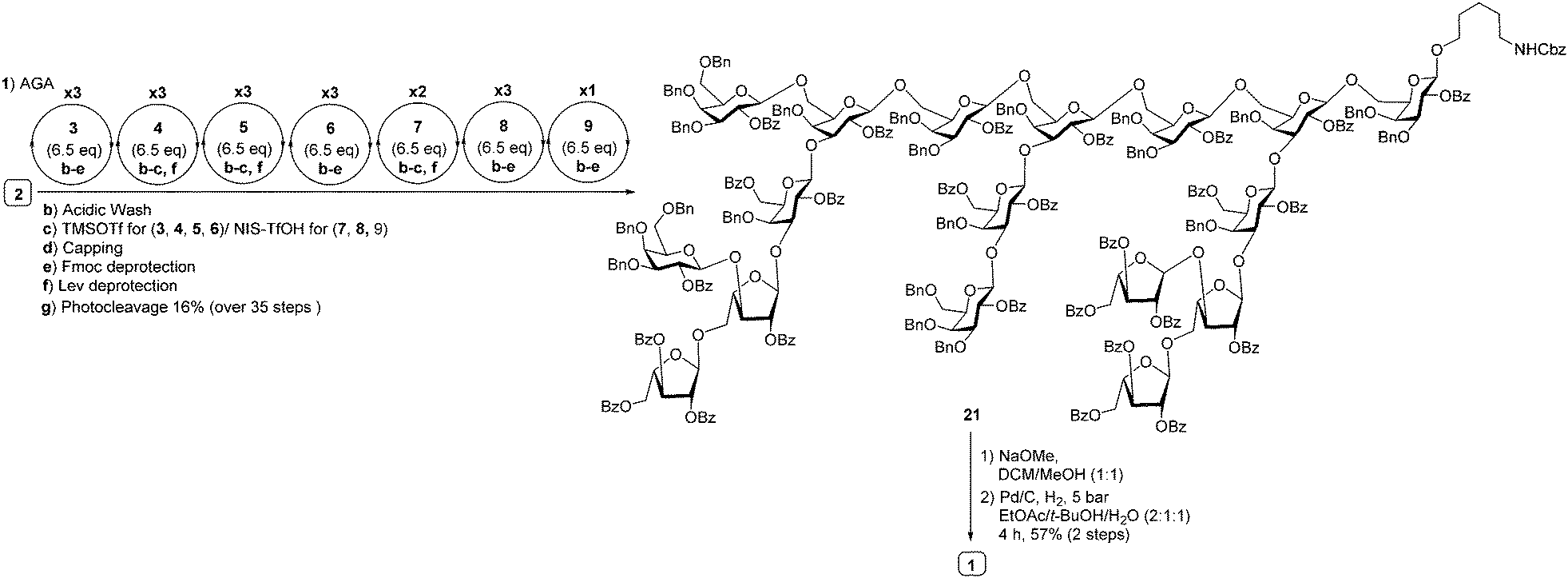 | ||
| Scheme 3 Synthesis of heptadecasaccharide repeating unit of arabinogalactan polysaccharide HH1-1 (1) using building blocks 3, 4, 5, 6, 7, 8 and 9. | ||
With the prerequisite building blocks 3–9 in hand, polystyrene resin containing the photocleavable aminopentanol linker 2 was placed in the reaction vessel of the automated synthesizer to prepare highly branched heptadecasaccharide 1. For the first time, pyranoside and furanosideglycosyl building blocks were used in concert in AGA to prepare highly branched oligomers. Starting from the reducing end, the highly branched side chains were appended first to the conserved hepadecasaccharide backbone. The initial linear trimer was assembled using glycosyl phosphate building blocks 3–5 on polystyrene resin equipped with photocleavable linker 2 at the reducing terminus. The linear trimer was assembled (see ESI†) and served as starting point for different modules such as acidic wash, glycosylation, capping to mask the unreacted nucleophile and deprotection to expose masked acceptor employing galactopyranosyl phosphate and thioarabinofuranosides to assemble a heptasaccharide. Galactopyranosyl phosphate building blocks 3–6 and L-arabinothiofuranoside donors 7–9 were strategically appended in appropriate positions during AGA to obtain fully protected highly branched heptadecasaccharide 21 (20 mg, 16%). Following AGA, the resin was subjected to UV irradiation using a continuous flow device13 to cleave protected heptadecasaccharide 21 from the resin that was subsequently purified by normal phase HPLC. Fully protected heptadecasaccharide 21 was treated with sodium methoxide to cleave all benzoate ester groups, followed by Pd(OH)2/C-catalyzed hydrogenolysis in the presence of hydrogen to furnish highly branched heptadecasaccharide 1 (4 mg) (Scheme 3). The identity and purity of heptadecasaccharide 1 was confirmed by extensive 2D-NMR spectroscopy analysis and the β-glycosidic linkages in 1 were confirmed by measuring the coupling constant between the anomeric carbon and proton (JC1–H1 = 160–163 Hz, in the case galctopyranosides; JC1–H1 = 170–175 Hz in the case of arabinofuranosides (see ESI†) and MALDI-TOF analysis.
In conclusion, we prepared the highly branched heptadecasaccharide repeating unit from Carthamus tinctorius L. arabinogalactan polysaccharide HH1-1 by AGA. Important for a robust AGA process are orthogonally protected thiofuranoside and galactopyranosyl phosphate building blocks that will be helpful for the synthesis of other arabinogalactan polysaccharides.
We gratefully acknowledge the generous financial support of the Max-Planck Society.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- Y. Yao, J. Yao, Z. Du, P. Wang and K. Ding, Carbohydr. Polym., 2018, 202, 134–142 CrossRef CAS PubMed.
- Y. Yao, L. Zhou, W. Liao, H. Chen, Z. Du, C. Shao, P. Wang and K. Ding, Carbohydr. Polym., 2019, 204, 111–123 CrossRef CAS PubMed.
- C. Hu, S. Wu, F. He, D. Cai, Z. Xu, W. Ma, Y. Liu, B. Wei, T. Li and K. Ding, Angew. Chem., Int. Ed., 2022, 61, e202202554 CAS.
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed.
- S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2015, 6, 6158–6164 RSC.
- Q. Zhu, Z. Shen, F. Chiodo, S. Nicolardi, A. Molinaro, A. Silipo and B. Yu, Nat. Commun., 2020, 11, 4142 CrossRef CAS PubMed.
- C.-C. Wang, J.-C. Lee, S.-Y. Luo, S. S. Kulkarni, Y.-W. Huang, C.-C. Lee, K.-L. Chang and S.-C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS PubMed.
- T. J. Boltje, J.-H. Kim, J. Park and G.-J. Boons, Nat. Chem., 2010, 2, 552–557 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- K. Naresh, F. Schumacher, H. S. Hahm and P. H. Seeberger, Chem. Commun., 2017, 53, 9085–9088 RSC.
- A. A. Joseph, A. Pardo-Vargas and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 8561–8564 CrossRef CAS PubMed.
- N. Xie and C. M. Taylor, Org. Lett., 2010, 12, 4968–4971 CrossRef CAS PubMed.
- M. Hurevich, J. Kandasamy, B. M. Ponnappa, D. Kopetzki, T. McQuade and P. H. Seeberger, Org. Lett., 2014, 16, 1794 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00687e |
| This journal is © The Royal Society of Chemistry 2023 |