
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambient temperature liquid salt electrolytes†
Sourav
Bhowmick
 *a,
Mukhtiar
Ahmed
a,
Andrei
Filippov
a,
Laura C.
Loaiza
b,
Faiz Ullah
Shah
*a and
Patrik
Johansson
*bc
*a,
Mukhtiar
Ahmed
a,
Andrei
Filippov
a,
Laura C.
Loaiza
b,
Faiz Ullah
Shah
*a and
Patrik
Johansson
*bc
aChemistry of Interfaces, Luleå University of Technology, SE-971 87 Luleå, Sweden. E-mail: sourav.bhowmick@associated.ltu.se; faiz.ullah@ltu.se
bDepartment of Physics, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden. E-mail: patrik.johansson@chalmers.se
cALISTORE-European Research Institute, CNRS FR 3104, Hub de l’Energie, 80039 Amiens, France
First published on 3rd February 2023
Abstract
Alkali metal salts usually have high melting points due to strong electrostatic interactions and solvents are needed to create ambient temperature liquid electrolytes. Here, we report on six phosphate-anion-based alkali metal salts, Li/Na/K, all of which are liquids at room temperature, with glass transition temperatures ranging from −61 to −29 °C, and are thermally stable up to at least 225 °C. While the focus herein is on various physico-chemical properties, these salts also exhibit high anodic stabilities, up to 6 V vs. M/M+ (M = Li/Na/K), and deliver some battery performance – at elevated temperatures as there are severe viscosity limitations at room-temperature. While the battery performance arguably is sub-par, solvent-free electrolytes based on alkali metal salts such as these should pave the way for conceptually different Li/Na/K-batteries, either by refined anion design or by using several salts to create eutectic mixtures.
To tackle climate change, we need to develop more efficient energy storage devices such as batteries1–4 for better use of renewable energy.1 Several alkali-ion batteries are currently put forth, the lithium-ion battery (LIB) being the dominant technology of today,5 the sodium-ion battery (SIB) being an emerging technology,6 and the potassium-ion battery (PIB) being conceptually very interesting.7 The liquid electrolytes used in these batteries are all based on salts dissolved in organic solvents, which renders a number of drawbacks, in particular in terms of thermal and chemical stability and safety, but also limited electrochemical stability windows (ESWs).8,9
Alternatives are either drastically different electrolytes, such as solid-state batteries based on solid polymer electrolytes (SPEs)10,11 or inorganic/ceramic electrolytes,12 or even (hybrid) metal–organic framework (MOF) electrolytes,13 or less so by keeping the electrolytes liquid and altering the nature of the solvent. One such path is to employ ionic liquids (ILs), with wide ESWs and liquid ranges, high ionic conductivities, negligible vapor pressures, and high thermal stabilities.14–20 However, this adds complexity, as another (organic) cation is introduced, and it also increases the cost of the electrolyte.
A more profound change is to use alkali metal salts that themselves are liquids at ambient temperature. Only a few such examples exist, e.g. the lithium aluminates21 and lithium borates22 containing two oligoether groups and two electron-withdrawing fluorinated moieties rendered viscous liquids at ambient temperature with ionic conductivities ranging from 10−5 to 10−4 S cm−1. The Li/Na-TOTO salts, where TOTO is 2,5,8,11-tetraoxatridecan-13-oate, exhibit glass transition temperatures at −53 and −57 °C, respectively (whereas K-TOTO is a white solid melting at +60 °C).23 The triethylene glycol-based 1,2,3-triazolate lithium salt revealed a glass transition temperature of −50 °C and ionic conductivity of 6.5 × 10−7 S cm−1 at 30 °C.24 None of these have been studied as electrolytes in batteries. The recently reported ambient temperature liquid salts based on fluorinated lithium borates, however, with ionic conductivities from 5.3 × 10−6 to 1.8 × 10−4 S cm−1 at 25 °C, show high compatibility with lithium metal electrodes with stable plating/stripping and overall promising battery performance.25
Apart from the uttermost need to be weakly coordinating anions (WCAs),26 the anions employed should be asymmetric and structurally flexible – to lower the melting point and maximize fluidity/ionic conductivity.27 A more common way to create solvent-free electrolytes is eutectic salt mixtures.28,29
With the goal to create ambient temperature liquid alkali metal salts, inspired by basic IL anion design criteria in general and the oligoether-based anions in particular, we created two different oligoether phosphate anions, bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl) phosphate [TEEP]− and bis(2-(2-ethoxyethoxy)ethyl) phosphate [DEEP]−, from their corresponding neutral tris(2-(2-(2-methoxyethoxy)ethoxy)ethyl) phosphate (TMOP) and tris(2-(2-ethoxyethoxy)ethyl) phosphate (TEOP), respectively. The phosphorus content might also bring flame-retardant properties30 and the long ethylene oxide-based side-chains should provide both asymmetry and structural flexibility.31
First brief descriptions of the synthesis and the characterization of the intermediate and final products are presented, followed by thermal behaviour, transport properties including ionic conductivity and ion diffusion data. The ion–ion interactions and their effects on the ion transport and dynamics are monitored using NMR and FTIR spectroscopies and, finally, systematic electrochemical assessments of the neat salts are discussed together with minor battery cycling tests.
In the first step, two neutral species, the trialkyl phosphates (TMOP and TEOP) are synthesized with reasonably high yields (>80%) by reacting the commercially available phosphoryl chloride and the corresponding primary alcohol without any solvent at an ambient temperature for 8 hours (Scheme 1). The 1H NMR spectrum of TMOP (Fig. S1, ESI†) shows a sharp singlet at 3.32 ppm, which is assigned to the aliphatic methoxy protons of the ether chain. The three sets of 1H resonance lines observed in the 1H NMR spectrum of TMOP are due to the three different kinds of chemically inequivalent ethoxy protons. Similarly, in the case of TEOP, the methyl protons of ethoxy groups appear as a triplet at 1.11 ppm and five sets of 1H resonance lines are observed because of the five different chemically inequivalent protons of the ether chain (Fig. S4, ESI†). All the characteristic 13C resonance lines corresponding to the trialkyl phosphates are observed in the 13C{1H} NMR spectra for both TMOP and TEOP (Fig. S2 and S5, ESI†). Finally, in the 31P NMR spectra, sharp singlet peaks are observed at −1.03 and −1.14 ppm for TMOP and TEOP, respectively (Fig. S3 and S6, ESI†).
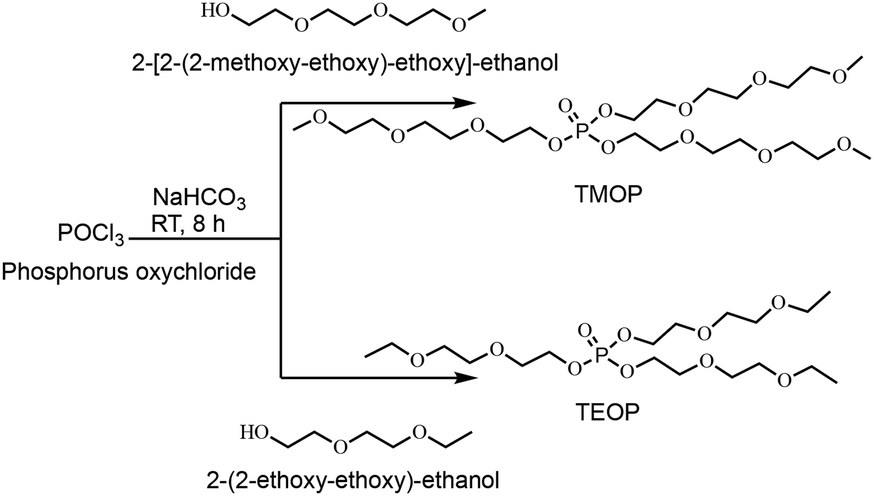 | ||
| Scheme 1 Synthesis of TMOP and TEOP trialkyl phosphates. | ||
In the second step, TMOP and TEOP trialkyl phosphates were reacted with metal iodide in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio to obtain the six liquid alkali metal salts (Scheme 2).
:1 stoichiometric ratio to obtain the six liquid alkali metal salts (Scheme 2).
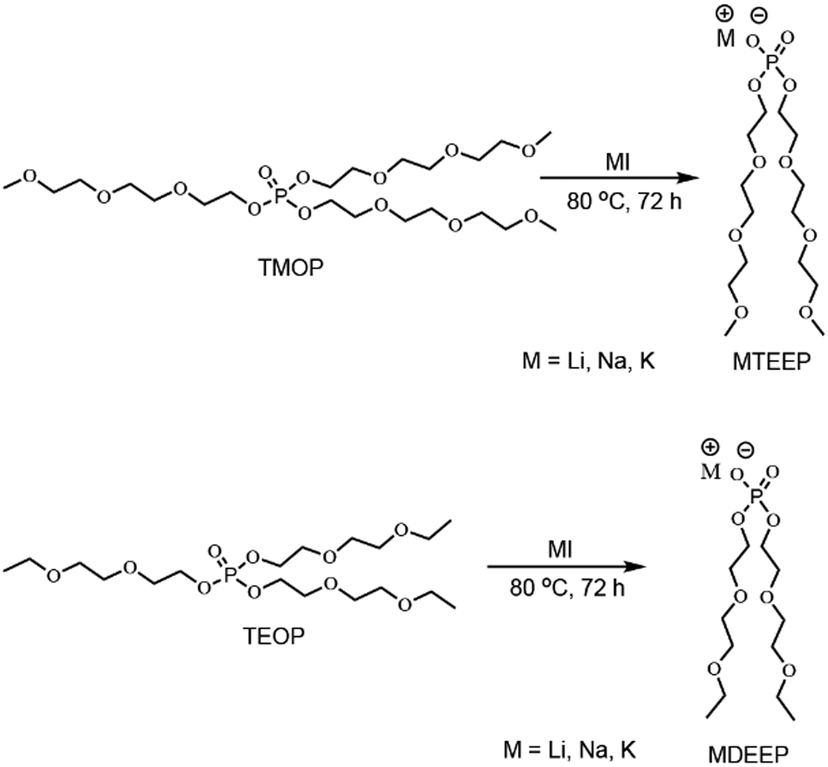 | ||
| Scheme 2 Synthesis of phosphate-based alkali metal salts. | ||
The 31P{1H} NMR spectra of these alkali metal salts all revealed single 31P resonance lines, confirming the purity of the final products. The 31P resonance lines of the salts are deshielded by 1 to 3 ppm as compared with those of the neutral TMOP and TEOP trialkyl phosphates. As expected, the 1H NMR spectrum of NaTEEP shows a sharp singlet at 3.39 ppm for the methoxy protons of the ether chain (Fig. S13, ESI†), while it is a triplet at 1.22 ppm in the case of NaDEEP because the –CH3 is directly attached to a –CH2– group (Fig. S16, ESI†). The methylene groups of NaTEEP give three different sets of 1H resonance lines (Fig. S13, ESI†), while in the case of NaDEEP five sets of 1H resonance lines are observed (Fig. S16, ESI†). Similar trends are found in the 1H NMR spectra of the other five salts. The 13C{1H} NMR spectra of all these salts show all the characteristic resonance lines. 7Li and 23Na NMR show single resonance lines for all salts. All the NMR spectra are shown in the ESI.†
The dynamic TGA data revealed the decomposition temperatures (Td) to be in the range from 225 to 292 °C (Fig. S31a and Table 1, ESI†). The influence of the alkali metal cation is clear as the Li/K-salts are more stable. The reason being that the Na-salts have a two-step decomposition path, <250 °C and >300 °C, while the Li/K-salts have all their weight loss in a single step. Ultimately this must be due to differences in the ion–ion interactions.
| Salt | M W (g mol−1) | T d (°C) | T g (°C) | σ 30 °C (mS cm−1) | σ 60 °C (mS cm−1) |
|---|---|---|---|---|---|
| LiTEEP | 396.30 | 292 | −58 | 0.04 | 0.23 |
| LiDEEP | 336.24 | 289 | −61 | 0.01 | 0.08 |
| NaTEEP | 412.35 | 245 | −29 | 0.04 | 0.14 |
| NaDEEP | 352.29 | 225 | −41 | 0.01 | 0.04 |
| KTEEP | 428.45 | 277 | −39 | 0.03 | 0.24 |
| KDEEP | 368.40 | 268 | −46 | 0.02 | 0.14 |
The DSC traces of all show the presence of glass transition temperatures (Tgs), confirming all salts to be glass forming liquids (Fig. S31b, ESI†) that are significantly affected by the nature of the alkali cation, likely originating from different ion–ion interactions (Table 1). The lower Tgs of the Li-salts indicate slower crystallization rates and stable supercooled states. Overall, these salts present significantly lower Tgs than those of the popular M-TFSI salts with Tgs > 60 °C.32
The alkali metal salts have ionic conductivities >10−5 S cm−1 at 30 °C (Fig. 1a and Table 1), which are slightly lower than those of common fluorinated IL-based electrolytes.33 Specifically, this is higher than for the 1,2,3-triazolate lithium salt24 and comparable with lithium aluminates21 and borates,22,25 which also are liquids at ambient temperature. The TEEP-based salts exhibit the higher ionic conductivities, while decreasing as Na+ > Li+ > K+ in the lower temperature range, and vice-versa in the higher temperature region. This suggests that the K-salts are more easily dissociated as compared to the Li/Na-salts. The VFT parameters show no significant differences in T0, while both σ0 and Eσ are higher for the DEEP-based salts, in accordance with the DSC data and show a higher thermal energy to be required for the same ionic mobility.
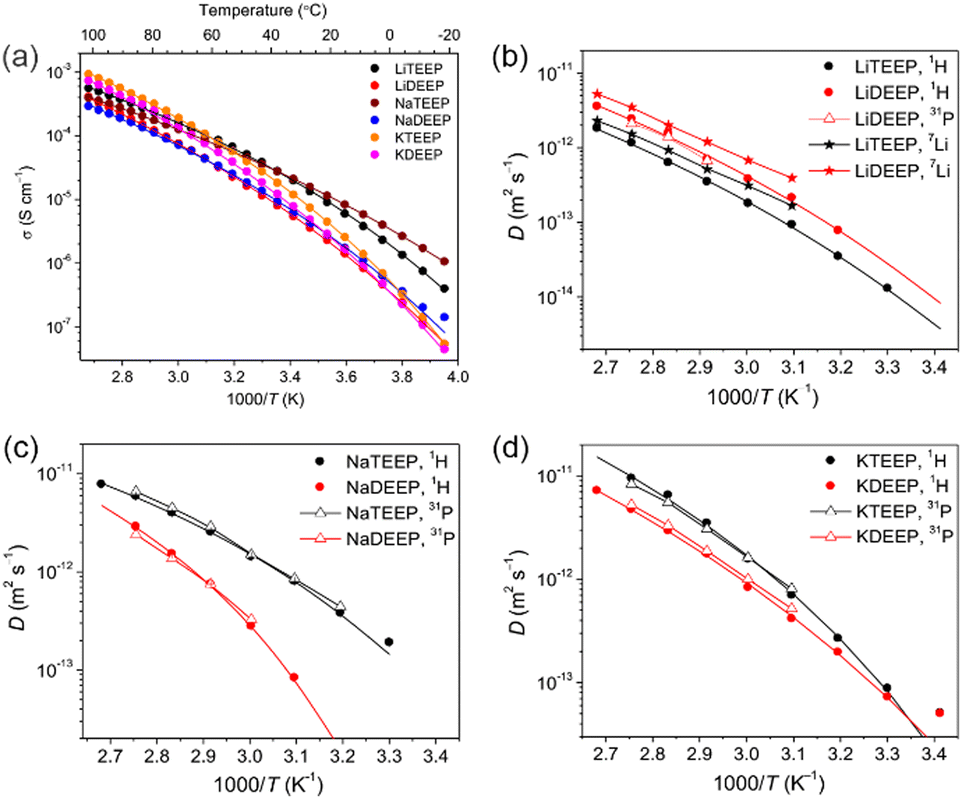 | ||
| Fig. 1 Temperature-dependent ionic conductivities (a), and diffusion coefficients for (b) Li-, (c) Na- and (d) K-salts as measured by 1H, 7Li and 31P NMR spectroscopy. The lines are best VFT fits. | ||
The 1H and 31P NMR spectroscopy diffusion decays (DDs) are close to a single-component form for all our salts and the diffusion coefficients (Ds) obtained match very well throughout the whole temperature range (Fig. 1 and 2). For the Li-salts, the DEEP anion diffuses faster than the TEEP anion (Fig. 1b), although the latter salt exhibits higher ionic conductivity. In addition, the Li+ ions diffuse faster in LiDEEP than in LiTEEP, but vice versa for the Na- and K-salts (Fig. 1c and d), which is in accordance with the ionic conductivities. The unusual mobility of the DEEP anion in the LiDEEP salt might be due to its complicated structure, which is evident from the 31P and 7Li spectra (Fig. S33 and S35, ESI†) and is further outlined below. The nature of alkali metal cations significantly affects the diffusivity of the anions; the TEEP anion diffuses slower in combination with Li+, while the TEEP anion has comparable diffusivities when coupled with Na+ or K+ (Fig. 2a).
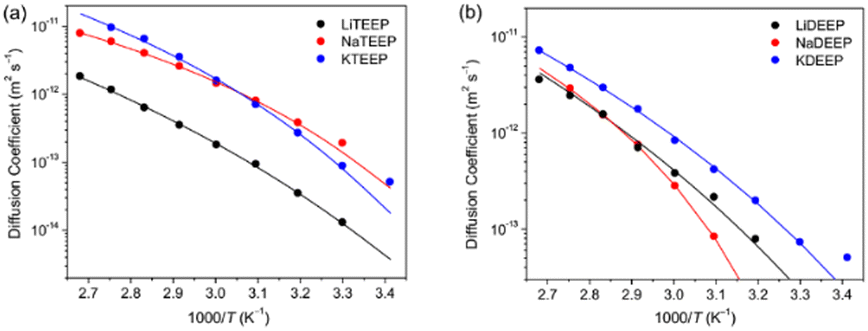 | ||
| Fig. 2 Effect of alkali metal cations on the diffusion coefficients of the anions (a) TEEP and (b) DEEP, as derived by 1H NMR spectroscopy. | ||
On the other hand, the DEEP anion combined with K+ diffuses faster than when combined with Li+ or Na+ (Fig. 2b), which is in accordance with simulations of both aqueous and non-aqueous Li+, Na+ and K+ electrolytes.34,35
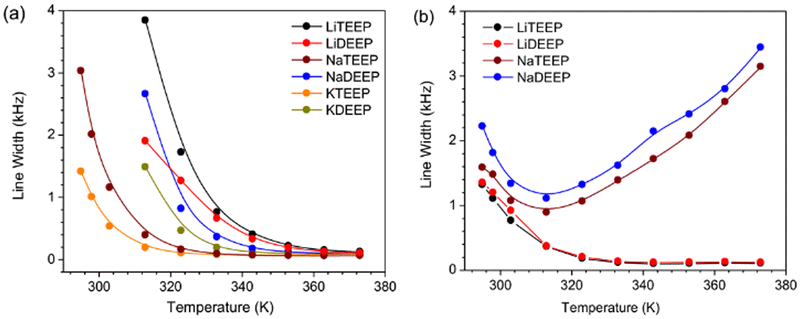 | ||
| Fig. 3 Line widths (FWHM) of (a) the 31P NMR and (b) 7Li and 23Na NMR spectra of our six salts as a function of temperature. | ||
From the multinuclear (31P, 7Li, and 23Na) NMR spectra we first find that the full width at half-maximum (FWHM) of the 31P NMR spectra are broader for all salts at lower temperatures and narrower with increasing temperature, suggesting acceleration of reorientations (Fig. 3a), while the same behaviour for the 7Li NMR spectra (Fig. 3b) suggests faster Li+ ions and, possibly, changes in the local environments of both the 31P and 7Li nuclei.
The 23Na NMR spectra, however, show a sharp FWHM decrease followed by an increase, which indicates a strong influence of quadrupolar interactions leading to ion dynamics, as 23Na has a larger quadrupolar moment than 7Li. A similar trend for NaTFSI containing IL-based electrolytes was suggested to be due to motional broadening.33 The sharp up field chemical shift in the 31P NMR spectra as a function of temperature (Fig. S32 and S33, ESI†) suggests weakened ion–ion interactions and increased mobility. In contrast, no significant changes are observed in the 7Li NMR spectra (Fig. S32b and S35, ESI†), while there are substantial down-shifts in the 23Na NMR spectra (Fig. S32b and S34, ESI†). All combined, this suggests that the Na-salts are readily dissociated at increased temperatures. LiDEEP shows single resonance lines at lower temperatures, but an additional resonance line in both the 7Li and the 31P NMR spectra at higher temperatures, indicative of two phases being present (Fig. S33 and S35, ESI†). Subsequently dissolving LiDEEP in CDCl3 renders only single resonance lines confirming that the additional peaks are neither from any impurity nor any decomposition product.
Turning to the FTIR spectra, these foremost show significant changes in the characteristic stretching frequencies of the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, PO3, O–P–O and P–O–C groups, when comparing the salts with the neutral intermediates and as a function of cation (Fig. S36, ESI†). The symmetric stretching band of the PO group is shifted to lower wavenumbers upon cation interactions of different strengths (Fig. S36a, ESI†). A similar trend is observed for the DEEP-based salts (Fig. S36d, ESI†). Similarly, the PO3 stretching band at 1026 cm−1 shifts based on the cation and from a neutral compound to a salt (Fig. S36b and e, ESI†). Finally, the band at 978 cm−1 (O–P–O stretching mode) for TMOP and TEOP down-shifts similarly, as well as the weak bands at 849 and 835 cm−1 ((P–O)–C stretching modes) (Fig. S36b and e, ESI†). Notably, both the O–P–O and (P–O)–C shifts indicate stronger interactions with Na+ and K+ cations (Fig. S36c and f, ESI†).
O, PO3, O–P–O and P–O–C groups, when comparing the salts with the neutral intermediates and as a function of cation (Fig. S36, ESI†). The symmetric stretching band of the PO group is shifted to lower wavenumbers upon cation interactions of different strengths (Fig. S36a, ESI†). A similar trend is observed for the DEEP-based salts (Fig. S36d, ESI†). Similarly, the PO3 stretching band at 1026 cm−1 shifts based on the cation and from a neutral compound to a salt (Fig. S36b and e, ESI†). Finally, the band at 978 cm−1 (O–P–O stretching mode) for TMOP and TEOP down-shifts similarly, as well as the weak bands at 849 and 835 cm−1 ((P–O)–C stretching modes) (Fig. S36b and e, ESI†). Notably, both the O–P–O and (P–O)–C shifts indicate stronger interactions with Na+ and K+ cations (Fig. S36c and f, ESI†).
For basic assessment of the ESW, cathodic and anodic LSV scans were performed to determine both the reduction of the alkali cations and the oxidation of the phosphate anions. No cathodic peaks are observed for the former, while the latter reveal excellent resistance to oxidation (Fig. 4 and Table S3, ESI†). That the M-DEEP salts outperform the M-TEEP salts might be due to the stronger resonance or mesomeric effect ability of the ethoxy group, as opposed to the methoxy group of TEEP, attached to the oligoether chain, as the higher the mesomeric effect, the higher the electrochemical stability.36 The effect is somewhat less pronounced for NaDEEP when using GC electrodes and overall all the salts exhibit significantly higher oxidative stability using the GC electrodes.
 | ||
| Fig. 4 Anodic LSV scans for our six salts using Pt (a–c) and GC (d–f) WEs, respectively, and a scan rate of 1 mV s−1 at 20 °C. | ||
Moving to the single, proof-of-concept, alkali battery assessment, the Li/LiTEEP/LTO cell shows the characteristic plateau of LTO at 1.5 V vs. Li+/Li° (Fig. 5). Clearly, the full capacity of the electrode cannot be reached, likely due to viscosity-limited penetration of the electrode by the electrolyte, and hence only a small portion of the electrode is active. The capacity increases as a function of temperature (60 °C), but while we can run 10 cycles, this results in large capacity fading – either due to side-reactions, such as electrolyte decomposition, or to clogging of the electrode pores, leading to even less accessibility to the active material.
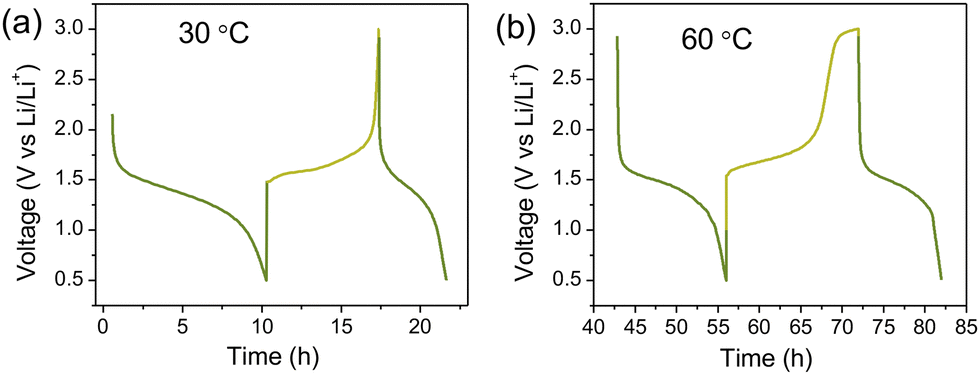 | ||
| Fig. 5 Galvanostatic cycling of Li/LiTEEP/LTO cells using 0.025 mA at (a) 30 °C and (b) 60 °C. | ||
To conclude, these ambient temperature liquid alkali metal salts present unique possibilities for fluorine-free and solvent-free electrolytes. The salts provide appreciable thermal and electrochemical stabilities, low glass transition temperatures, and moderate ionic conductivities – which do increase with temperature. The Na+ cation seems to interact differently with the anions as compared to both Li+ and K+. There is some fundamental promise for the salts to be applicable to Li/Na/K batteries. A caveat is that they all contain more water than most inorganic salts used for batteries and this, alongside the stability vs. reduction and stripping/plating behaviour, is an area for further development and study.
S. B.: synthesis, characterization, writing original draft. M. A.: characterization, editing. A. F.: NMR diffusometry, editing. L. C. L.: electrochemistry, editing. F. U. S.: supervision, conceptualization, methodology, editing. P. J.: conceptualization, methodology, editing. The Kempe Foundation in Memory of J. C. and Seth M. Kempe are gratefully acknowledged for the stipend for S. B. (grant number: SMK-1945). The Swedish Energy Agency (project number: 48194-1) is acknowledged for supporting this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Zaghib and C. M. Julien, Rechargeable lithium batteries for energy storage in smart grids in Woodhead Publishing Series in Energy 81, Elsevier, 2015 Search PubMed.
- P. J. Loftus, A. M. Cohen, J. C. S. Long and J. D. Jenkins, Wiley Interdiscip. Rev.: Clim. Change, 2015, 6, 93 CrossRef PubMed.
- L. S. Martins, L. F. Guimaraes, A. B. B. Junior, J. A. S. Tenorio and D. C. R. Espinosa, J. Environ. Manage., 2021, 295, 113091 CrossRef PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- Y. Liu, R. Zhang, J. Wang and Y. Wang, iScience, 2021, 24, 102332 CrossRef CAS PubMed.
- N. Tapia-Ruiz, et al. , JPhys Energy, 2021, 3, 031503 CrossRef CAS.
- V. Anoopkumar, B. John and T. D. Mercy, ACS Appl. Eng. Mater., 2020, 3, 9478 CrossRef CAS.
- C. Jiang, H. Li and C. Wang, Sci. Bull., 2017, 62, 1473 CrossRef CAS PubMed.
- W. Teng, et al. , Energy Environ. Mater., 2022, 1–28 Search PubMed.
- Y. Gao, et al. , Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- R. Zettl, S. Lunghammer, B. Gadermaier, A. Boulaoued, P. Johansson, H. M. R. Wilkening and I. Hanzu, Adv. Energy Mater., 2021, 11, 2170060 CrossRef CAS.
- E. Knipping, C. Aucher, G. Guirado and L. Aubouya, New J. Chem., 2018, 42, 4693–4699 RSC.
- J. Zeng, J. R. Nair, C. Francia, S. Bodoardo and N. Penazzi, Int. J. Electrochem. Sci., 2013, 8, 3912–3927 CAS.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- S.-M. Han, J.-H. Kim and D.-W. Kim, J. Electrochem. Soc., 2015, 162, A3103–A3109 CrossRef CAS.
- S. Ferrari, et al. , J. Electrochem. Soc., 2014, 162, A3001–A3006 CrossRef.
- C. J. Allen, et al. , J. Phys. Chem. C, 2012, 116, 20755–20764 CrossRef CAS.
- D. Bresser, E. Paillard and S. J. Passerini, Electrochem. Sci. Technol., 2014, 5, 37–44 CAS.
- T. Fujinami and Y. Buzoujima, J. Power Sources, 2003, 119–121, 438–441 CrossRef CAS.
- H. Shobukawa, H. Tokuda, S. I. Tabata and M. Watanabe, Electrochim. Acta, 2004, 50, 1–5 CrossRef.
- O. Zech, M. Kellermeier, S. Thomaier, E. Maurer, R. Klein, C. Schreiner and W. Kunz, Chem. – Eur. J., 2009, 15, 1341–1345 CrossRef CAS PubMed.
- D. Flachard, J. Rolland, M. M. Obadia, A. Serghei, R. Bouchet and E. Drockenmuller, Chem. Commun., 2018, 54, 9035–9038 RSC.
- G. Guzmán-González, M. Alvarez-Tirado, J. L. Olmedo-Martínez, M. L. Picchio, N. Casado, M. Forsyth and D. Mecerreyes, Adv. Energy Mater., 2022, 2202974 Search PubMed.
- S. H. Strauss, Chem. Rev., 1993, 93, 927–942 CrossRef CAS.
- F. Philippi and T. Welton, Phys. Chem. Chem. Phys., 2021, 23, 6993–7021 RSC.
- J. Forero-Saboya, E. Hosseini-Bab-Anari, M. E. Abdelhamid, K. Moth-Poulsen and P. Johansson, Chem. Commun., 2019, 55, 632 RSC.
- M. J. Marczewski, B. Stanje, I. Hanzu, M. Wilkening and P. Johansson, Phys. Chem. Chem. Phys., 2014, 16, 12341–12349 RSC.
- P. Shi, et al. , Chem. Commun., 2018, 54, 4453–4456 RSC.
- F. U. Shah, O. I. Gnezdilov, I. A. Khan, A. Filippov, N. A. Slad and P. Johansson, J. Phys. Chem. B, 2020, 124, 9690–9700 CrossRef CAS PubMed.
- R. Hagiwara, K. Matsumoto, K. Tamaki, T. Nohira and T. Goto, US. Pat., 2009/0212743 A1, 2009 Search PubMed.
- M. Hilder, M. Gras, C. R. Pope, M. Kar, D. R. MacFarlane, M. Forsyth and L. A. O’Dell, et al. , Phys. Chem. Chem. Phys., 2017, 19, 17461–17468 RSC.
- T. A. Pham, K. E. Kweon, A. Samanta, V. Lordi and J. E. Pask, J. Phys. Chem. C, 2017, 121, 21913–21920 CrossRef CAS.
- T. Ikeda, M. Boero and K. Terakura, J. Chem. Phys., 2007, 126, 034501 CrossRef PubMed.
- K. L. Browning, R. L. Sacci and G. M. Veith, J. Electrochem. Soc., 2017, 164, A580–A586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00318c |
| This journal is © The Royal Society of Chemistry 2023 |