
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable regulation of translation by harnessing the CRISPR-Cas13 system†
Roser
Montagud-Martínez
,
Rosa
Márquez-Costa
and
Guillermo
Rodrigo
 *
*
Institute for Integrative Systems Biology (I2SysBio), CSIC – University of Valencia, 46980, Paterna, Spain. E-mail: guillermo.rodrigo@csic.es
First published on 30th January 2023
Abstract
The ability to control protein expression at both the transcriptional and post-transcriptional levels is instrumental for the cell to integrate multiple molecular signals and then reach high operational sophistication. Although challenging, fully artificial regulations at different levels are required for boosting systems and synthetic biology. Here, we report the development of a novel framework to regulate translation by repurposing the CRISPR-Cas13 immune system, which uses an RNA-guided ribonuclease. By exploiting a cell-free expression system for prototyping gene regulatory structures, our results demonstrate that CRISPR-dCas13a ribonucleoproteins (d means catalytically dead) can be programmed to repress or activate translation initiation. The performance assessment of the engineered systems also revealed guide RNA design principles. Moreover, we show that the system can work in vivo. This development complements the ability to regulate transcription with other CRISPR-Cas systems and offers potential applications.
Living organisms of ranging complexity have evolved to not only rely on transcription regulation, but on different mechanisms acting after RNA is made, in order to fine-tune protein expression1 and, perhaps more importantly, get access to gene regulatory architectures of high integration capacity.2 The latter means architectures to facilitate the combination of external and internal signals in the circuits, as well as to enable the divergence of co-regulated gene expressions at the transcriptional level.3 In this regard, to boost the development of transformative technologies through the application of synthetic gene circuits,4 a combination of reliable control mechanisms at different points of the genetic information flow is required to achieve the degree of integrability, and then functional complexity, found in nature. However, much of the work in the field has been focused on transcription regulation. Certainly, the repurposing of the CRISPR-Cas9 system (CRISPR stands for clustered regularly interspaced short palindromic repeats)5 to regulate transcription in a programmable way, for repression and activation, constituted a major breakthrough.6,7 Yet, counterparts at other points (e.g., to regulate translation) combining RNA programmability with protein stability are lacking.
Of note, the engineering of small RNAs (sRNAs) able to regulate gene expression at a post-transcriptional level in prokaryotes has reached great success in recent years,8–11 such as to end with circuits with sufficient operational sophistication.12,13 These sRNAs control trans ribosome binding and are easily combinable with transcription factors to facilitate combinatorial regulation. In addition, by taking advantage of a singular versatility, signal ribotransducers have been implemented through the use of aptamers responding to small molecules14 and novel transcriptional riboregulators have been developed.15,16 Nevertheless, these elements often require a great excess of expression to function, require genetic engineering to be interfaced with natural gene expression programs, and are difficult to export to eukaryotic contexts.
Given that CRISPR-Cas systems represent paradigmatic examples of concurrent programmability, stability, and specificity, together with high functional diversity,17 we decided to exploit a suitable immune system of very recent discovery (viz., CRISPR-Cas13)18 to develop a new framework for the programmable regulation of translation. To this end, we worked with a catalytically dead version of a Cas13a protein (dCas13a). In particular, we chose the RNA-guided ribonuclease (RNase) from Leptotrichia wadei, as this has been shown to be one of the most effective for RNA targeting and independent of a protospacer flanking site.19 This way, the resulting ribonucleoprotein formed by the small guide RNA (sgRNA) and dCas13a can be programmed to target any messenger RNA (mRNA) without cleavage. To test this designer gene regulatory mechanism, we took advantage in this work of a cell-free expression system derived from Escherichia coli, which includes the core machineries for transcription, translation, and macromolecular degradation.20 This allowed us to assess the performance of the system from a chemical biology perspective (i.e., with a mechanistic focus) by a rapid and controlled characterization of multiple sgRNAs.21 We also transformed E. coli cells with suitable plasmids to show the engineered regulation in vivo through colony assays.
We first programmed the CRISPR-dCas13a ribonucleoprotein to repress the translation initiation of an active mRNA (Fig. 1a). In this case, we used a superfolder green fluorescent protein (sfGFP) as a reporter element.22 For that, the spacer of the sgRNA was designed to target the Shine–Dalgarno region and the start codon of the sfGFP mRNA, thereby preventing ribosome binding (Fig. 1b).10 We found a significant reduction in protein expression (about 50–80% depending on the experience, as we noted variability in the readout) as a consequence of the mRNA targeting by the ribonucleoprotein (Fig. 1c; see also Fig. S1, ESI† where the impact of in vitro dCas13a expression on regulation was assessed and Note S1, ESI†). The sgRNA was expressed from a constitutive promoter (J23119). In the absence of dCas13a, the naked sgRNA was not able to repress translation, despite having complementarity with the mRNA (ΔG = −57.3 kcal mol−1, predicted with ViennaRNA23). This suggests that the resulting ribonucleoprotein displays the required stability to interfere with the ribosome, and that dCas13a might then be seen as an RNA chaperone independent of the cell machinery.24 We also noticed that the repression degree scales with the sgRNA expression level (modulated by playing with different plasmid concentrations; Fig. 1d). Indeed, because the mRNA is constantly produced, the regulation is only revealed when the ribonucleoprotein is in sufficient excess to compete with the ribosome and RNases for targeting the mRNA; a more challenging scenario than in the case of transcriptional repression.6 In addition, to assess the specificity of the regulation, we introduced a series of point mutations in the spacer of the sgRNA. Together with a non-targeting sgRNA (having a random spacer), we found a significant tolerance to mismatches (Fig. 1e; this tolerance appears to be lesser in the seed region). Previous studies of mRNA knocking down with the CRISPR-Cas13a ribonucleoprotein (catalytically active) also revealed some tolerance to single mismatches, but not to multiple.19 To get mechanistic insight about the functioning, we used an RNA beacon appropriately labelled with a fluorophore and a dark quencher,25 mimicking the mRNA. This allowed us to directly prove the interaction between the ribonucleoprotein and its target nucleic acid (Fig. 1f).
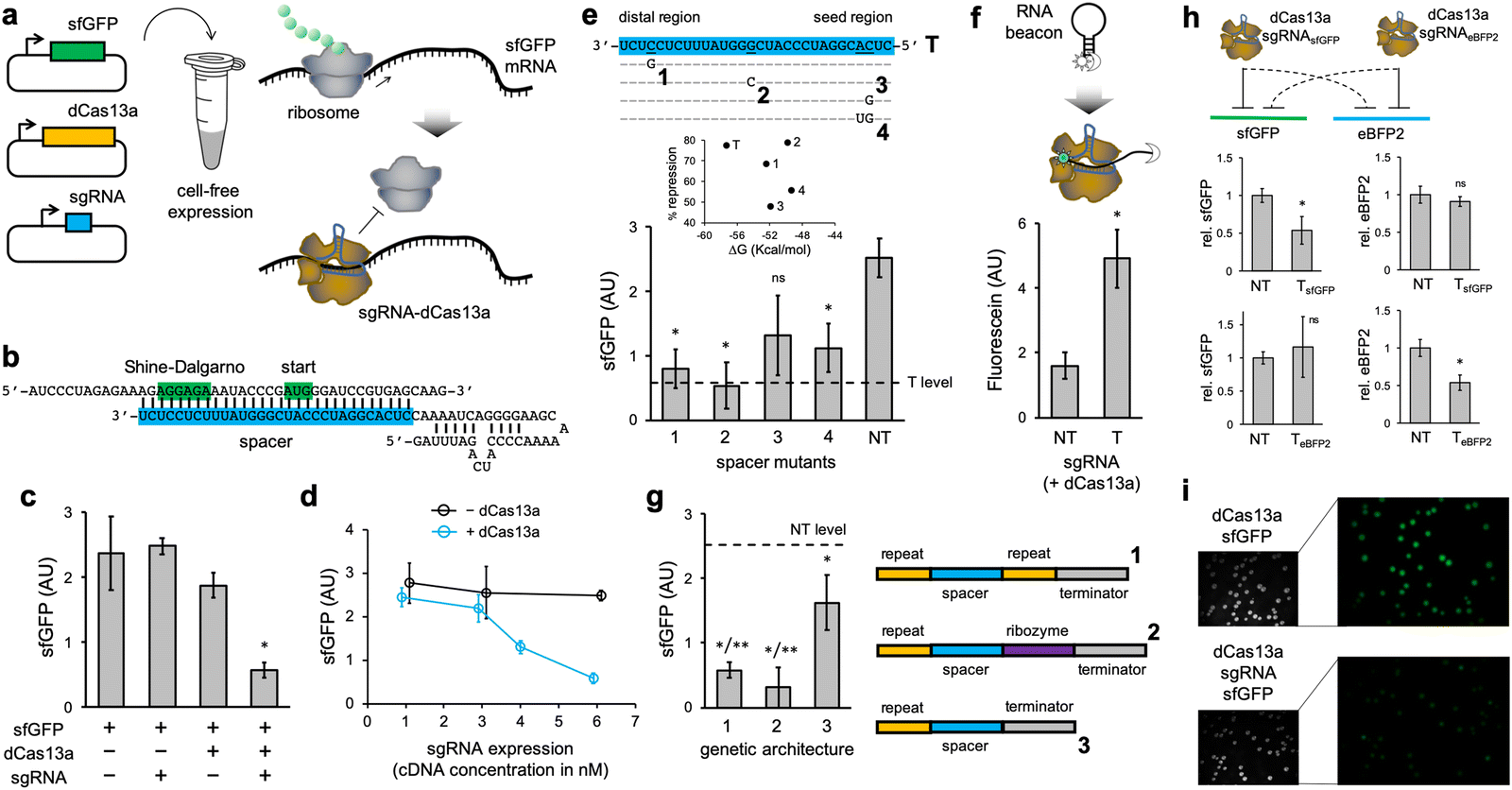 | ||
| Fig. 1 Repression of translation initiation with a programmed CRISPR-dCas13a ribonucleoprotein. (a) Schematics of the gene expression system employed, together with an illustration of the intended regulation. The ribonucleoprotein interacts with the 5′ UTR of the mRNA to prevent ribosome binding and then translation initiation. (b) RNA sequence and structure details of the sgRNA–mRNA interaction. The Shine–Dalgarno box and the start codon are shown in green, while the spacer of the sgRNA is shown in blue. (c) sfGFP expression for different combinations of elements, showing translation initiation control (*with respect to any other condition). (d) Regulatory activity for different sgRNA expression levels, with and without dCas13a. (e) Effect of point mutations in the spacer of the sgRNA (*with respect to T, nsnot significant). On top, sequence details. The subplot relates the percentage of repression with the free energy of the sgRNA–mRNA interaction. T means targeting spacer with perfect complementarity, while NT means non-targeting spacer. (f) Characterization of the interaction through the use of an RNA beacon labelled with a fluorophore (sun icon) and a dark quencher (moon icon; *with respect to NT). (g) Effect of the genetic architecture employed for sgRNA expression (*with respect to NT, **with respect to architecture 3). (h) Characterization of a new designer system using eBFP2 as a reporter. sfGFP or eBFP2 expression for different conditions, showing orthogonal regulation with different sgRNAs (*with respect to NT). (i) In vivo results. Images of E. coli colonies harboring plasmids to express the sfGFP, the dCas13a, and the sgRNA (where indicated). Fluorescence and bright field images are shown. Error bars correspond to standard deviations (n = 3). Statistical significance assessed in all cases by a Welch's t-test (two-tailed P < 0.05). | ||
To produce those sgRNAs, the corresponding transcriptional units were implemented with two dCas13a recognition motifs (direct repeats), as it occurs in nature. Whilst the wild-type CRISPR protein has two distinct RNase activities, one to process the sgRNA precursor and another to cleave the target mRNA,26 dCas13a is only catalytically dead in terms of mRNA cleavage. We then decided to inspect the effect on the regulatory activity of the particular genetic architecture to produce a mature sgRNA (Fig. 1g). Our results show that the use of the Hepatitis delta virus ribozyme27 at the 3′ end of the sgRNA leads to a fully functional element. Of note, self-processing ribozymes have already been used to get spotless Cas9-related sgRNAs.28 However, a sgRNA with no 3′ end processing loses regulatory power, although it is still able to exert some repression. Arguably, the transcriptional terminator hinders the ability of the spacer to guide the ribonucleoprotein to its target.
To test the ability of exerting orthogonal regulation, we engineered a new system using the enhanced blue fluorescent protein 2 (eBFP2) as a reporter.29 The 5′ untranslated region (UTR) and the spacer of the sgRNA were redesigned (Fig. S2a, ESI†). We found that the ribonucleoproteins only repressed in a significant manner their cognate mRNAs (Fig. 1h). In addition, we expressed the sfGFP-based regulatory system in E. coli from plasmids. The fluorescence of E. coli cells expressing the sgRNA was lower than that of cells not expressing it, as imaged in colony assays, thereby indicating translation repression (Fig. 1i). Fluorescence was quantified using an image processing software (Fig. S3a, ESI†). These results show the utility of the CRISPR-Cas13 system to program gene expression in vivo.
Subsequently, we programmed the ribonucleoprotein to activate the translation initiation of a cis-repressed mRNA (Fig. 2a). That is, an mRNA with a structured 5′ UTR that sequesters the Shine–Dalgarno region and the start codon, hence preventing ribosome binding. Here, we considered a structured 5′ UTR previously designed (dubbed toehold switch),9 which was cloned to control the expression of sfGFP. The spacer of the sgRNA was designed to induce a conformational change in the 5′ UTR upon interaction, resulting in the exposition of the Shine–Dalgarno region and the start codon to the solvent (Fig. 2b). We ensured that the free energy release associated with the sgRNA–mRNA interaction was sizeable enough (ΔG = −27.8 kcal mol−1). In this case, the distal region of the spacer nucleates the interaction, as it is complementary to a region initially unpaired in the 5′ UTR. The regulatory mode determines such a free energy release, which is lesser in the case of activation of translation than of repression as a consequence of a structured 5′ UTR. Importantly, we found a significant increase in protein expression (about 200%) as a consequence of the action of the ribonucleoprotein (Fig. 2c; see also Fig. S4, ESI† where the maximal possible expression was measured). The sgRNA was again expressed from a constitutive promoter. In contrast to the case of repression of initiation, the naked sgRNA was now able to regulate expression, although to a lower extent than with dCas13a (about 80% increase). We attribute this fact to the lack of competition with the ribosome for binding, as the mRNA is produced to be translationally off (Fig. 2d).24 Of note, our results indicate that it is possible to target highly structured mRNA regions with CRISPR-Cas13 systems, in contrast to previous results with a catalytically active ribonucleoprotein,18 provided energetic criteria are met.
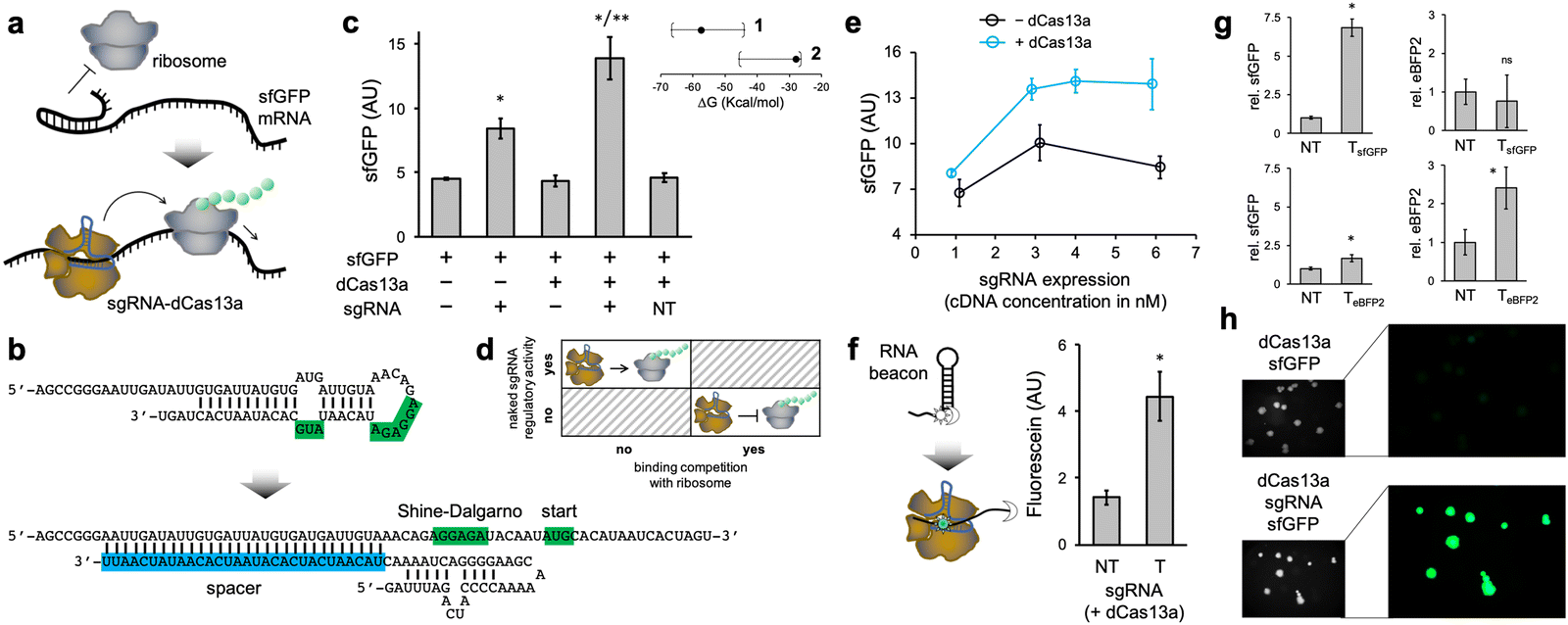 | ||
| Fig. 2 Activation of translation initiation with a programmed CRISPR-dCas13a ribonucleoprotein. (a) Illustration of the intended regulation. The ribonucleoprotein interacts with the structured 5′ UTR of the mRNA to promote ribosome binding and then translation initiation. (b) RNA sequence and structure detail of the sgRNA–mRNA interaction. The Shine–Dalgarno box and the start codon are shown in green, while the spacer of the sgRNA is shown in blue. (c) sfGFP expression for different combinations of elements, showing translation initiation control (*with respect to NT, **with respect to the condition with naked sgRNA). T means targeting spacer, while NT means non-targeting spacer. The subplot shows the distribution of the free energy of the sgRNA–mRNA interaction (95% confidence interval, from randomly generated sequences) for each mechanism of translation control (1: repression of initiation, 2: activation of initiation). Points indicate the actual values of the designed systems. (d) Summary of fundamental differences between the repression and activation mechanisms. (e) Regulatory activity for different sgRNA expression levels, with and without dCas13a. (f) Characterization of the interaction through the use of an RNA beacon labelled with a fluorophore (sun icon) and a dark quencher (moon icon; *with respect to NT). (g) Characterization of a new designer system using eBFP2 as a reporter. sfGFP or eBFP2 expression for different conditions, showing rough orthogonal regulation with different sgRNAs (*with respect to NT). (h) In vivo results. Images of E. coli colonies harboring plasmids to express the sfGFP, the dCas13a, and the sgRNA (where indicated). Fluorescence and bright field images are shown. Error bars correspond to standard deviations (n = 3). Statistical significance assessed in all cases by a Welch's t-test (two-tailed P < 0.05). | ||
In addition, we evaluated the dependence on the sgRNA expression level (again by playing with different plasmid concentrations), finding a lower requirement to achieve significant regulatory activity than in the case of repression (Fig. 2e). Next, we used a new, more structured RNA beacon appropriately labelled with a fluorophore and a dark quencher, mimicking the mRNA, to mechanistically substantiate the interaction between the ribonucleoprotein and its target nucleic acid (Fig. 2f).
Lastly, we engineered a new system using eBFP2 to test the orthogonal regulation in the case of activation, redesigning the 5′ UTR and the sgRNA (Fig. S2b, ESI†). We found that the ribonucleoproteins substantially activated their cognate mRNAs (Fig. 2g). We also observed that the eBFP2-targeting ribonucleoprotein was able to faintly activate sfGFP expression, but such a cross-regulation was not noticed in the opposite case (i.e., the sfGFP-targeting ribonucleoprotein was really orthogonal to eBFP2 expression). In addition, we expressed the sfGFP-based activation system in E. coli from plasmids. The fluorescence of E. coli cells expressing the sgRNA was higher than that of cells not expressing it, as imaged in colony assays, thereby indicating translation activation (Fig. 2h; see also Fig. S3b, ESI† for quantitative results).
Beyond its native function of RNA silencing, the CRISPR-Cas13 system has already been repurposed for subcellular RNA tracking,19 precise RNA edition,30 reading RNA methylation (with impact on protein expression),31 and ultrasensitive RNA detection.32 This work represents an expansion of this CRISPR universe aimed at controlling ribosome activity (to program gene expression at the post-transcriptional level). Hence, together with established regulators of transcription, this development shows great promise for systems and synthetic biology. For instance, we may expect the implementation of synthetic circuits that are functionally complex but have a reduced number of genes, noting that with only transcriptional regulations multiple layers are required to achieve combinatorial logic behaviors.33,34 Furthermore, we may anticipate an increased ability to successfully fine tune gene expression to obtain functional circuits, beyond the genetic engineering required to find the appropriate cis-regulatory elements, such as ribosome binding sites.35,36 In addition, to implement designer circuits, a programmed CRISPR-dCas13a ribonucleoprotein might be used to systematically perturb natural gene networks in order to discover fundamental principles of biological organization,37 as it has been done with CRISPR interference.38 There are several scenarios in which a regulation of translation with a removable element acting in trans would be more adequate, such as to reveal the role (or the optimal expression level) of a specific gene within a bacterial operon, of special importance in metabolic engineering frameworks,39 or to discover whether a transcript is functional beyond being a mere template for protein production.40
Our results come from the use of a cell-free expression system, allowing the regulatory mechanism to be assessed in a controlled way. Designer systems were also assayed in living cells (Note S2, ESI†), so the implementation of functional circuits including these elements represents subsequent steps. Nonetheless, the expression of some Cas13 proteins may be toxic for the cells,41 which would enforce the adoption of rational design measures to reduce the imposed burden42 or to perform a functional screening of different class 2, type VI systems.17 It is important to also note that CRISPR-Cas systems require an excess of the ribonucleoprotein with respect to the target nucleic acid to work.43 In this regard, the regulation with CRISPR-Cas13 would be more efficient on genes with lower transcription and higher translation rates than on genes with the opposite expression pattern. In any case, a moderate dynamic regulatory range (i.e., the fold change in protein expression) is expected with respect to transcriptional control systems, as the target is constantly produced and degraded. Overall, as we successfully repurpose key molecular mechanisms from which living systems are built off, we are better positioned to offer innovative solutions in biotechnology and biomedicine.44
We thank V. Noireaux (U. Minnesota) for useful help with the myTXTL system. This work was supported by the grants PGC2018-101410-B-I00 from the Spanish Ministry of Science, Innovation, and Universities (co-financed by the European Regional Development Fund) and SEJI/2020/011 from the Regional Government of Valencia (to GR). RMC held a predoctoral fellowship (PRE2019-088531). Publication fees covered by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Vogel and E. M. Marcotte, Nat. Rev. Genet., 2012, 13, 227–232 CrossRef CAS PubMed.
- N. J. Martinez, et al. , Genes Dev., 2008, 22, 2535–2549 CrossRef CAS PubMed.
- S. Balaji, et al. , J. Mol. Biol., 2006, 360, 213–227 CrossRef CAS PubMed.
- W. Weber and M. Fussenegger, Nat. Rev. Genet., 2012, 13, 21–35 CrossRef CAS PubMed.
- M. Jinek, et al. , Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- L. S. Qi, et al. , Cell, 2013, 152, 1173–1183 CrossRef CAS PubMed.
- J. G. Zalatan, et al. , Cell, 2015, 160, 339–350 CrossRef CAS PubMed.
- G. Rodrigo, et al. , Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15271–15276 CrossRef CAS.
- A. A. Green, et al. , Cell, 2014, 159, 925–939 CrossRef CAS.
- A. Ghodasara and C. A. Voigt, Nucleic Acids Res., 2017, 45, 8116–8127 CrossRef CAS.
- J. Kim, et al. , Nat. Chem. Biol., 2019, 15, 1173–1182 CrossRef CAS PubMed.
- A. E. Friedland, et al. , Science, 2009, 324, 1199–1202 CrossRef CAS PubMed.
- A. Rosado, et al. , PLoS Genet., 2018, 14, e1007548 CrossRef PubMed.
- S. Shen, et al. , Nucleic Acids Res., 2015, 43, 5158–5170 CrossRef CAS PubMed.
- C. C. Liu, et al. , Nat. Methods, 2012, 9, 1088–1094 CrossRef CAS PubMed.
- J. Chappell, et al. , Nat. Chem. Biol., 2015, 11, 214–220 CrossRef CAS PubMed.
- K. S. Makarova, et al. , Nat. Rev. Microbiol., 2020, 18, 67–83 CrossRef CAS PubMed.
- O. O. Abudayyeh, et al. , Science, 2016, 353, aaf5573 CrossRef PubMed.
- O. O. Abudayyeh, et al. , Nature, 2017, 550, 280–284 CrossRef PubMed.
- J. Garamella, et al. , ACS Synth. Biol., 2016, 5, 344–355 CrossRef CAS PubMed.
- R. Marshall, et al. , Mol. Cell, 2018, 69, 146–157 CrossRef CAS PubMed.
- J. D. Pédelacq, et al. , Nat. Biotechnol., 2006, 24, 79–88 CrossRef PubMed.
- A. R. Gruber, et al. , Nucleic Acids Res., 2008, 36, W70–W74 CrossRef CAS PubMed.
- G. Desnoyers, et al. , Trends Genet., 2013, 29, 92–98 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS PubMed.
- A. East-Seletsky, et al. , Nature, 2016, 538, 270–273 CrossRef CAS PubMed.
- A. R. Ferré-D’Amaré, et al. , Nature, 1998, 395, 567–574 CrossRef PubMed.
- Y. Gao and Y. Zhao, J. Integr. Plant Biol., 2014, 56, 343–349 CrossRef CAS PubMed.
- H. W. Ai, et al. , Biochemistry, 2007, 46, 5904–5910 CrossRef CAS PubMed.
- D. B. T. Cox, et al. , Science, 2017, 358, 1019–1027 CrossRef CAS PubMed.
- S. Rauch, et al. , J. Am. Chem. Soc., 2018, 140, 11974–11981 CrossRef CAS PubMed.
- J. S. Gootenberg, et al. , Science, 2017, 356, 438–442 CrossRef CAS PubMed.
- A. A. Nielsen, et al. , Science, 2016, 352, aac7341 CrossRef PubMed.
- M. W. Gander, et al. , Nat. Commun., 2017, 8, 15459 CrossRef CAS PubMed.
- H. M. Salis, et al. , Nat. Biotechnol., 2009, 27, 946–950 CrossRef CAS PubMed.
- R. G. Egbert and E. Klavins, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16817–16822 CrossRef CAS PubMed.
- C. J. Bashor and J. J. Collins, Annu. Rev. Biophys., 2018, 47, 399–423 CrossRef CAS.
- J. M. Peters, et al. , Cell, 2016, 165, 1493–1506 CrossRef CAS.
- B. F. Pfleger, et al. , Nat. Biotechnol., 2006, 24, 1027–1032 CrossRef CAS.
- L. Poliseno, et al. , Nature, 2010, 465, 1033–1038 CrossRef CAS PubMed.
- K. Zhang, et al. , Front. Bioeng. Biotechnol., 2020, 8, 856 CrossRef PubMed.
- O. Borkowski, et al. , Curr. Opin. Microbiol., 2016, 33, 123–130 CrossRef CAS.
- D. L. Jones, et al. , Science, 2017, 357, 1420–1424 CrossRef CAS.
- W. Weber and M. Fussenegger, Nat. Rev. Genet., 2012, 13, 21–35 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc00058c |
| This journal is © The Royal Society of Chemistry 2023 |