
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical C(sp3)–H functionalization of ethers via hydrogen-atom transfer by means of cathodic reduction†
Leonardo
Rapisarda
a,
Andrea
Fermi
 *ab,
Paola
Ceroni
ab,
Riccardo
Giovanelli
ab,
Giulio
Bertuzzi
*ab and
Marco
Bandini
*ab
*ab,
Paola
Ceroni
ab,
Riccardo
Giovanelli
ab,
Giulio
Bertuzzi
*ab and
Marco
Bandini
*ab
aDipartimento di Chimica “Giacomo Ciamician”, Alma Mater Studiorum – Università di Bologna, via Selmi 2, 40126, Bologna, Italy. E-mail: marco.bandini@unibo.it
bCenter for Chemical Catalysis – C3, Alma Mater Studiorum – Università di Bologna Via Selmi 2, 40126, Bologna, Italy
First published on 6th February 2023
Abstract
The chemo- and stereoselective electrochemical allylation/alkylation of ethers is presented via a C(sp3)–H activation event. The electrosynthetic protocol enables the realization of a large library of functionalized ethers (35 examples) in high yields (up to 84%) via cathodic activation of a new type of redox-active carbonate (RAC), capable of triggering HAT (Hydrogen-Atom-Transfer) events through the generation of electrophilic oxy radicals. The process displayed high functional group tolerance and mild reaction conditions. A mechanistic elucidation via voltammetric analysis completes the study.
The chemical manipulation of unreactive C(sp3)–H bonds is among the most rapid synthetic tools to achieve key building blocks from the chemical feedstock. It also represents an extraordinary synthetic challenge, given the inertness of the C–H bonds towards selective functionalizations.1 In this landscape, the “radical approach”, based on Hydrogen-Atom-Transfer (HAT) methodologies, is currently paralleling the well consolidated transition metal catalyzed “two-electron manifold” strategies.2 As a matter of fact, HAT can effectively combine pivotal aspects such as selectivity, simplicity, and sustainability in site-selective C(sp3)–H functionalizations.3
In very recent times, the organic synthetic community has faced the (re)emerging of organic electrosynthesis (i.e. eChem) for the generation and functionalization of radical species.4 However, despite its undoubted advantages in terms of rapid and productive chemical diversification, eChem has been rarely adopted in HAT-based C(sp3)–H functionalizations. As a matter of fact, the field is dominated by halogenation, oxygenation and azidation reactions via anodic Shono oxidation of (mostly) amines (Fig. 1, top a).5 On the contrary, direct intermolecular HAT processes, for the production of key reactive intermediates and subsequent nucleophilic,6 or, more rarely, electrophilic7 trapping have been scarcely documented (Fig. 1, top b). In addition, the few reported strategies proceed through anodic oxidation for the formation of the hydrogen-atom abstractor.
 | ||
| Fig. 1 State of the art in the eChem promoted C–H activation procedures (i.e. direct and indirect anodic oxidation (top)). The present electroreductive methodology (bottom). | ||
The development of complementary electrochemical functionalization of unactivated C–H bonds, triggered by cathodic reduction, would expand significantly the portfolio of chemical diversity accessible through eChem.8 The challenge in this strategy lies in the intrinsic difficulty towards the generation and productive employment of oxidant species as hydrogen atom abstractors in a strongly reductive environment.
Inspired by our recent results on selective radical-based transformations9 and discoveries on the suitability of Morita–Baylis–Hillman (MBH) acetates 1 as electrophilic radical acceptors in eChem allylation strategies,10 we introduce an unprecedented electrochemical allylation/alkylation of simple and abundant ether feedstocks 2, proceeding under cathodic reduction. The strategy relies on a HAT manifold and leads to the discovery of a novel precursor of hydridic hydrogen atom abstractors, prone to HAT events on simple ethers (α-oxy radical A) and capable of overriding further reduction and direct addition to electrophilic MBH 1 (Fig. 1 bottom i vs. ii and iii).11 Importantly, the application of a sacrificial anode strategy would effectively suppress the oxidation of A to the corresponding oxonium cations B.
To primarily test the feasibility of our hypothesis, we selected N-acetoxy-phthalimide 3a as the model HAT reagent.12 Encouragingly, when 3a was subjected to a constant current electrolysis (4 mA, TEABF4 as electrolyte, Cgraph. cathode and Zn anode, 2.5 F mol3a−1), in the presence of 1a and a 2a/DMF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solvent mixture, the desired product 4aa was isolated in 18% yield as a single E isomer (Table 1, entry 1). However, 4aa was obtained in combination with 5aa, arising from the direct addition of the methyl radical (from 3a) onto 1a (35% yield), along with 6a (21% yield), as the result of an undesired reduction of 1a.
:2) solvent mixture, the desired product 4aa was isolated in 18% yield as a single E isomer (Table 1, entry 1). However, 4aa was obtained in combination with 5aa, arising from the direct addition of the methyl radical (from 3a) onto 1a (35% yield), along with 6a (21% yield), as the result of an undesired reduction of 1a.
|
|
||||
|---|---|---|---|---|
| Entry |
2a:cosolvent |
3:electrolyte |
Electrolysis | Yieldb [%] |
|
a All reactions were carried in the Electrasyn 2.0 apparatus (undivided cell, see ESI for details).
b Isolated yields after flash chromatography. In brackets yields of 5 and 6a, respectively (1H NMR by internal standard). E/Z ratios were determined via1H-NMR spectroscopy on the reaction crude mixtures (>25:1). CCE: constant current electrolysis; CVE: constant voltage electrolysis.
|
||||
| 1 | 1:2 (DMF) |
3a:TEABF4 |
CCE (I = 4 mA) | 18 (35/21) |
| 2 | 1:2 (DMF) |
3b:TEABF4 |
CCE (I = 4 mA) | — (—/17) |
| 3 | 1:2 (DMF) |
3c:TEABF4 |
CCE (I = 4 mA) | — (—/13) |
| 4 | 1:2 (DMF) |
3d:TEABF4 |
CCE (I = 4 mA) | 34 (—/12) |
| 5 | 1:2 (DMF) |
—:TEABF4 |
CCE (I = 4 mA) | — (—/13) |
| 6 | 5:1 (DMF) |
3d:TBAPF6 |
CCE (I = 4 mA) | 46 (—/11) |
| 7 | 5:1 (DMF) |
3d:LiBF4 |
CCE (I = 4 mA) | 50 (—/8) |
| 8 | 5:1 (ACN) |
3d:LiBF4 |
CCE (I = 4 mA) | 53 (—/—) |
| 9 | 5:1 (ACN) |
3d:LiBF4 |
CCE (I = 2 mA) | 63 (—/—) |
| 10 |
5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 (ACN) :1 (ACN)
|
3d
:LiBF
4
|
CVE (V = 5 V) | 75 (—/—) |
| 11 | 5:1 (ACN) |
3d:LiBF4 |
CVE (V = 3 V) | 30 (—/—) |
| 12 | 5:1 (ACN) |
3d:LiBF4 |
CVE (V = 7 V) | 63 (—/—) |
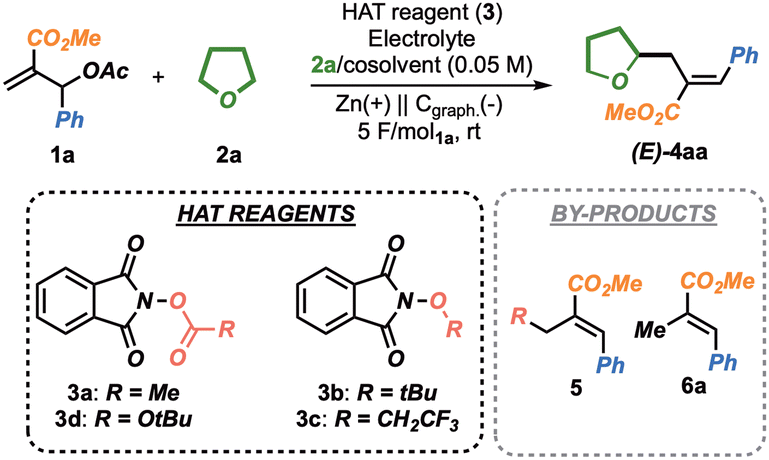
To validate the hypothesis that an electrophilic radical precursor could improve the reaction outcomes, N-tert-butoxyphthalimide 3b (entry 2) and N-trifluoroethoxyphthalimide 3c (entry 3) were tested under the conditions described in entry 1. Disappointingly, no product was formed in both cases. We thus speculated that, moving from ether- to more reactive carbonate-derivatives could facilitate the reduction-β-scission of the phthalimide adduct. Accordingly, we synthesized carbonate 3d, for which we propose the acronym RAC, standing for Redox-Active-Carbonate.13 Delightfully, the employment of this RAC in the eChem protocol led to the isolation of 4aa in 34% yield (entry 4) along with minor quantities of 6a (12%), as an indication that the cathodic events involved mainly 3d. As expected, the use of electrophilic alkoxy radicals completely suppressed the formation of 5ad. Interestingly, RAC 3d represents a valuable complement to peroxide-based reagents, intrinsically more difficult to reduce (see Fig. S2, ESI†), providing new opportunities within the electrochemical HAT scenario.
Importantly, a blank experiment in the absence of 3d was shown not to produce 4aa, even in trace amounts (entry 5). Then, higher amounts of THF in the solvent mixture (entry 6) increased the yield (46% yield), by likely facilitating the capture of the electrophilic radical generated by 3d. For solubility reasons, electrolytes such as TBAPF6 (entry 6) or LiBF4 (entry 7) were preferred, with the latter being optimal. Interestingly, a co-solvent switch from DMF to ACN was found to suppress the formation of 6a (entry 8). Finally, if lowering the operating current from 4 mA to 2 mA was already found beneficial (63% yield, entry 9),14 a switch to constant voltage electrolysis (CVE, 5 V) allowed us to reach the optimal 75% yield in 4aa (entry 10, Conditions A). Further tuning of the reaction voltage was found to be detrimental (entries 11 and 12).
The generality of the methodology was first evaluated by subjecting a series of MBH derivatives (1b–v) to the optimal allylation of THF by means of the eChem HAT protocol (Scheme 1). Within the series of aromatic/heteroaromatic acetates (1a–m), we were pleased to record good to excellent yields (up to 84%) obtained on the corresponding cinnamates 4 regardless of both electronic properties and position of substituents such as halogens, trifluoromethyl-, cyano-, alkyl- and methoxy-groups. Subjection of 1n and 1o to the same protocol resulted in products 4na (52% yield) and 4oa (38% yield), featuring a conjugated diene or ene–yne moiety, respectively. In addition, aliphatic MBH acetates 1p, 1q and citronellal-derived 1r were also productively engaged in the disclosed process (38–56% yield). Variation of the electron-withdrawing group to introduce radical-sensitive moieties such as benzylic (1s) and propargylic esters (1t) or to produce α,β-unsaturated ketones (4ua) and nitriles (4va) were adequately tolerated (36–81% yield). Importantly, the synthetic relevance of the methodology was verified on late-stage functionalization of derivatized naturally occurring scaffolds, such as L-valine derivative 1w and 5α-cholestanol derivative 1x. A survey of ethers 2 in the allylation reaction was then undertaken. This stage posed a significant challenge, since, for each entry, the polarity and the conductivity of the reaction mixture changed markedly. Unfortunately, Conditions A proved too sensitive to the reaction medium to be employed. A small re-optimization led us to identify another set of parameters (i.e. CCE electrolysis, Table 1, entry 6, Conditions B).
 | ||
| Scheme 1 Scope of the protocol for MBH acetates (1, Conditions A: Table 1, entry 10) and ethers (2, Conditions B, Table 1, entry 6). E/Z ratios were determined via1H-NMR spectroscopy on the reaction crude mixtures and were found to be ≥13:1 (see ESI† for details). a Reaction performed on 1.0 mmol scale of 1a (see ESI† for details). b The Z isomer was isolated as the major product (E/Z = 1:5). In some cases, variable amounts of starting MBH adducts were recovered untouched. | ||
Therefore, a series of 9 different ethers (2b–j) was productively functionalized with acetate 1a (Conditions B). When 2-MeTHF 2b was engaged in the process, isomers 4ab and 4ab′ were isolated (55% combined yield, 2.5:1 ratio). Dioxane 2f, 1,3-benzodioxole 2g and 1,3-dioxolane 2h underwent the desired transformation smoothly (50–61% yield). Importantly, in the case of the more reactive 2g and 2h, the amount of ether could be decreased to as low as 20 equiv. Finally, acyclic ethers such as Et2O (2i) and 1,2-dimethoxyethane (2j) could also be engaged in the present process, although with moderate yields (44% and 39%, respectively).
Interestingly, the protocol was effectively extended also to electron-poor olefins 7a and 7b (Giese-type addition, Conditions B) that provided the desired products 8a and 8b in up to 55% yield.15 Furthermore, since the preparation of both starting materials 1 and RAC 3d relies on the activation of hydroxy moieties, we also demonstrated that compound 4aa can be isolated in 41% yield (Conditions B), via in situ activation of both MBH alcohols and N-hydroxyphthalimide under Boc2O/DMAP/THF conditions (see ESI† for details).
Mechanistically, the machinery depicted in Scheme 2a is postulated. In particular, cathodic reduction of 3d would lead to the tBuOCO2˙ C16 and phthalimide anion. The alternative fragmentation of 3d to give a phthalimido radical and tertbutylcarbonate anion is unlikely, due to the non-productivity of N-trifluoroacetoxyphthalimide in the present protocol (see Table S1, ESI†).17 Subsequently, the radical C could undergo direct HAT with ether 2 resulting in the α-oxy radical A (path b) or first decompose to the strong electrophilic tertbutoxyl radical D that would then be responsible for the HAT step (path a).18 Subsequently, the α-oxy radical A is postulated to be intercepted, regioselectively, by the electrophilic β-carbon position of 1, followed by a second monoelectronic cathodic reduction of the so-formed radical intermediate E10 leading to the final α,β-unsaturated ester 4via elimination of the acetate anion. Here, (i) the absence of compound 5 that would result from the methyl radical trapping of 1 (β-fragmentation of D to acetone and Me˙) and (ii) the higher stability of (alkoxycarbonyl)oxyl radicals with respect to alkoxy ones would suggest path b as the most likely one,19 although the concomitant formation of D from the partial decomposition of 3d cannot be completely excluded.20 Additionally, dedicated labelling studies (THF and THF-d8) and ON–OFF experiments emphasized the role of the HAT process in the rate-determining-step and underlined the non-prevalence of active background radical chains (see ESI†).
 | ||
| Scheme 2 (a) Tentative reaction mechanism. (b) Electrochemical carboxylation of 4aa. | ||
Cyclic voltammetry experiments were then carried out (Fig. S2, ESI†). Both RAC 3d and RAE 3a showed very similar redox behaviour, with a first chemically irreversible reduction process with cathodic peaks (Epc) at −1.26 and −1.24 V vs. SCE, respectively. In agreement with literature reports,21 this is likely localized on the phthalimide fragment, and it is followed by the N–O bond cleavage with the formation of a phthalimide anion and neutral radicals tBuOCO2˙ (3d) and Me˙ (3a). On the other hand, ether 3b is characterized by a first reduction process at E1/2 = −1.43 V vs. SCE that is not followed by a chemical reaction. Therefore, 3b is not suitable for its application in the described reaction protocol, not delivering the desired alkoxy radical, useful for the HAT process. Furthermore, MBH acetate 1a shows a more negative and chemically irreversible reduction process (Epc = −2.08 V vs. SCE) and it is therefore out of the available range of applied potentials to perform a redox-driven chemical initiation, in competition with 3.
Finally, the synthetic versatility of compound 4 was demonstrated by subjecting 4aa to electrolytic conditions in the presence of 1 atm CO2.22 Monomethyl malonate 10 was isolated as the only regioisomer (1.8:1 dr) in 54% yield (Scheme 2b).
In conclusion, in the present investigation we have documented eChem C(sp3)–H activation of ethers under cathodic reduction by means of a new redox-active-carbonate (RAC) as an efficient HAT promoter. The use of MBH acetates as electrophilic partners resulted in a regio- and stereoselective protocol for the allylation/alkylation of ethers (35 examples).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Yamaguchi, A. T. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (b) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed.
- (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC.
- For representative review articles on visible-light photoredox promoted HAT methodologies, see: (a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875 CrossRef CAS PubMed ; For recent relevant contributions see: ; (b) M. H. Shaw, V. W. Shurtleff, J. A. Terrett, J. D. Cuthbertson and D. W. C. MacMillan, Science, 2016, 352, 1304 CrossRef CAS PubMed; (c) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218 CrossRef CAS PubMed.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) E. C. R. McKenzie, S. Hosseini, A. G. Couto Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2022, 122, 3292 CrossRef CAS PubMed; (c) M. C. Leech and K. Lam, Nat. Rev. Chem., 2022, 6, 275 CrossRef; (d) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317 CrossRef CAS PubMed; (e) N. E. S. Tai, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487 CrossRef PubMed; (f) Science of Synthesis: Electrochemistry in Organic Synthesis, ed. L. Ackermann, Thieme, Stuttgart, 2021, p. 573 Search PubMed.
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC.
- (a) E. J. Horn, B. R. Rosen, Y. Chen, J. Tang, K. Chen, M. D. Eastgate and P. S. Baran, Nature, 2016, 533, 57 CrossRef PubMed; (b) S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578 CrossRef CAS PubMed; (c) P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275 CrossRef CAS PubMed; (d) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. Dutta Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693 CrossRef CAS PubMed ; For examples of intramolecular HAT, see: ; (e) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385 CrossRef CAS PubMed.
- (a) H. Huang, Z. M. Strater and T. H. Lambert, J. Am. Chem. Soc., 2020, 142, 1698 CrossRef CAS PubMed; (b) J. Sim, B. Ryou, M. Choi, C. Lee and C.-M. Park, Org. Lett., 2022, 24, 4264 CrossRef CAS PubMed.
- B. Huan, Z. Sun and G. Sun, eScience, 2022, 2, 243 CrossRef.
- (a) Y. Liu, S. Battaglioli, L. Lombardi, A. Menichetti, G. Valenti, M. Montalti and M. Bandini, Org. Lett., 2021, 23, 4441 CrossRef CAS PubMed; (b) S. Battaglioli, G. Bertuzzi, R. Pedrazzani, J. Benetti, G. Valenti, M. Montalti, M. Monari and M. Bandini, Adv. Synth. Catal., 2022, 364, 720 CrossRef CAS; (c) L. Lombardi, A. Cerveri, R. Giovanelli, M. Castiñeira Reis, C. Silva López, G. Bertuzzi and M. Bandini, Angew. Chem., Int. Ed., 2022, 61, e202211732 CrossRef CAS PubMed.
- (a) G. Bertuzzi, G. Ombrosi and M. Bandini, Org. Lett., 2022, 24, 4354 CrossRef PubMed; (b) A. Brunetti, G. Bertuzzi and M. Bandini, Synthesis DOI:10.1055/a-2029-0488.
- (a) F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier and P. Geerlings, Org. Lett., 2007, 9, 2721 CrossRef CAS PubMed; (b) F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486 CrossRef CAS.
- I. N.-M. Leibler, M. A. Tekle-Smith and A. G. Doyle, Nat. Commun., 2021, 12, 6950 CrossRef CAS PubMed.
- RAC 3d has been utilized as a synthetic alternative to Boc-anhydride, see: J. R. Tagat, R. W. Steensma, S. W. McCombie, D. V. Nazareno, S.-I. Lin, B. R. Neustadt, K. Cox, S. Xu, L. Wojcik, M. G. Murray, N. Vantuno, B. M. Baroudy and J. M. Strizki, J. Med. Chem., 2001, 44, 3343 CrossRef CAS PubMed.
- Under these conditions, the reaction voltage was monitored to be around 5 V.
- (a) D. Ravelli, M. Zoccolillo, M. Mella and M. Fagnoni, Adv. Synth. Catal., 2014, 356, 2781 CrossRef CAS; (b) B. Niu, B. G. Blackburn, K. Sachidanandan, M. V. Cooke and S. Laulhé, Green Chem., 2021, 23, 9454 RSC.
- J. Chateauneuf, J. Lusztyk, B. Maillard and K. U. Prelog, J. Am. Chem. Soc., 1988, 110, 6727 CrossRef CAS.
- L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607 CrossRef CAS PubMed.
- (a) D. E. Edge and J. K. Kochi, J. Am. Chem. Soc., 1973, 95, 2635 CrossRef CAS; (b) Y.-H. Fu, G.-B. Shen, K. Wang and X.-Q. Zhu, ACS Omega, 2022, 7, 25555 CrossRef CAS PubMed.
- (a) M. Bühl, P. DaBell, D. W. Manley, R. P. MacCaughan and J. C. Walton, J. Am. Chem. Soc., 2015, 137, 16153 CrossRef PubMed; (b) S.-Q. Lai, B.-Y. Wei, J.-W. Wang, W. Yu and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21997 CrossRef CAS PubMed; (c) L. Quach, S. Dutta, P. M. Pflüger, F. Sandfort, P. Bellotti and F. Glorius, ACS Catal., 2022, 12, 2499 CrossRef CAS.
- (a) M. Galeotti, M. Salamone and M. Bietti, Chem. Soc. Rev., 2022, 51, 2171 RSC; (b) Y. Gong, L. Su, Z. Zhu, Y. Ye and H. Gong, Angew. Chem., Int. Ed., 2022, 61, e202201662 CAS.
- M. A. Syroheshkin, I. B. Krylov, A. M. Hughes, I. V. Alabugin, D. V. Nasybullina, M. Y. Sharipov, V. P. Gultyai and A. O. Terent’ev, J. Phys. Org. Chem., 2017, 30, 3744 CrossRef.
- (a) H. Wang, Y.-F. Du, M.-Y. Lin, K. Zhang and J.-X. Lu, Chin. J. Chem., 2008, 26, 1745 CrossRef CAS; (b) H. Wang, K. Zhang, Y.-Z. Liu, M.-Y. Lin and J.-X. Lu, Tetrahedron, 2008, 64, 314 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06999g |
| This journal is © The Royal Society of Chemistry 2023 |