
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of vinylidene-π-allyl palladium(II) species†
Can
Li
ab,
Zhengnan
Zhou
ab,
Yuling
Li
ab,
Yinlong
Guo
 *ab and
Shengming
Ma
*ac
*ab and
Shengming
Ma
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: ylguo@mail.sioc.ac.cn; masm@sioc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P. R. China
First published on 25th February 2023
Abstract
The reactivity of a new type of organometallic intermediate, vinylidene-π-allyl palladium species, has been demonstrated: the reaction between 4-alken-2-ynyl carbonates and stabilized carbon nucleophiles afforded functionalized 1,2,3,-butatriene compounds in moderate to high yields and excellent regioselectivities.
π-Allyl palladium chemistry (Scheme 1a) has been well-established1,2 and has become a powerful protocol for the formation of carbon–carbon and carbon–heteroatom bonds. In analogy, methylene-π-allyl Pd species (Scheme 1b) also show attractive reactivity towards different types of nucleophiles affording allenes3 or 1,3-dienes,4–9 respectively. Here, we wish to report the first example of the reactivity of vinylidene-π-allyl palladium species (Scheme 1c) formed from 4-alken-2-ynyl carbonates with nucleophiles.
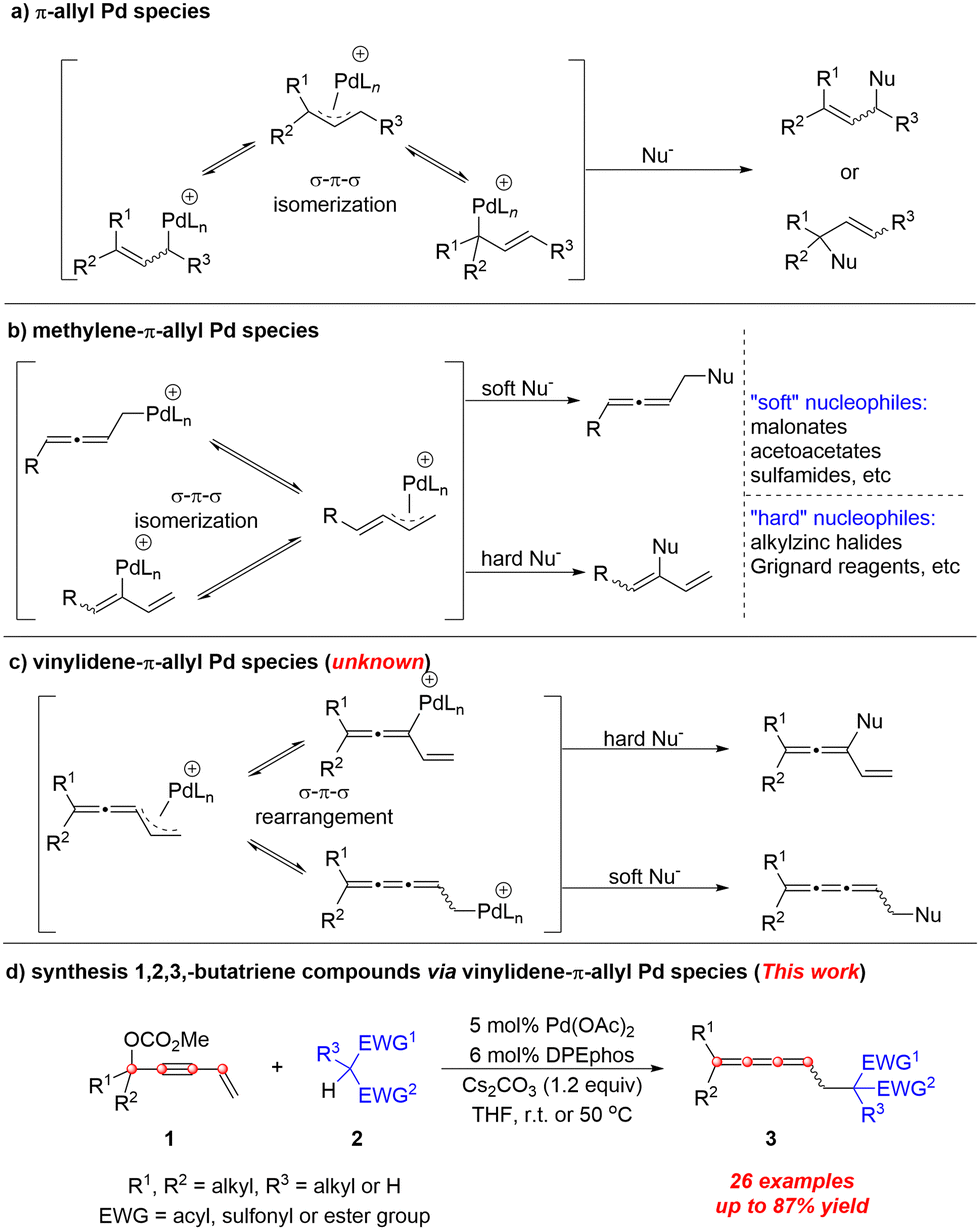 | ||
| Scheme 1 Profiles of π-allyl palladium species. | ||
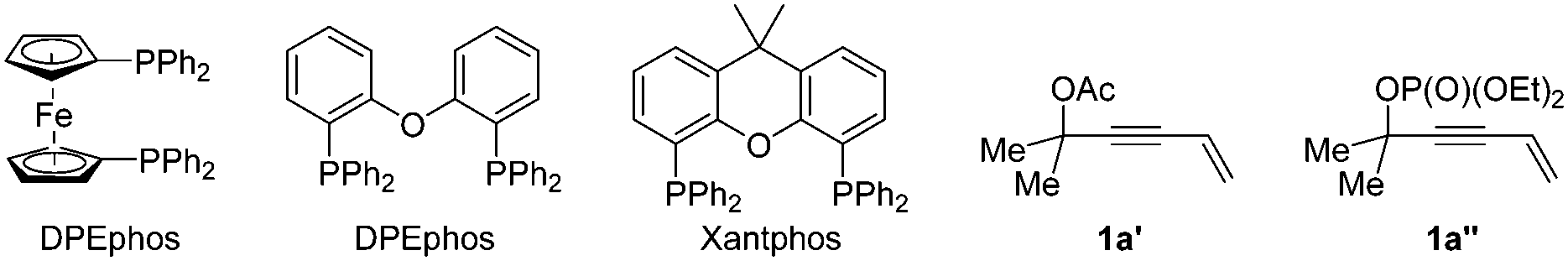
In our initial studies, the reaction of 4-alken-2-ynyl carbonate 1a and ethyl 2-benzoylpropionate 2a catalyzed by Pd(OAc)2 (5 mol%) and PPh3 (12 mol%) in THF at room temperature for 12 h afforded no product (Table 1, entry 1). Subsequent screening of various bisphosphine ligands (Table 1, entries 2–5) led to the formation of cumulated triene 3aa as the major product together with a small amount of vinylallene 4aa as determined by 1H NMR analysis of the crude reaction mixture. The reaction with DPEphos gave product 3aa in 60% yield with only 4% yield of 4aa (Table 1, entry 4). To improve the yield of product 3aa, a series of inorganic bases were screened (Table 1, entries 6–9) and Cs2CO3 was found to promote the reaction in 81% yield of 3aa and 3% yield of 4aa. Although tBuOLi provided a higher yield than Cs2CO3, the regioselectivity was lower (Table 1, entry 9). As a comparison, the corresponding acetate 1a′ showed lower reactivity (entry 10) and the corresponding phosphate 1a′′ was incompatible with this catalytic system (entry 11). Under the catalysis of Pd(OAc)2 (5 mol%) and DPEphos (6 mol%), replacing THF with other representative solvents such as CH3CN, EtOAc, DME, 1,4-dioxane and n-hexane resulted in poor yields and low regioselectivities (Table 1, entries 12–16). A reaction on the 0.5 mmol scale at a concentration of 0.1 M afforded product 3aa in a lower yield and regioselectivity (56% of 3aa with 6% of 4aa) (Table 1, compare entry 7 with entry 17). After further optimization we observed that the reaction at a concentration of 0.05 M could improve the yield and regioselectivity (Table 1, entry 18). Thus, the reaction parameters for entry 18 have been defined as the standard conditions. Besides, the structure of the regioisomer 4aa was confirmed by isolation based on a large scale experiment (entry 19).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Base | Solvent | Recovery of 1ab (%) | Yield of 3aab (%) | Yield of 4aab (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (1.2 equiv.), Pd(OAc)2 (5 mol%), ligand (6 mol%), and base (1.2 equiv.) in solvent (2 mL) unless otherwise noted. b Determined by 1H-NMR analysis with CH3NO2 as the internal standard. c 12 mol% of PPh3 was used. d The corresponding 1a′ was used instead of 1a. e Recovery of 1a′. f The corresponding 1a′′ was used instead of 1a. g Recovery of 1a′′. h The reaction was carried out on a 1 mmol scale in 10 mL of THF. i The reaction was carried out on a 0.5 mmol scale in 10 mL of THF for 24 h. j The reaction was carried out on a 1 mmol scale for 24 h. k Isolated yield. | ||||||
| 1c | PPh3 | — | THF | 80 | — | — |
| 2 | DPPE | — | THF | 83 | — | — |
| 3 | DPPF | — | THF | — | 53 | 5 |
| 4 | DPEphos | — | THF | — | 60 | 4 |
| 5 | Xantphos | — | THF | — | 17 | 24 |
| 6 | DPEphos | K2CO3 | THF | — | 56 | 4 |
| 7 | DPEphos | Cs2CO3 | THF | — | 81 | 3 |
| 8 | DPEphos | K3CO4 | THF | — | 72 | 3 |
| 9 | DPEphos | tBuOLi | THF | — | 88 | 11 |
| 10d | DPEphos | Cs2CO3 | THF | 78e | 8 | — |
| 11f | DPEphos | Cs2CO3 | THF | 2g | 1 | — |
| 12 | DPEphos | Cs2CO3 | CH3CN | — | 75 | 4 |
| 13 | DPEphos | Cs2CO3 | EtOAc | — | 69 | 4 |
| 14 | DPEphos | Cs2CO3 | DME | — | 75 | 3 |
| 15 | DPEphos | Cs2CO3 | Dioxane | — | 58 | 3 |
| 16 | DPEphos | Cs2CO3 | n-Hexane | — | 69 | 3 |
| 17h | DPEphos | Cs2CO3 | THF | — | 56 | 6 |
| 18i | DPEphos | Cs2CO3 | THF | — | 80 | 4 |
| 19j | Xantphos | — | THF | — | 10 | 23(20k) |
|
||||||
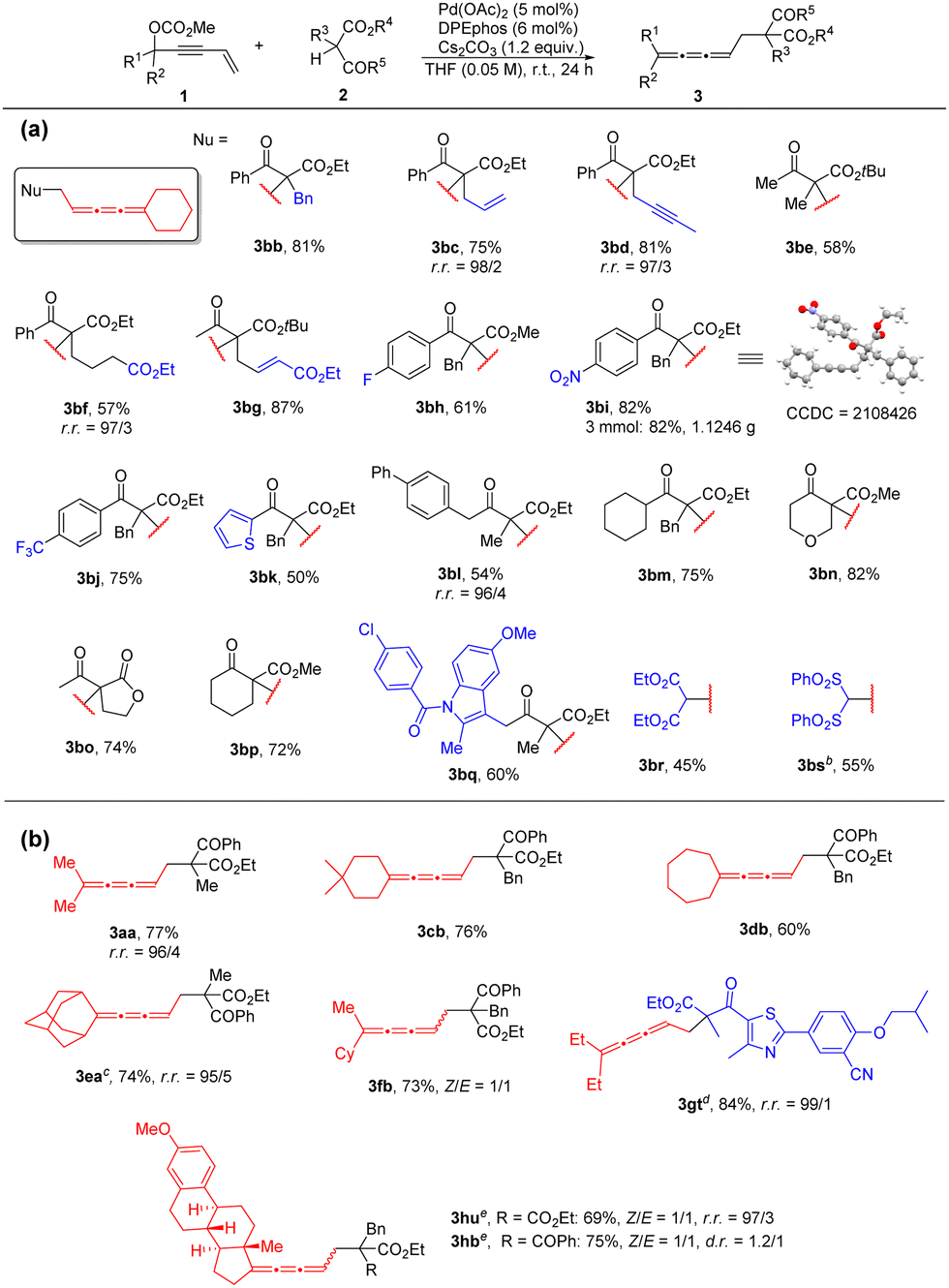
With the optimal conditions in hand, we chose 1,1-pentamethylenepent-4-en-2-ynyl carbonate 1b as a model substrate to explore the scope of β-ketocarbonyls 2 (Table 2a). Firstly, the substrates with substitution on the α-position of β-ketocarbonyls (R3) such as alkyl, alkenyl, alkynyl and ester groups all demonstrated high reactivity to afford the corresponding 1,2,3-butatrienes 3bb–3bg in 57–87% yields. R5 of the phenyl group with an electron withdrawing group (fluoro (2h), nitro (2i), trifluoromethyl (2j)) may also be tolerated generating the products 3bh–3bj smoothly in 61%–82% yields. Three mmol scale reaction of 1b with 2i afforded 1.1246 g (82%) of product 3bi, whose structure was further confirmed by single-crystal X-ray diffraction. When the R5 group is a heteroaromatic group such as 2-thienyl, the reaction afforded 3bk in 50% yield. Moreover, the R5 group may also be an alkyl group affording the corresponding 1,2,3-butatriene products 3bl and 3bm. In addition, the carbon- and oxygen-containing cyclic β-ketoesters 2n–2p reacted smoothly with 72–82% yields. To our delight, commercially available drugs, such as Indomethacin and Febuxostat derived β-ketoesters 2q and 2t could also be incorporated into this reaction, resulting in the desired cumulated butatrienes 3bq and 3gt. In addition, diethyl malonate (2r) and bis(phenylsulfonyl)methane (2s) may also react with 1b to afford the corresponding 1,2,3-triene products 3br and 3bs in moderate yields.
| a Unless otherwise indicated, the reaction was performed with 0.5 mmol of 1, 1.2 equiv. of 2, 5 mol% of Pd(OAc)2, 6 mol% of DPEphos and 1.2 equiv. of Cs2CO3 in THF (0.05 M) at room temperature for 24 h on a 0.5 mmol scale. Yields of isolated products are given. r.r. refers to the regioselectivity of the 1,2,3,-butatriene product vs. vinylallene, which is determined by 1H-NMR analysis of the crude product. b The reaction was carried out for 36 h. c The reaction was carried out at 50 °C for 42 h. d The reaction was carried out for 33 h. e The reaction was carried out at 50 °C for 48 h. |
|---|
|
Next, we investigated the scope of 3-vinyl propargylic carbonates 1 (Table 2b). Cyclic ketone derived 3-vinyl propargylic carbonates 1b–1e or acyclic ketone derived 3-vinyl propargylic carbonates 1a and 1g worked successfully under the standard reaction conditions. Non-symmetrical ketones such as cyclohexyl methyl ketone and estrone derived substrates 1f and 1h also gave the cumulated butatrienes 3fb, 3hu and 3hb in decent yields.
We proposed a possible mechanism as shown in Scheme 2a: Firstly, Pd(0)Ln would undergo SN2′-type oxidative addition to form intermediate Int I.10 Followed by releasing one molecule of CO2, vinylidene-π-allyl palladium species Int II was generated. Subsequently, the enolate was formed with the help of MeO− or Cs2CO3. Then the carbon nucleophile would attack the terminal carbon atom viaInt III to generate the linear selective products 3aa and Pd(0)Ln was regenerated. In order to identify possible intermediates in the reaction process, we carried out solvent-assisted electrospray ionization-mass spectrometry (SAESI-MS) and SAESI-MS/MS analysis (Scheme 2b).11 A solution of 1a (0.2 mmol), 2a (0.24 mmol), Pd(OAc)2 (0.01 mmol), DPEphos (0.012 mmol), and Cs2CO3 (0.24 mmol) in THF (2 mL) was stirred at room temperature. After 2.5 hours, the reaction mixture was analyzed. Ints I–IV have been detected and further confirmed by a SAESI-MS/MS experiment (see ESI†), which firmly supports the above-mentioned mechanism.
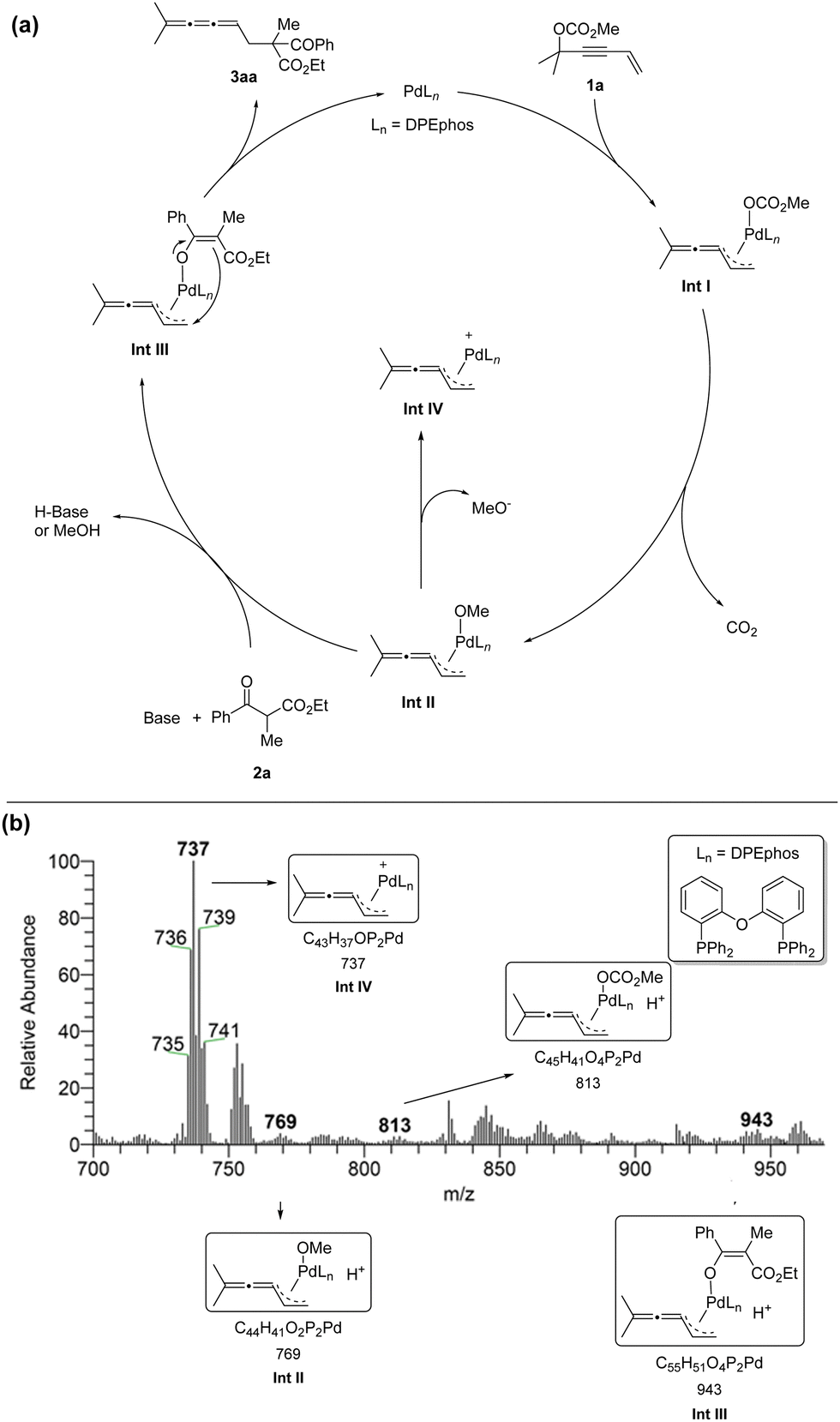 | ||
| Scheme 2 Plausible mechanism and SAESI-MS studies. | ||
In conclusion, we have developed a new strategy for the construction of functionalized 1,2,3-butatriene compounds via a new vinylidene-π-allyl palladium species, which was formed from the oxidative addition reaction of the Pd-DPEphos complex with 4-alken-2-ynyl carbonates. We are actively pursuing other reactivity of this new vinylidene-π-allyl palladium species.
Financial support from the National Key R&D Program of China (No. 2021YFA1500100) is greatly appreciated. We also thank Jie Wang in our group for reproducing the results of 3aa, 3be, and 3br presented in Table 2.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Tsuji, H. Takahashi and M. Morikawa, Tetrahedron Lett., 1965, 6, 4387 CrossRef; (b) B. M. Trost and T. J. Dietsch, J. Am. Chem. Soc., 1973, 95, 8200 CrossRef CAS.
- For selected reviews on π-allyl Pd species, see: (a) B. M. Trost and D. L. V. Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS; (c) B. M. Trost, M. R. Machacek and A. Aponick, Acc. Chem. Res., 2006, 39, 747 CrossRef CAS PubMed; (d) Z. Lu and S. Ma, Angew. Chem., Int. Ed., 2008, 47, 258 CrossRef CAS PubMed; (e) T. Jensen and P. Fristrup, Chem. – Eur. J., 2009, 15, 9632 CrossRef CAS PubMed; (f) M. Diéguez and O. Pàmies, Acc. Chem. Res., 2010, 43, 312 CrossRef PubMed; (g) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467 RSC; (h) B. M. Trost, Org. Process Res. Dev., 2012, 16, 185 CrossRef PubMed; (i) N. A. Butta and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929 RSC; (j) B. M. Trost, Tetrahedron, 2015, 71, 5708 CrossRef CAS PubMed; (k) Y. Liu, S. Han, W. Liu and B. M. Stoltz, Acc. Chem. Res., 2015, 48, 740 CrossRef CAS PubMed; (l) N. Butt, G. Yang and W. Zhang, Chem. Rec., 2016, 16, 2687 CrossRef; (m) R. L. Grangea, E. A. Clizbeb and P. A. Evans, Synthesis, 2016, 2911 Search PubMed; (n) Y. Wang, L. Lu and W. Xiao, Chem. – Asian J., 2018, 13, 2174 CrossRef CAS; (o) O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, Jan-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373 CrossRef PubMed ; For books, see: ; (p) Transition Metal Catalyzed Enantioselective Allylic Substitution, in Organic Synthesis, ed. U. Kazmaier, M. Beller, J. M. Brown, P. H. Dixneuf, A. Fürstner, L. Gooßen, L. S. Hegedus, P. Hofmann, T. Ikariya, L. A. Oro and Q.-L. Zhou, Springer, Heidelberg, 2012 Search PubMed; (q) J. Tsuji, Palladium reagents and catalysts: New perspectives for the 21st century, Wiley, 2004 CrossRef.
- For the synthesis of allenes via methylene-π-allyl Pd species using “soft” nucleophiles, see: (a) M. Ogasawara, H. Ikeda, T. Nagano and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 2089 CrossRef CAS PubMed; (b) Y. Imada, K. Ueno, K. Kutsuwa and S.-I. Murahashi, Chem. Lett., 2002, 140 CrossRef CAS; (c) M. Ogasawara, K. Ueyama, T. Nagano, Y. Mizuhata and T. Hayashi, Org. Lett., 2003, 5, 217 CrossRef CAS PubMed; (d) M. Ogasawara, H. L. Ngo, T. Sakamoto, T. Takahashi and W. B. Lin, Org. Lett., 2005, 7, 2881 CrossRef CAS PubMed; (e) M. Ogasawara, Y. H. Ge, K. Uetake and T. Takahashi, Org. Lett., 2005, 7, 5697 CrossRef CAS; (f) M. Ogasawara, H. L. Ngo, T. Sakamoto and T. Takahashi, J. Org. Chem., 2005, 70, 5764 CrossRef CAS PubMed; (g) B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186 CrossRef CAS PubMed; (h) Y. Imada, M. Nishida, K. Kutsuwa, S.-I. Murahashi and T. Naota, Org. Lett., 2005, 7, 5837 CrossRef CAS PubMed; (i) M. Ogasawara, L. Y. Fan, Y. H. Ge and T. Takahashi, Org. Lett., 2006, 8, 5409 CrossRef CAS PubMed; (j) Y. Imada, M. Nishida and T. Naota, Tetrahedron Lett., 2008, 49, 4915 CrossRef CAS; (k) T. Nemoto, M. Kanematsu, S. Tamura and Y. Hamada, Adv. Synth. Catal., 2009, 351, 1773 CrossRef CAS; (l) B. Q. Wan and S. Ma, Angew. Chem., Int. Ed., 2013, 52, 441 CrossRef CAS PubMed; (m) Z. Wu, F. Berhal, M. M. Zhao, Z. G. Zhang, T. Ayad and V. Ratovelomanana-Vidal, ACS Catal., 2014, 4, 44 CrossRef CAS; (n) J. X. Dai, X. Y. Duan, J. Zhou, C. L. Fu and S. Ma, Chin. J. Chem., 2018, 36, 387 CrossRef CAS; (o) B. M. Trost, J. E. Schultz, T. W. Chang and M. R. Maduabum, J. Am. Chem. Soc., 2019, 141, 9521 CrossRef CAS PubMed; (p) H. C. Liu, Y. Z. Hu, Z. F. Wang, H. Y. Tao and C. J. Wang, Chem. – Eur. J., 2019, 25, 8681 CAS; (q) N. J. Adamson, H. Jeddi and S. J. Malcolmson, J. Am. Chem. Soc., 2019, 141, 8574 CrossRef CAS; (r) H. Tsukamoto, T. Konno, K. Ito and T. Doi, Org. Lett., 2019, 21, 6811 CrossRef CAS PubMed; (s) S. H. Song, J. Zhou, C. L. Fu and S. Ma, Nat. Commun., 2019, 10, 507 CrossRef PubMed; (t) S. H. Song and S. Ma, Chin. J. Chem., 2020, 38, 1233 CrossRef CAS; (u) S. Q. Yang, Y. F. Wang, W. C. Zhao, G. Q. Lin and Z. T. He, J. Am. Chem. Soc., 2021, 143, 7285 CrossRef CAS PubMed; (v) J. Z. Xiao, H. B. Xu, X. H. Huo, W. B. Zhang and S. Ma, Chin. J. Chem., 2021, 39, 1958 CrossRef CAS; (w) Y. C. Zhang, X. Zhang and S. Ma, Nat. Commun., 2021, 12, 2416 CrossRef CAS PubMed; (x) C. F. Huang, F. C. Shi, Y. F. Cui, C. Li, J. Lin, Q. Liu, A. N. Qin, H. N. Wang, G. L. Wu, P. L. Wu, J. Z. Xiao, H. B. Xu, Y. Yuan, Y. Z. Zhai, W. F. Zheng, Y. Zheng, B. Yu and S. Ma, Chem. Sci., 2021, 12, 9347 RSC; (y) J. C. Zhang, X. H. Huo, J. Z. Xiao, L. Zhao, S. Ma and W. B. Zhang, J. Am. Chem. Soc., 2021, 143, 12622 CrossRef CAS PubMed.
- For the synthesis of 1,3-dienes via methylene-π-allyl Pd species using organic metal reagents as nucleophiles, see: (a) Y. D. Djahanbini, B. Cazes and J. Gore, Tetrahedron Lett., 1984, 25, 203 CrossRef; (b) J. S. Schneekloth Jr., M. Pucheault and C. M. Crews, Eur. J. Org. Chem., 2007, 40 CrossRef.
- For the synthesis of 1,3-dienes via methylene-π-allyl Pd species using Organoboronic acids as nucleophiles, see: (a) M. Yoshida, T. Gotou and M. Ihara, Chem. Commun., 2004, 1124 RSC; (b) T. Liu, J. Dong, S. J. Cao, L. C. Guo and L. Wu, RSC Adv., 2014, 4, 61722 RSC; (c) D. J. Lippincott, R. T. H. Linstadt, M. R. Maser and B. H. Lipshutz, Angew. Chem., Int. Ed., 2017, 56, 847 CrossRef CAS PubMed; (d) D. J. Lippincott, R. T. H. Linstadt, M. R. Maser, F. Gallou and B. H. Lipshutz, Org. Lett., 2018, 20, 4719 CrossRef CAS PubMed; (e) R. W. Brown, F. Zamani, M. G. Gardiner, H. B. Yu, S. G. Pyne and C. J. T. Hyland, Chem. Sci., 2019, 10, 9051 RSC.
- For the synthesis of 1,3-dienes via carbonylation of methylene-π-allyl Pd species, see: (a) J. Nokami, A. Maihara and J. Tsuji, Tetrahedron Lett., 1990, 31, 5629 CrossRef CAS; (b) K. Uemura and Y. Inoue, Appl. Organomet. Chem., 2000, 14, 8 CrossRef CAS.
- For the synthesis of 1,3-dienes via methylene-π-allyl Pd species using terminal alkynes as nucleophiles, see: (a) Y. Tao Xia, J. J. Wu, C. Y. Zhang, M. Mao, Y. G. Ji and L. Wu, Org. Lett., 2019, 21, 6383 CrossRef PubMed; (b) M. J. Sowden, J. S. Ward and M. S. Sherburn, Angew. Chem., Int. Ed., 2020, 59, 4145 CrossRef CAS PubMed.
- M. Mao, L. Zhang, Y. Z. Chen, J. Zhu and L. Wu, ACS Catal., 2017, 7, 181 CrossRef CAS.
- J. Lin, T. H. Zhu, M. Q. Jia and S. Ma, Chem. Commun., 2019, 55, 4523 RSC.
- (a) Y. Wang, W. Zhang and S. Ma, J. Am. Chem. Soc., 2013, 135, 11517 CrossRef CAS PubMed; (b) H. N. Wang, H. W. Luo, Z. M. Zhang, W. F. Zheng, Y. Yin, H. Qian, J. L. Zhang and S. Ma, J. Am. Chem. Soc., 2020, 142, 9763–9771 CrossRef CAS PubMed.
- J. T. Zhang, H. Y. Wang, W. Zhu, T. T. Cai and Y. L. Guo, Anal. Chem., 2014, 86, 8937 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, characterization data, and copies of NMR spectra. CCDC 2108426. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06871k |
| This journal is © The Royal Society of Chemistry 2023 |