
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative insertion of amines into conjugated macrocycles: transformation of antiaromatic norcorrole into aromatic azacorrole†
Sha
Li
a,
Yahan
Sun
a,
Xiaofang
Li
*a,
Oskar
Smaga
 b,
Sebastian
Koniarz
b,
Miłosz
Pawlicki
c and
Piotr J.
Chmielewski
*b
b,
Sebastian
Koniarz
b,
Miłosz
Pawlicki
c and
Piotr J.
Chmielewski
*b
aKey Laboratory of Theoretical Organic Chemistry and Functional Molecules, Ministry of Education, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology Xiangtan, Hunan 411201, China. E-mail: lixiaofang@hnust.edu.cn
bDepartment of Chemistry, University of Wrocław, F. Joliot-Curie 14, Wrocław 50 383, Poland. E-mail: piotr.chmielewski@chem.uni.wroc.pl
cFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, Kraków 30 387, Poland
First published on 10th March 2023
Abstract
A new group of aromatic porphyrinoids was obtained by an oxidative insertion of primary amines into the antiaromatic ring of 5,14-dimesityl-norcorrolatonickel(II) activated by iodosobenzene. The substituted 10-azacorroles thus formed were characterized by spectroscopic and electrochemical methods, and XRD analysis. Protonated forms of azacorroles were shown to remain aromatic despite disconnection of the original π-electron delocalization path.
The aromaticity and antiaromaticity of complex polycyclic systems have attracted the attention of chemists of various fields owing to theoretical interest in these phenomena as well as a potential applicability.1–17 Porphyrinoids constitute a class of macrocyclic systems that include both aromatic and antiaromatic species.18–24 The prominent representatives of the planar tetrapyrrolic porphyrinoids, i.e. aromatic corrole (Corr)25,26 and antiaromatic norcorrole (NCorr)27–29 both possess twenty π-electron pools but differences in the macrocyclic scaffolds allow 18πe porphyrin-like (Por) delocalization in the former and 16πe delocalization in the latter (Fig. 1). An interplay between the aromaticity and antiaromaticity of porphyrinoids involves oxidation/reduction processes30–35 or alteration of the π-electron delocalization path achieved by the macrocycle skeleton modifications.19–21,28,31,32,36–50
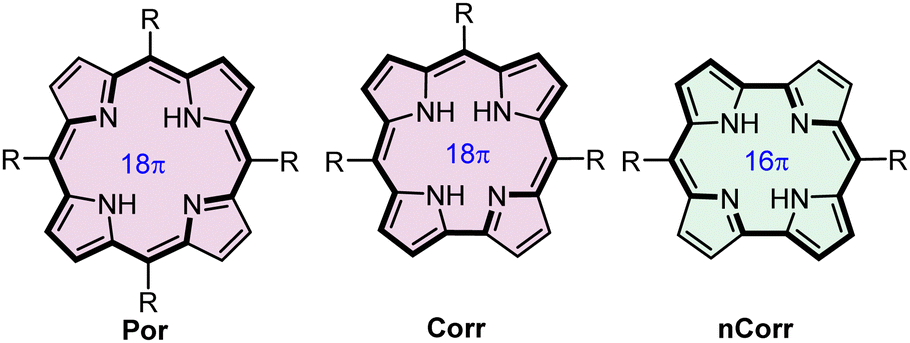 | ||
| Fig. 1 Schematic representations of the π-electron delocalization paths in selected tetrapyrroles. | ||
Originally reported 10-azacorroles have been obtained by annulation of the predefined linear dipyrrin precursors,37,39,40 and likewise the N,N′-substituted diazaporphyrins and their complexes.31,32,38,51 We found out that a broad family of N-substituted azacorroles can be synthesized in a one-step one-pot reaction of readily obtainable 5,14-dimesitylnorcorrolatonickel(II) 1 with primary amines (Table 1).52,53 In the present paper, the structural, spectroscopic, redox, and acid–base characteristics of this novel group of porphyrinoids are presented.
|
|
||||
|---|---|---|---|---|
| Entry | Product | R | Time (h) | Yielda (%) |
| a Isolated yields, reaction conditions: 1 (0.052 mmol, 1 equiv.), 2 (0.234 mmol, 4.5 equiv.), PhIO (0.18 mmol, 3.5 equiv.), CH2Cl2 (5 mL), room temperature. b The product was obtained with isoamyl nitrite instead of PhIO with 30% yield after 30 min. | ||||
| 1 | 3a | 4-ClPh | 2.0 | 61 |
| 2 | 3b | 4-BrPh | 2.0 | 54 |
| 3 | 3c | 4-FPh | 2.0 | 65 |
| 4 | 3d | 4-NO2Ph | 1.0 | 71 |
| 5 | 3e | 4-CNPh | 1.5 | 81 |
| 6 | 3f | 4-CHOPh | 2.5 | 84 |
| 7 | 3g | 4-CO2MePh | 2.5 | 72 |
| 8 | 3h | 3-ClPh | 1.0 | 80 |
| 9 | 3i | 3-BrPh | 1.0 | 82 |
| 10 | 3j | 3-FPh | 2.5 | 82 |
| 11 | 3k | 3-MePh | 2.5 | 78 |
| 12 | 3l | 3-OMePh | 2.0 | 74 |
| 13 | 3m | 2,6-Cl2Ph | 4.0 | 26 |
| 14 | 3n | 2,6-Br2Ph | 4.0 | 24 |
| 15 | 3o | 4-MePh | 4.0 | 52 |
| 16 | 3p | Ph | 4.0 | 55 |
| 17 | 3q | Et | 20 | 26 |
| 18 | 3r | Pr | 7.0 | 39 |
| 19 | 3s | 4-OMePh | 12 | N.D.b |
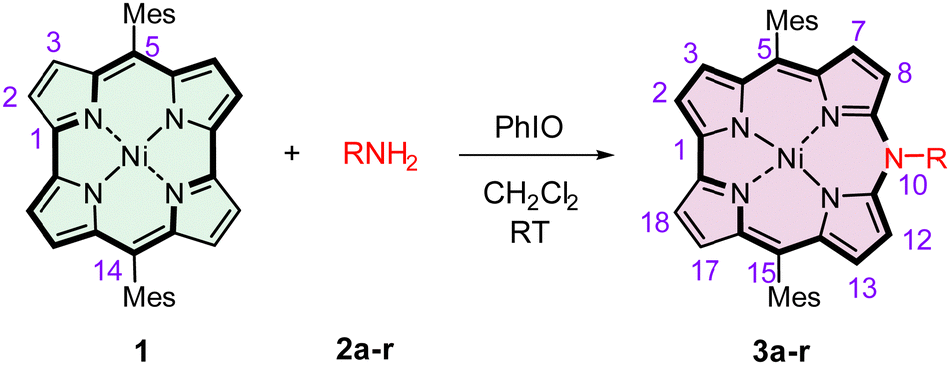
In the early stage of our studies, we reacted 1 with aniline, o-, m-, and p-toluidine, or m- and p-anisidine in the presence of an excess of isoamyl nitrite previously used as a nitrating agent.30 The reaction gave rise to insertion products, i.e., 10-N-(aryl)-5,15-dimesityl-10-azacorrolatonickel(II) with about 30% yield after 30 min of reaction time at room temperature in dichloromethane. No reaction, however, was observed for amines with strongly electron-withdrawing substituents on the amine aryl rings. Thus, we tested several catalysts with rational anticipation that they should act as oxidants generating an active form of the norcorrole. Our survey involved FeCl3, MnO2, PbO2, Mn2(OAc)3, PIFA (PhI(OCOCF3)2), mCPBA, PhI(Oac)2, PhIO, p-chloranil, and DDQ, which were applied to the reaction of 1 with 4-chloroaniline. Only for iodosylbenzene, the reaction ran effectively yielding 61% of 10-N-(4-chlorophenyl)-5,15-dimesityl-10-azacorrole 3a (Table 1), and for p-chloranil as the oxidant the yield was 8%. A 4.5 times excess of the amine over the norcorrole in the presence of 3.5 equiv. of PhIO allowed the highest yield of the azacorrole to be reached. The reaction carried out under the same conditions for other amines gave rise to azacorrole products with 24–84% yields (Table 1). No azacorrole was formed for p-anisidine, p-aminophenol, 1,4-diaminobenzene, or 3,4,5-trimethoxyaniline, likely due to oxidation of these amines by PhIO. Significantly, the reaction of 1 with another hypervalent iodine oxidant PIFA has been shown54 to result in a regioselective macrocyclic ring fission at the bipyrrole α–α′ bond leading in the presence of alcohols to linear tetrapyrroles terminated symmetrically with alkoxy-groups.54 On the other hand, reactions of 1 with primary amines have been reported to result in mono- or bis-aminated norcorroles.55 Apparently, a mild oxidant (PhIO) is sufficient to produce a cationic radical [1]˙+ that reacts with the amine molecule at the initial step of the reaction.
The new compounds were characterized by high-resolution mass spectrometry, 1H and 13C NMR, optical spectroscopy, and for selected systems, also by single crystal X-ray diffraction analyses (Fig. 2 and Tables S6 and S7, ESI†).56 HRMS confirmed the compositions of all azacorrole products, while four doublets of the pyrrole β-protons in the region of δ 7.2–8.3 ppm in 1H NMR spectra clearly indicated the effective twofold symmetry of the molecule and aromatic character of these systems. The obvious differences among the spectra of 3a–s arose from the presence of various substituents at N10. The crystal structures (Fig. 2) revealed the planarity of the macrocyclic rings with a marginal ruffling distortion in 3d, 3e, and 3h and slight saddling distortion in 3s (Fig. S114–S117, ESI†).57 The absorption spectra of all the systems were similar with a Soret-like band at 395 nm and three weaker Q-bands at 563–565 nm, 586–592 nm and 630–643 nm. These spectra were also similar to that of unsubstituted or N-acetylated azacorroles37 and strongly differed from that of the parent norcorrolatonickel(II) 1 reflecting a change from antiaromaticity of 1 to aromaticity of the azacorrole ring. The cyclic voltammetry (Fig. S118–S136 and Table S1, ESI†) showed two reversible oxidation processes (ox1, ox2) for all the systems with potentials Eox1 ranging from 0.13 to 0.22 V and Eox2 from 0.67 to 0.75 V (all potentials vs. Fc/Fc+ internal standard). Apparently, the oxidations occurred at potentials similar to those reported for the parent system 1. The potentials of reversible first reductions Ered1 (from −2.15 to −2.05 V) were similar to those observed previously for other 10-azacorrolatonickel(II) complexes37 but about 1.1 V lower than the first reduction potential of 1.28,30 The TD DFT calculated HOMO–LUMO energy gap (2.21 eV for 3d) was close to the difference between the first oxidation and first reduction potentials (e(Eox1 – Ered1) = 2.36 eV for 3d, Table S1, ESI†). Thin-layer spectroelectrochemical measurements in DCM indicated a ligand-centered oxidation (Fig. S144 in ESI†) with a significant decrease of the Soret-like band intensity, hypsochromic shift of this band (from 396 to 360 nm) as well as broadening and bathochromic shift in the Q-band region (to 667 and 702 nm). These changes indicated loss of the aromatic character of the macrocycle in 3p+˙ upon removal of the first electron58–61 likely due to involvement of the meso-nitrogen electron lone pair in the delocalization path of 3. Chemical oxidation of 3p with tris(4-bromophenyl)ammoniumyl hexachloroantimonate (BAHA, Magic Blue)62,63 resulted in similar spectral changes (Fig. S145 and S146 in ESI†). A BAHA-generated EPR spectrum of 3p+˙ confirmed the radical character of the mono-oxidized species64–67 with an isotropic signal at g0 = 2.0198 (RT, DCM, Fig. S147, ESI†) and frozen-solution spectrum of orthorhombic symmetry and small anisotropy of the Zeeman tensor (g1 = 2.0295, g2 = 2.0193, g3 = 2.0042, see Fig. S149 in ESI†) at 77 K.47,60,64–67
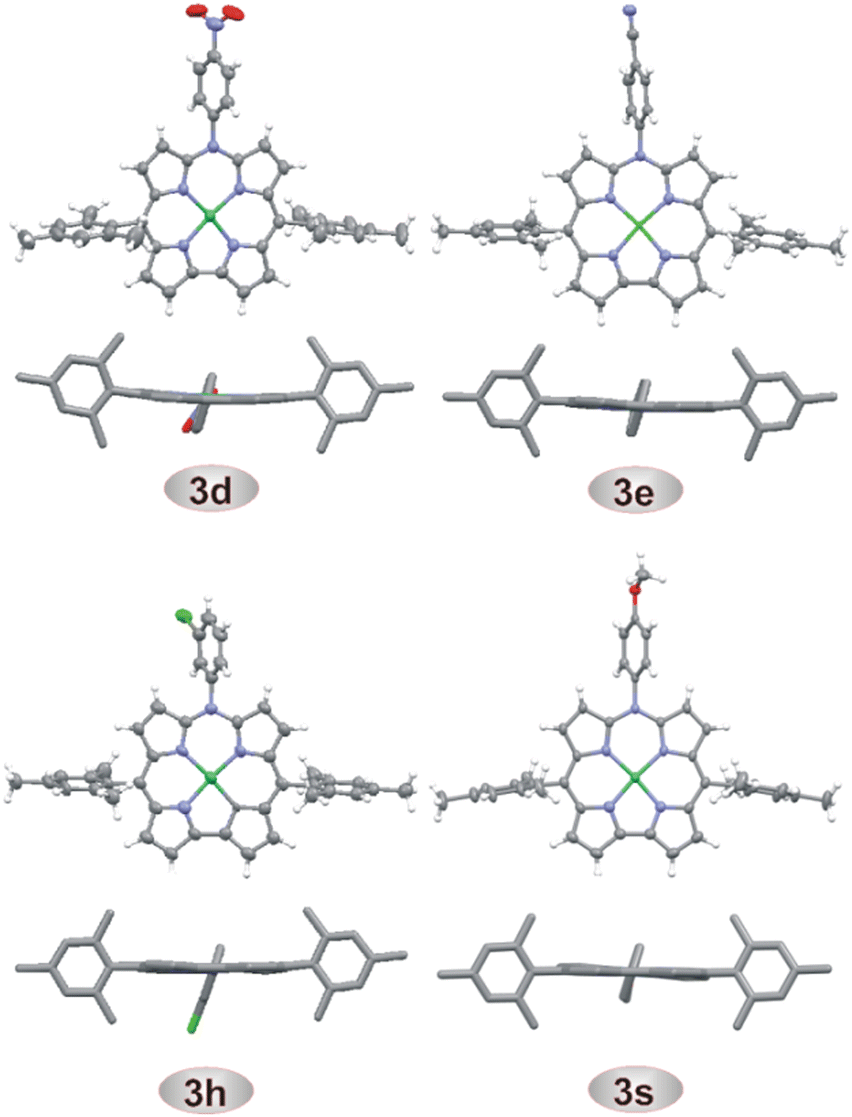 | ||
| Fig. 2 Thermal ellipsoid (50% probability level, front projections) and stick representations (side views with all hydrogens omitted) of 10-(p-NO2Ph)-, 10-(p-CNPh)-, 10-(m-ClPh)-, and 10-(p-OMePh)-5,15-dimesityl-10-azacorrole nickel(II) complexes. | ||
Addition of trifluoroacetic acid (TFA) to a solution of 3 in CH2Cl2 or CHCl3 resulted in reversible spectral changes (Fig. 3A and Fig. S140–S142, ESI†). The full conversion into the protonated form required high excesses of the acid, i.e. 2–5% (v/v) of TFA. These changes can be fully reversed by the addition of the equivalent amount of triethylamine. The protonation was also monitored by low-temperature 1H NMR (CDCl3, 220 K) to slow down chemical exchange. Addition of 1% of TFA resulted in the appearance of a new set of sharp signals attributed to the protonated species [3iH]+ (Fig. 3B). The 2D homo- and heteronuclear NMR experiments indicated pyrrole β-carbon C3 as an exclusive protonation site (Fig. 3B and Fig. S150–S154, ESI†) despite the apparent lack of any steric factors discriminating among the four distinct targets. However, deuteration by means of TFA-d (CDCl3, 220 K) revealed that all pyrrole β-CH's, except equivalent sites at C7 and C13 were subjected to the isotope exchange (Fig. S155 in ESI†). The DFT-determined relative stability of [3dH]+ tautomers unequivocally indicated C3/17 as the most, while C7/C13 as the least favorable protonation sites among pyrrole β-carbons, in line with the experimental results (Fig. 3C and Table S17, ESI†). The sp3 hybridization of C3 revealed by its chemical shift in the 13C NMR spectrum (δC 48.1 ppm) indicated disconnection of the original π-electron conjugation path at the protonated pyrrole. Despite that, the macrocyclic aromaticity of azacorrole in the protonated state was retained as it could be inferred from chemical shifts of the pyrrole β-protons in an aromatic region of δ 7.2–7.7 ppm (Fig. 3B). The GIAO-calculated proton chemical shifts for [3dH]+, negative sign of all NICS(1) values determined over the centroids of each ring, and anisotropy of the induced current density (AICD) were in line with the aromaticity of 3-protonated azacorrolatonickel(II) (Fig. 3D). To account for this, canonical structures with positive charge localized at the pyrrole β-carbon adjacent to the protonation site, thus allowing 18π-electron conjugation, are needed to be involved in the resonance (Fig. 3D).
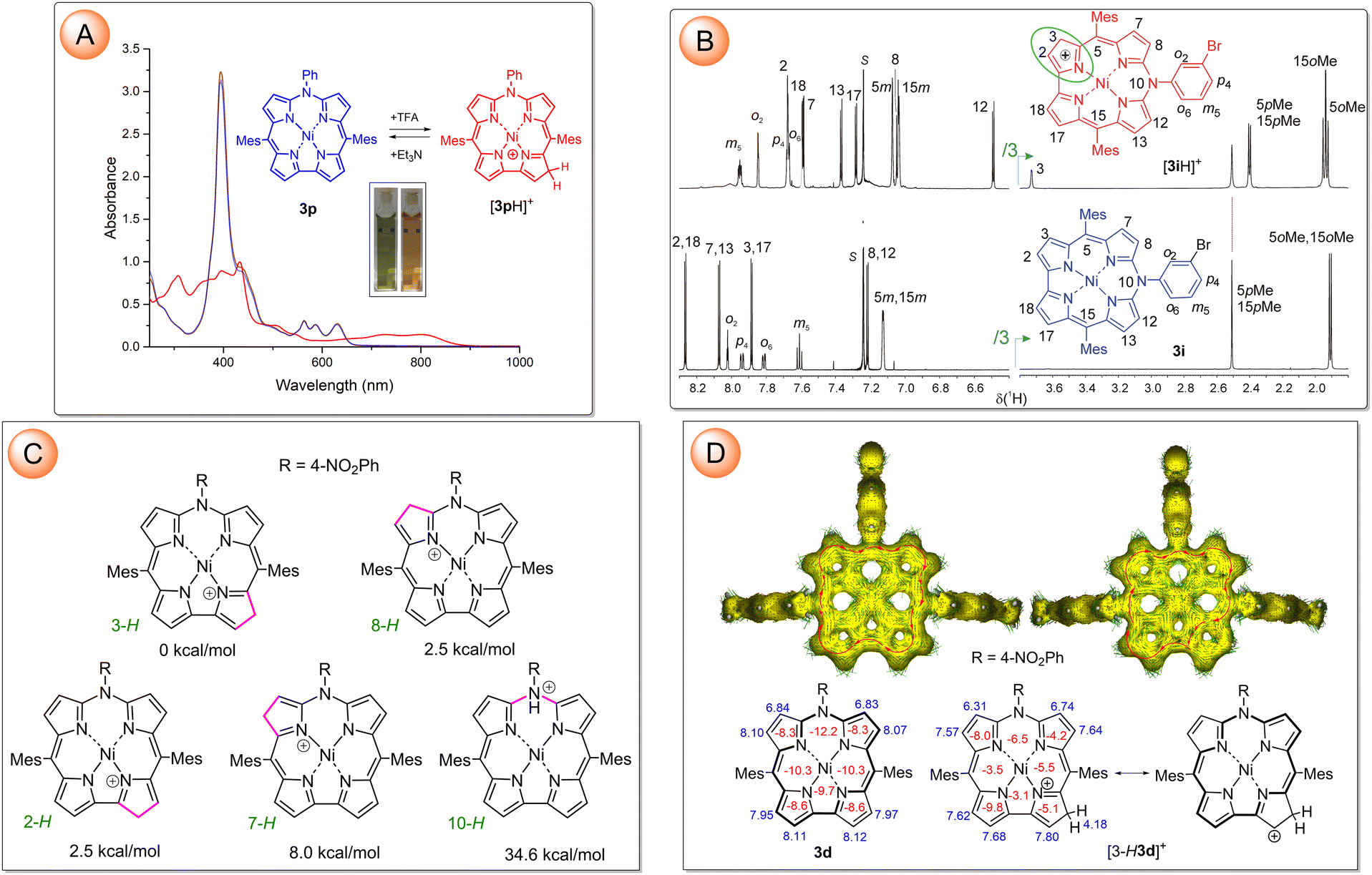 | ||
| Fig. 3 (A) Superimposed UV-vis-NIR spectra of the 3p solution in DCM (brown trace), that was recorded upon addition of TFA (10 equiv. red trace), and after neutralization with Et3N (10 equiv., blue trace). (B) Expansions of selected regions of 1H NMR spectra of 3i (lower trace) and its protonated derivative [3iH]+ (upper trace) with the signal assignments (CDCl3, 220 K). Note that in the high-field region, the intensities of the signals are divided by the factor of 3. (C) Structures of possible tautomers of [3dH]+ with relative energies of the DFT-optimized models. (D) GIAO-calculated chemical shifts (blue numbers), NICS(1) indices (red numbers), and AICD plots (0.03 isosurface) calculated for 3d (left column) and [3-H3d]+ (right column). | ||
In conclusion, a series of nickel(II) complexes of N-substituted 10-azacorroles were obtained using oxidative insertion of primary amines into oxidant-activated antiaromatic norcorrolatonickel(II). The compounds obtained in this way were diamagnetic, aromatic, and electron-donating. Despite neutral character and the lack of an obvious protonation site, nickel(II) azacorroles appeared to be proton acceptors. Macrocycles protonated at one of the pyrrole β-carbons remained aromatic. The electron donating properties of the azacorrolatonickel(II) are reflected also by relatively low oxidation potentials allowing facile generation of the radical species. Further study on these compounds will involve exploration of their potential in organic electronic devices.
This work was supported by the National Natural Science Foundation of China (No. 22171076), the Science and Technology Innovation Program of Hunan Province (2021RC5028), and the Polish National Science Center (2018/29/B/ST5/00280). Quantum chemical calculations were performed in the Wrocław Centers for Networking and Supercomputing. Publication of this article was supported by the Excellence Initiative – Research University program for the University of Wrocław.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. K. Cyrański, T. M. Krygowski, M. Wisiorowski, N. J. R. van Eikema Hommes and P. v R. Schleyer, Angew. Chem., Int. Ed., 1998, 37, 177 CrossRef.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
- T. M. Krygowski, H. Szatyłowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383 CrossRef CAS PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed.
- R. Breslow and S. T. Schneebeli, Tetrahedron, 2011, 67, 10171 CrossRef CAS.
- T. Nishinaga, T. Ohmae, K. Aita, M. Takase, M. Iyoda, T. Arai and Y. Kunugi, Chem. Commun., 2013, 49, 5354 RSC.
- K. Aita, T. Ohmae, M. Takase, K. Nomura, H. Kimura and T. Nishinaga, Org. Lett., 2013, 15, 3522 CrossRef CAS PubMed.
- M. Schmidt, D. Wassy, M. Hermann, M. T. Gonzáles, N. Agräit, L. A. Zotti, B. Esser and E. Leary, Chem. Commun., 2021, 57, 745 RSC.
- R. Breslow, Acc. Chem. Res., 1973, 6, 393 CrossRef CAS.
- P. V. R. Schleyer and H. Jiao, Pure Appl. Chem., 1996, 68, 209 CrossRef CAS.
- P. v R. Schleyer, C. Meaerker, A. Dransfeld, H. Jiao and N. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed.
- P. v R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. van Eikema Hommes, Org. Lett., 2001, 3, 2465 CrossRef CAS PubMed.
- M. Randić, Chem. Rev., 2003, 103, 3449 CrossRef PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed.
- A. D. Allen and T. T. Tidwell, Chem. Rev., 2001, 101, 1333 CrossRef CAS PubMed.
- B. Szyszko, M. J. Białek, E. Pacholska-Dudziak and L. Latos-Grażyński, Chem. Rev., 2017, 117, 2839 CrossRef CAS PubMed.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed.
- R. Orłowski, D. Gryko and D. T. Gryko, Chem. Rev., 2017, 117, 3102 CrossRef PubMed.
- Y. Matano, Chem. Rev., 2017, 117, 3138 CrossRef CAS PubMed.
- M. Pawlicki and L. Latos-Grażyński, Chem. Asian J., 2015, 10, 1438 CrossRef CAS PubMed.
- J. Klajn, W. Stawski, P. J. Chmielewski, J. Cybińska and M. Pawlicki, Chem. Commun., 2019, 55, 4558 RSC.
- K. Wypych, M. Dimitrova, D. Sundholm and M. Pawlicki, Org. Lett., 2022, 24, 4876 CrossRef CAS PubMed.
- Z. Gross, N. Galili and I. Saltsman, Angew. Chem., Int. Ed., 1999, 38, 1427 CrossRef CAS PubMed.
- D. T. Gryko, Eur. J. Org. Chem., 2002, 1735 CrossRef CAS.
- M. Broering, S. Köhler and C. Kleeberg, Angew. Chem., Int. Ed., 2008, 47, 5658 CrossRef CAS PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542 CrossRef CAS PubMed.
- T. Yonezawa, S. A. Shafie, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2017, 56, 11822 CrossRef CAS PubMed.
- Z. Deng, X. Li, M. Stępień and P. J. Chmielewski, Chem. – Eur. J., 2016, 22, 4231 CrossRef CAS PubMed.
- T. Satoh, M. Minoura, H. Nakano, K. Furukawa and Y. Matano, Angew. Chem., Int. Ed., 2016, 55, 2235 CrossRef CAS PubMed.
- K. Sudoh, T. Satoh, T. Amaya, K. Furukawa, M. Minoura, H. Nakano and Y. Matano, Chem. – Eur. J., 2017, 23, 16364 CrossRef CAS PubMed.
- A. Yamaji, H. Tsurugi, Y. Miyake, K. Mashima and H. Shinokubo, Chem. – Eur. J., 2016, 22, 3956 CrossRef CAS PubMed.
- T. Yoshida, S. A. Shafie, H. Kawashima, N. Fukui and H. Shinokubo, Org. Lett., 2021, 23, 2826 CrossRef CAS PubMed.
- S. Ukai, N. Fukui, T. Ikeue and H. Shinokubo, Chem. Lett., 2022, 51, 182 CrossRef CAS.
- A. Nishiyama, M. Fukuda, S. Mori, K. Furukawa, H. Fliegl, H. Furuta and S. Shimizu, Angew. Chem., Int. Ed., 2018, 57, 9728 CrossRef CAS PubMed.
- M. Horie, Y. Hayashi, S. Yamaguchi and H. Shinokubo, Chem. – Eur. J., 2012, 18, 5919 CrossRef CAS PubMed.
- Y. Satoh, Y. Fujita, N. Muramatsu, K. Furukawa, T. Ikoma, M. Minoura, H. Nakano and Y. Matano, ChemPlusChem, 2021, 86, 1476 CrossRef CAS PubMed.
- H. Omori, S. Hiroto and H. Shinokubo, Chem. Commun., 2016, 52, 3540 RSC.
- H. Omori, S. Hiroto and H. Shinokubo, Org. Lett., 2016, 18, 2978 CrossRef CAS PubMed.
- H. Omori, S. Hiroto, Y. Takeda, H. Fliegl, S. Minakata and H. Shinokubo, J. Am. Chem. Soc., 2019, 141, 4800 CrossRef CAS PubMed.
- M. Bröring, F. Brégier, E. C. Tejero, Ch Hell and M. C. Holthausen, Angew. Chem., Int. Ed., 2007, 46, 445 CrossRef PubMed.
- D. Sakow, B. Böker, K. Brandhorst, O. Burghaus and M. Bröring, Angew. Chem., Int. Ed., 2013, 52, 4912 CrossRef CAS PubMed.
- D. Sakow, D. Baabe, B. Böker, O. Burghaus, M. Funk, C. Kleeberg, D. Menzel, C. Pietzonka and M. Brőring, Chem. – Eur. J., 2014, 20, 2913 CrossRef CAS PubMed.
- H. Omori and H. Shinokubo, Organometallics, 2019, 38, 2878 CrossRef CAS.
- S.-Y. Liu, T. Fukuoka, N. Fukui, J.-Y. Shin and H. Shinokubo, Org. Lett., 2020, 22, 4400 CrossRef CAS PubMed.
- D. Ren, O. Smaga, X. Fu, X. Li, M. Pawlicki, S. Koniarz and P. J. Chmielewski, Org. Lett., 2021, 23, 1032 CrossRef CAS PubMed.
- B. Liu, T. Yoshida, X. Li, M. Stępień, H. Shinokubo and P. J. Chmielewski, Angew. Chem., Int. Ed., 2016, 55, 13142 CrossRef CAS PubMed.
- S.-Y. Liu, H. Tanaka, R. Nozawa, N. Fukui and H. Shinokubo, Chem. – Eur. J., 2019, 25, 7618 CrossRef CAS PubMed.
- A. Yagi, N. Okada, N. Fukui, H. Tanaka, T. Hatakeyama and H. Shinokubo, Chem. Lett., 2022, 51, 321 CrossRef CAS.
- M. Kawamata, T. Sugai, M. Minoura, Y. Maruyama, K. Furukawa, C. Holstrom, V. N. Nemykin, H. Nakano and Y. Matano, Chem. Asian J., 2017, 12, 816 CrossRef CAS PubMed.
- X. Li, Y. H. Sun, X. Y. Yu and J. X. Tan, Azacaroline compounds and process for their preparation, CN Pat., 112174972-A, Hunan University of Science and Technology, 2021 Search PubMed.
- S. Li, Y. Sun, Y. Meng, X. Li and S. Zhang, Chin. J. Org. Chem., 2022, 42, 2390 CrossRef.
- A. A. Shafie, H. Kawashima, Y. Miyake and H. Shinokubo, ChemPlusChem, 2019, 84, 623 CrossRef PubMed.
- T. Yoshida and H. Shinokubo, Mater. Chem. Front., 2017, 1, 1853 RSC.
- CCDC documents 2217087, 2217088, 2217094, and 2217098 contain crystallographic data for this paper†.
- J. Krumsieck and M. Bröring, Chem. – Eur. J., 2021, 27, 11580 CrossRef CAS PubMed.
- P. Schweyen, K. Brandhorst, R. Wicht, B. Wolfram and M. Bröring, Angew. Chem., Int. Ed., 2015, 54, 8213 CrossRef CAS PubMed.
- D. Shimizu and A. Osuka, Chem. Sci., 2018, 9, 1409 Search PubMed.
- D. Dolphin, T. Niem, R. H. Felton and I. Fujita, J. Am. Chem. Soc., 1975, 97, 5288 CrossRef CAS PubMed.
- A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed.
- K. Miyagawa, I. Hisaki, N. Fukui and H. Shinokubo, Chem. Sci., 2021, 12, 5224 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- F. V. Lovecchio, E. S. Gore and D. H. Busch, J. Am. Chem. Soc., 1974, 96, 3109 CrossRef CAS.
- P. J. Chmielewski and L. Latos-Grażyński, Inorg. Chem., 1997, 36, 840 CrossRef CAS.
- I. Schmidt and P. J. Chmielewski, Inorg. Chem., 2003, 42, 5579 CrossRef CAS PubMed.
- P. J. Chmielewski, Inorg. Chem., 2007, 46, 1617 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and separation details, spectroscopic characterization of the compounds, electrochemistry, and crystallographic data. CCDC 2217087, 2217088, 2217094, and 2217098. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06648c |
| This journal is © The Royal Society of Chemistry 2023 |