
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling DNA nanodevices with light-switchable buffers†
Valentin Jean
Périllat‡
a,
Erica
Del Grosso‡
b,
Cesare
Berton
 a,
Francesco
Ricci
*b and
Cristian
Pezzato
*ac
a,
Francesco
Ricci
*b and
Cristian
Pezzato
*ac
aInstitut des Sciences et Ingénierie Chimiques École Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
bDepartment of Chemistry, University of Rome Tor Vergata Via della Ricerca Scientifica, 00133 Rome, Italy. E-mail: francesco.ricci@uniroma2.it
cDepartment of Chemical Sciences, University of Padua Via Marzolo 1, 35131 Padua, Italy. E-mail: cristian.pezzato@unipd.it
First published on 2nd February 2023
Abstract
Control over synthetic DNA-based nanodevices can be achieved with a variety of physical and chemical stimuli. Actuation with light, however, is as advantageous as difficult to implement without modifying DNA strands with photo-switchable groups. Herein, we show that DNA nanodevices can be controlled using visible light in photo-switchable aqueous buffer solutions in a reversible and highly programmable fashion. The strategy presented here is non-invasive and allows the remote control with visible light of complex operations of DNA-based nanodevices such as the reversible release/loading of cargo molecules.
Synthetic DNA/RNA strands are excellent engineering materials to develop nanodevices and nanomachines that can find applications in sensing,1 drug delivery,2 imaging3 and molecular transport.4 The high programmability of Watson–Crick–Franklin base-pairing, together with the reversible nature of the interaction and the possibility to use it as a versatile molecular scaffold, make synthetic DNA particularly suited to design precise nanoscale structures.2b,5,6 Responsive DNA-based nanodevices are usually developed by rationally designing programmable nucleic acid domains that can recognize specific molecular inputs like nucleic acids,7 small molecules8 or proteins.9 Actuation of such nanodevices has been achieved with a variety of exogenous stimuli including temperature10 and electric4 and magnetic11 fields. To date, several strategies have been proposed for the photo-regulation of oligonucleotide structure and function.12–21 Photocleavable groups (i.e. ortho-nitrobenzyl moieties) can be introduced into DNA strands to induce structural reconfiguration upon their cleavage at specific wavelengths.15 More recently, a novel strategy demonstrates how thymidine residues placed in close proximity in DNA nanostructures can, upon UV-light (310 nm) absorption, form stable cyclobutane pyrimidine dimers and generate covalent bridges.16 However, the cleavage and formation of covalent bonds are irreversible, yielding DNA nanostructures that remain permanently non-responsive. Reversible control has been achieved by introducing azobenzene molecular photoswitches into DNA strands.17–21 Although, azobenzene-modified DNA strands are tedious and costly to synthesize. In addition, their actuation is generally slow and requires exposure to UV-damaging light. Herein, motivated by the above considerations, we propose an alternative strategy to control the functionalities of several DNA-based systems using visible light. To do so we propose here the use of indolinospirobenzopyrans (BIPSs),22 whose open protonated merocyanine forms can dissociate protons persistently and reversibly following visible light absorption (Fig. 1a).23 For this reason, BIPSs are suitable to regulate proton transfer reactions24 in molecular machines,25 supramolecular systems,26 and materials.27 We have shown that the ground state equilibrium composition of BIPSs in water can be tuned according to the Henderson–Hasselbalch equation, and that the corresponding light-triggered pH changes can be significantly expanded up to 3 pH units.28 Recently, it was shown that a similar approach can be used for controlling the organization of DNA fibres and for inducing chirality changes in DNA-origami-based plasmonic assemblies.29 Here we employ a methoxy-substituted BIPS (hereafter simply referred to as photoacid) as a light-switchable buffering agent28b for DNA nanodevices, showing that visible light-triggered pH changes can be programmed to control complex operations such as the release/loading of cargo molecules in a completely non-invasive fashion (Fig. 1b).
 | ||
| Fig. 1 (a) (left) Schematic representation of the reversible proton release/uptake of indolinospirobenzopyrans in water and (right) corresponding visible light-triggered pH change. (b) Schematic illustration of the remote and reversible control over the functionalities of DNA-based systems achieved in this study. | ||
The photoacid employed in this study can be actuated from physiological conditions, giving rise to pH jumps from pH 7.4 to 4.8 under 500 nm light irradiation. We first demonstrate the possibility of using such visible light-triggered reversible pH changes to control the operation of DNA-based pH-dependent unimolecular switches (i.e., pH nanoswitches) (Fig. 2a).30 These switches are single stranded DNA sequences that are rationally designed to form an intramolecular triplex structure between a hairpin duplex domain and a single-strand triplex-forming portion. By exploiting the pH-dependence of the Hoogsteen interactions in the triplex structure, we and others have demonstrated the possibility to control the folding/unfolding (triplex/duplex) process with pH.31 Of note, the sequence of these switches can also be conveniently programmed to show different pKa and thus folding/unfolding over different pH windows.
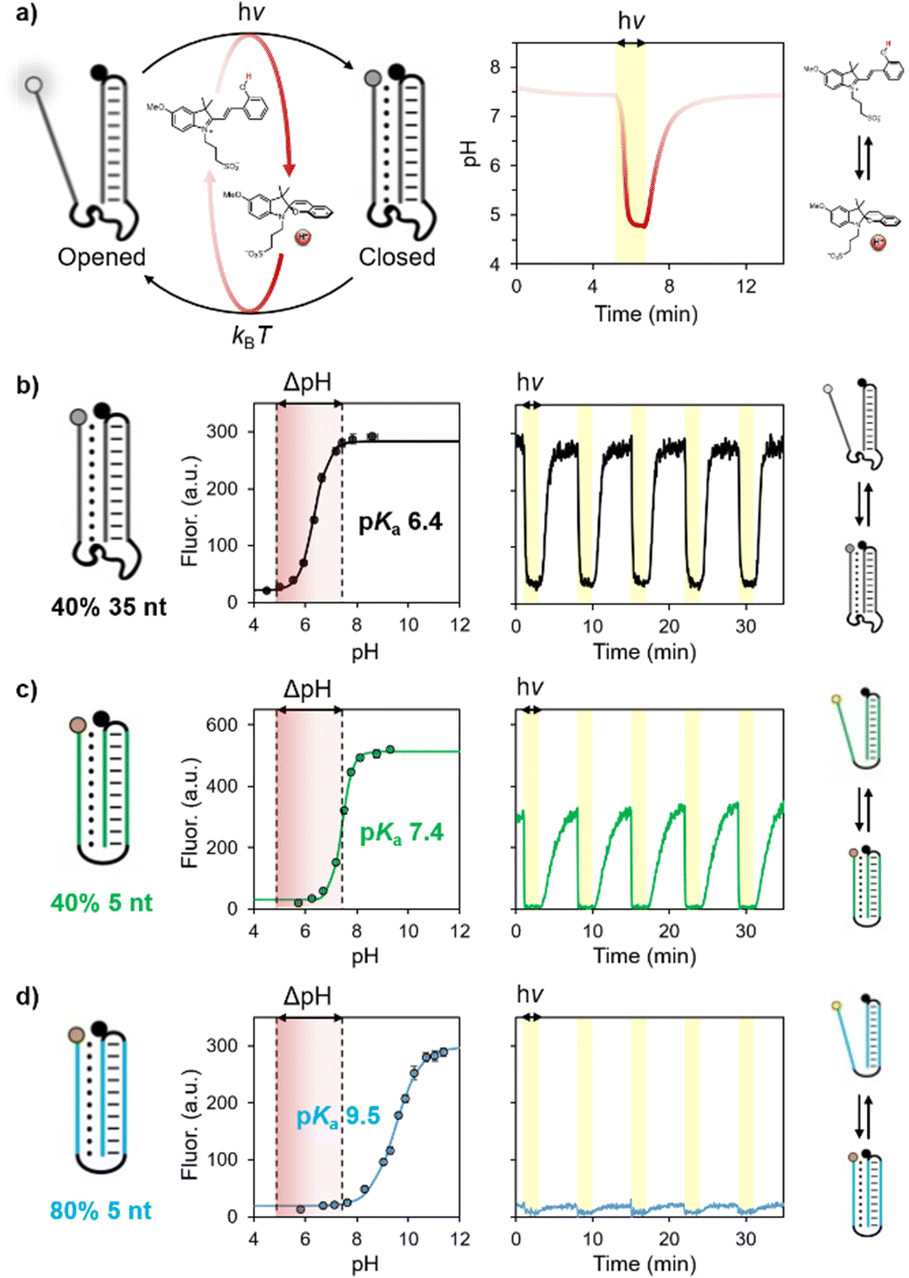 | ||
| Fig. 2 (a) Control over the folding/unfolding process of a pH-dependent DNA nanoswitch by a light-switchable buffer system that can be actuated at physiological pH. Upon light-irradiation (yellow region), the merocyanine converts into the corresponding spiropyran causing a rapid decrease of the pH from 7.4 to 4.8. Once the light irradiation is stopped, the medium returns to its original pH value. (b–d) pH titration curves and kinetic traces showing the reversibility of three different pH nanoswitches with pKa of 6.4, 7.4 and 9.5. Experimental conditions: [nanoswitch] = 15 nM, [photoacid] = 0.5 mM in an aqueous solution of NaCl (20 mM); see ESI† for more details. | ||
More specifically, in this work we employed three different DNA-based pH nanoswitches designed to show three different pH sensitivities over a wide range of pH. To measure the apparent pKa of these switches we have labelled them with pH-insensitive fluorophore/quencher pairs so that the folding/unfolding of the triplex structure can be easily followed by a change in fluorescence signal. To avoid possible optical interferences with the photoacid, whose absorption arrives up to 600 nm,31 we employed fluorophores that operate in the NIR region. The apparent pKa of the three different pH nanoswitches was probed by fluorescence spectroscopy, monitoring the progressive increase in fluorescence signal (due to triplex–duplex transition) upon consecutive additions of sodium hydroxide to a solution containing the nanoswitch and the photoacid, which act as a buffering agent in the dark (Fig. 2b–d, left). Apparent pKa values were estimated by non-linear least square fitting as described previously.30 The obtained values correlate with the different structures of the pH nanoswitches, as the increase of the pKa (from 6.4 to 9.5) can be ascribed to the shorter linker connecting the triplex-forming domains and the lower content of C–G–C Hoogsteen interactions.30c
We then moved on investigating the actuation of these pH nanoswitches with visible light. In a typical experiment, the pH nanoswitch is added to a solution containing the photoacid carefully neutralized to pH 7.4. Irradiation was provided from the top of the spectrofluorometer through a fiber-coupled LED light source (see ESI† for more details). Fluorescence time-course experiments were performed acquiring the fluorescence signal during repetitive light on/light off cycles (Fig. 2b–d, right). As expected, we found that the folding/unfolding behavior of the nanoswitches varies depending on the corresponding apparent pKa. At physiological pH, the pH nanoswitch featuring the lowest pKa (pKa = 6.4, represented in black) resides almost exclusively in its duplex conformation, as confirmed by the high fluorescence signal. Visible light irradiation (500 nm, 2 min, yellow regions) results in a rapid decrease of the solution's pH, which in turn induces the folding of the nanoswitch into its triplex conformation and a rapid decrease of the fluorescence signal. Of note, light-induced triplex formation occurs almost quantitatively as we observe more than 87% of the switch in its triplex folded conformation (see Fig. 2b). The other pH nanoswitches for which the corresponding apparent pKa equals (pKa = 7.4, green) or exceeds (pKa = 9.5, blue) the pH of the solution, display duplex/triplex distributions that are equimolar or almost totally shifted to the triplex conformation, respectively. It follows that light irradiation can shift their equilibrium distributions only to a lesser extent. Light/dark fluorescence intensities are fully compatible with those expected from the corresponding pH titration, considering the pH change of the solution from pH 7.4 to pH 4.8. In all cases, once the irradiation is stopped, a gradual increase of the fluorescence is observed, demonstrating the return of the nanoswitches to their corresponding equilibrium distributions due to the concomitant increase of the pH of the solution to the initial value in the dark. All the systems are fully reversible and display excellent stability over multiple cycles without a significant loss of efficiency (Fig. 2b–d).
To test whether we can achieve light-activated temporal control over the unfolding/folding process we decided to continue our studies with the pH nanoswitch for which the light-triggered duplex-to-triplex transition can be achieved quantitatively (pKa = 6.4, black). We first show that the pH recovery kinetics (i.e. the time needed for the solution's pH to return to the original value after stopping the light irradiation) depends on the initial pH of the solution, becoming progressively faster passing from acidic to basic pH values.28,32 This can be rationally controlled by partially neutralizing the photoacid solution to a different extent. By doing this we show that the half-life of pH recovery (t1/2) can be tuned from 8 to 1 minute by varying the neutralization degree (i.e. the equivalents of sodium hydroxide added in the solution, hereafter indicated with the Greek symbol α) from 0.19 to 0.74 (Fig. 3a). Using the same experimental conditions with our DNA pH nanoswitch we observed folding/unfolding kinetics that are fully consistent with the corresponding neutralization degree (Fig. 3b). Careful analysis of the half-life of the nanoswitch as a function of α reveals that the unfolding recovery is delayed by ca. 1 minute compared to the half-life of pH recovery (Fig. 3c). This result is consistent with the equilibration kinetics of this type of pH nanoswitch observed so far30 and indicates that our strategy is non-invasive. In addition, the actuation is highly modular, as both the extent and the duration of the folding/unfolding process can be tuned simply by varying the irradiation time (Fig. 3d).
 | ||
| Fig. 3 (a) pH changes as a function of the neutralization degree; from bottom to top, α = 0.19, 0.29, 0.37, 0.47, 0.62 and 0.74, respectively. (b) Corresponding time-course of the fluorescence intensity as a function of α. (c) Analysis of half-life as a function of α; the equilibration of the pH nanoswitch takes ca. 1 minute. (d) Switching experiment as a function of light irradiation (5 mW) time, t = 10, 5, 10 s and 1, 5, 10 and 20 minutes. Experimental conditions: [nanoswitch] = 15 nM, [photoacid] = 0.5 mM in an aqueous solution of NaCl (20 mM), T = 25 °C. | ||
Next, we wanted to see whether our approach can be extended to triplex-based receptors to achieve controlled loading/release of a DNA ligand. To do this we have employed a well-known pH-responsive receptor that allows to control the loading/release of a specific DNA ligand at different pHs.31a More specifically, we used a stem-loop structure re-engineered to contain a ligand binding site on the loop and a pH-dependent domain on the stem. At basic pH the receptor can efficiently load the ligand, whereas at acidic pH the Hoogsteen interactions of the triplex structure can stabilize the stem reducing the ability of the receptor to bind the ligand, thus inducing its release. The loading/release of the ligand can also be followed by labelling the ligand itself with a pH-independent optical pair (Fig. 4a).
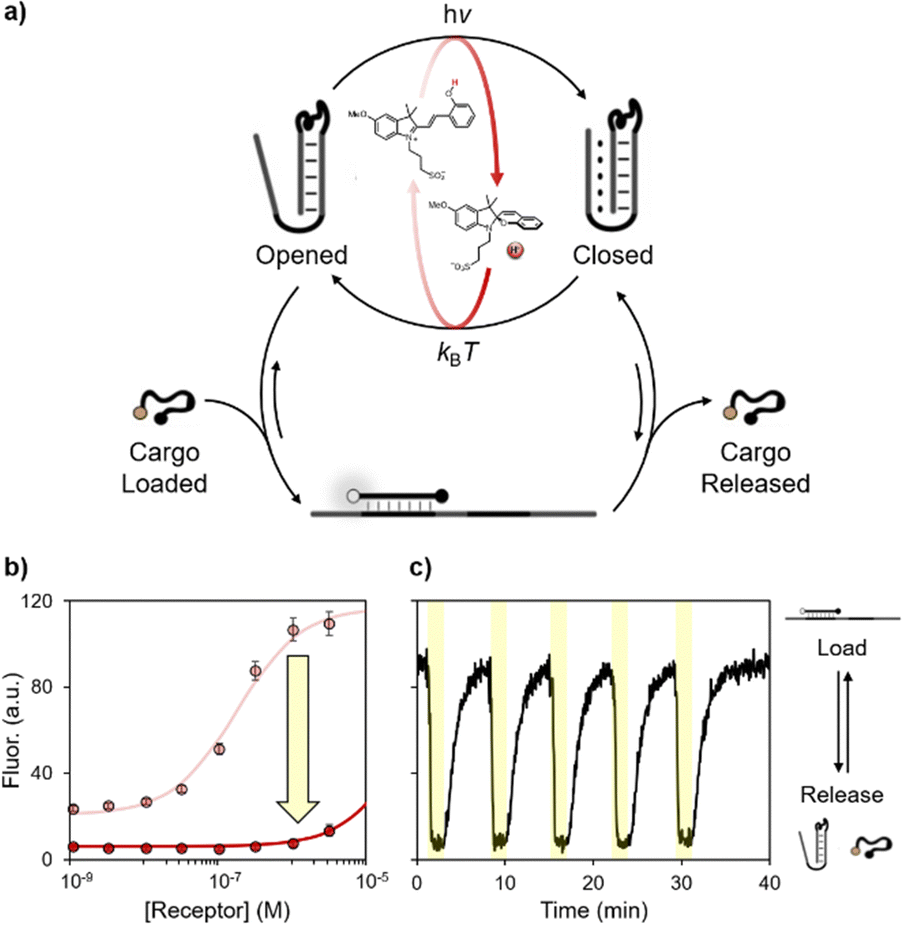 | ||
| Fig. 4 (a) Light-activated control over a DNA-based nanomachine for the loading/release of a DNA cargo. (b) Binding curves of the cargo (50 nM) obtained in the presence of the photoacid. (c) Kinetic trace of several on/off cycles obtained at a fixed concentration of receptor (320 nM) and cargo (50 nM) in the presence of the photoacid. Experimental conditions: [photoacid] = 1.0 mM in an aqueous solution of NaCl (150 mM) and MgCl2 (1 mM) neutralized until physiological pH or at pH 4.5, T = 25 °C. | ||
First, we investigated the capability of the DNA receptor to bind the ligand in our light-responsive medium neutralized at basic or acidic pHs (Fig. 4b). The apparent binding constant was estimated by non-linear least square fitting, as described previously.31a In accordance with previous studies, the apparent binding affinity decreases by ca. two orders of magnitude (Kd from 10−7 M at physiological pH to 10−5 M at pH 4.5). Finally, we tested the reversibility of this synthetic DNA receptor in our visible light-switchable buffer system (Fig. 4c). Under dark conditions and physiological pH, the high fluorescence signal indicates the loading of the ligand, which is achieved when the stem is in its duplex-structure. When the pH turns acidic after light-irradiation (500 nm, 2 minutes, yellow regions), the formation of the triplex structure induces the consequent release of the cargo, as witnessed by the rapid decrease of the fluorescence signal. Once the light irradiation is stopped, the pH of the solution gradually returns to its original value, promoting the transition to the duplex state and the re-uptake of the cargo. Also in this case the system displays excellent fatigue resistance, as the cargo can be reversibly released/loaded for at least 5 cycles.
In summary, we have described here the possibility to remotely and reversibly control the functionalities of pH-dependent DNA-based nanodevices by using visible light-switchable buffers. The majority of light-controlled DNA systems reported so far are based on azobenzene-modified oligonucleotides,17–21 which are tedious to realize and require UV-light irradiation. Given the simplicity, the (bio)compatibility, and the possibility to efficiently use visible light, we believe that the strategy presented here will open new horizons for controlling reversibly the behaviour of even more sophisticated DNA-based systems.
This research was supported by the Swiss National Science Foundation (SNSF “Ambizione” PZ00P2_180008). C. P. is thankful for support from the Italian Ministry of Education and Research (Rita Levi Montalcini Program). F. R. is thankful for support from the European Research Council, ERC (project no. 819160), Associazione Italiana per la Ricerca sul Cancro, AIRC (project no. 21965) and the Italian Ministry of University and Research (Project of National Interest, PRIN, 2017YER72K). E. D. G. acknowledges the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No 896962, “ENZYME-SWITCHES”.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840 CrossRef CAS PubMed; (b) M. Rossetti, E. Del Grosso, S. Ranallo, D. Mariottini, A. Idili, A. Bertucci and A. Porchetta, Anal. Bioanal. Chem., 2019, 411, 4293 CrossRef CAS PubMed.
- (a) C.-H. Lu, B. Willner and I. Willner, ACS Nano, 2013, 7, 8320 CrossRef CAS PubMed; (b) N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- Y. Zhang, V. Pan, X. Li, X. Yang, H. Li, P. Wang and Y. Ke, Small, 2019, 15, e1900228 CrossRef PubMed.
- E. Kopperger, J. List, S. Madhira, F. Rothfischer, D. C. Lamb and F. C. Simmel, Science, 2018, 359, 296 CrossRef CAS PubMed.
- (a) N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed; (b) Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124 CrossRef CAS PubMed.
- V. Linko and H. Dietz, Curr. Opin. Biotechnol, 2013, 24, 555 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605 CrossRef CAS PubMed.
- J. Deng and A. Walther, Adv. Mater., 2020, 32, e2002629 CrossRef PubMed.
- A. Bertucci, A. Porchetta, E. Del Grosso, T. Patiño, A. Idili and F. Ricci, Angew. Chem., Int. Ed., 2020, 59, 20577 CrossRef CAS PubMed.
- D. Gareau, A. Desrosiers and A. Vallée-Bélisle, Nano Lett., 2016, 16, 3976 CrossRef CAS PubMed.
- S. Lauback, K. R. Mattioli, A. E. Marras, M. Armstrong, T. P. Rudibaugh, R. Sooryakumar and C. E. Castro, Nat. Commun., 2018, 9, 1446 CrossRef PubMed.
- (a) A. S. Lubbe, W. Szymanski and B. L. Feringa, Chem. Soc. Rev., 2017, 46, 1052 RSC; (b) N. A. Simeth, P. de Mendoza, V. R. A. Dubach, M. C. A. Stuart, J. W. Smith, T. Kudernac, W. R. Browne and B. L. Feringa, Chem. Sci., 2022, 13, 3263 RSC.
- X. R. Liu, X. Hu, I. Y. Loh and Z. Wang, Nanoscale, 2022, 14, 5899 RSC.
- W. Chen, B. Willner and I. Willner, React. Funct. Polym., 2021, 104983 Search PubMed.
- (a) R. E. Kohman and X. Han, Chem. Commun., 2015, 51, 5747 RSC; (b) J. Deng, D. Bezold, H. J. Jessen and A. Walther, Angew. Chem., Int. Ed., 2020, 59, 12084 CrossRef CAS PubMed; (c) S. Yang, P. A. Pieters, A. Joesaar, B. W. A. Bögels, R. Brouwers, I. Myrgorodska, S. Mann and T. F. A. de Greef, ACS Nano, 2020, 14, 15992 CrossRef CAS PubMed; (d) I. Seitz, H. Ijäs, V. Linko and M. A. Kostiainen, ACS Appl. Mater. Interfaces, 2022, 14, 38515 CrossRef CAS PubMed.
- T. Gerling, M. Kube, B. Kick and H. Dietz, Sci. Adv., 2018, 4, eaau1157 CrossRef CAS PubMed.
- Y. Yang, M. Endo, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2012, 134, 20645 CrossRef CAS PubMed.
- Y. Kamiya and H. Asanuma, Acc. Chem. Res., 2014, 47, 1663 CrossRef CAS PubMed.
- P. Li, G. Xie, X.-Y. Kong, Z. Zhang, K. Xiao, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 15637 CrossRef CAS PubMed.
- A. Kuzyk, Y. Yang, X. Duan, S. Stoll, A. O. Govorov, H. Sugiyama, M. Endo and N. Liu, Nat. Commun., 2016, 7, 10591 CrossRef CAS PubMed.
- J. Wang, Z. Li, Z. Zhou, Y. Ouyang, J. Zhang, X. Ma, H. Tian and I. Willner, Chem. Sci., 2021, 12, 11204 RSC.
- L. Kortekaas and W. R. Browne, Chem. Soc. Rev., 2019, 48, 3406 RSC.
- (a) K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 7854 CrossRef CAS; (b) Z. Shi, P. Peng, D. Strohecker and Y. Liao, J. Am. Chem. Soc., 2011, 133, 14699 CrossRef CAS PubMed.
- (a) Y. Liao, Acc. Chem. Res., 2017, 50, 1956 CrossRef CAS PubMed; (b) Y. Liao, Phys. Chem. Chem. Phys., 2022, 24, 4116 RSC.
- (a) S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378 CrossRef CAS PubMed; (b) L. A. Tatum, J. T. Foy and I. Aprahamian, J. Am. Chem. Soc., 2014, 136, 117438 CrossRef PubMed; (c) L. P. Yang, F. Jia, J. S. Cui, S. B. Lu and W. Jiang, Org. Lett., 2017, 19, 2945 CrossRef CAS PubMed; (d) Q. Shi, Z. Meng, J. F. Xiang and C. F. Chen, Chem. Commun., 2018, 54, 3536 RSC; (e) N. Rad and V. Sashuk, Chem. Sci., 2022, 13, 12440 CAS.
- (a) P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Borner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646 CrossRef CAS PubMed; (b) Q. Shi and C. F. Chen, Org. Lett., 2017, 19, 3175 CrossRef CAS PubMed; (c) H. Zhang, J. Muhammad, K. Liu, R. H. A. Ras and O. Ikkala, Nanoscale, 2019, 11, 14118 RSC.
- (a) C. Maity, W. E. Hendriksen, J. H. Van Hesch and R. Eelkema, Angew. Chem., Int. Ed., 2015, 54, 998 CrossRef CAS PubMed; (b) X. Li, J. Fei, Y. Xu, D. Li, T. Yuan, G. Li, C. Wang and J. Li, Angew. Chem., Int. Ed., 2018, 57, 1903 CrossRef CAS PubMed; (c) C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447 CrossRef CAS PubMed; (d) R. J. Li, C. Pezzato, C. Berton and K. Severin, Chem. Sci., 2021, 12, 4981 RSC.
- (a) C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457 RSC; (b) C. Berton, D. M. Busiello, S. Zamuner, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Angew. Chem., Int. Ed., 2021, 60, 21737 CrossRef CAS PubMed.
- (a) F. J. Rizzuto, C. M. Platnich, X. Luo, Y. Shen, M. D. Dore, C. Lachance-Brais, A. Guarné, G. Cosa and H. F. Sleiman, Nat. Chem., 2021, 13, 843 CrossRef CAS PubMed; (b) J. Ryssy, A. K. Natarajan, J. Wang, A. J. Lehtonen, M.-K. Nguyen, R. Klajn and A. Kuzyk, Angew. Chem., Int. Ed., 2021, 60, 5859 CrossRef CAS PubMed.
- (a) Y. Hu, A. Cecconello, A. Idili, F. Ricci and I. Willner, Angew. Chem., Int. Ed., 2017, 56, 15210 CrossRef CAS PubMed; (b) A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836 CrossRef CAS PubMed; (c) D. Mariottini, A. Idili, M. A. D. Nijenhuis, G. Ercolani and F. Ricci, J. Am. Chem. Soc., 2019, 141, 11367 CrossRef CAS PubMed.
- (a) A. Porchetta, A. Idili, A. Vallée-Bélisle and F. Ricci, Nano Lett., 2015, 15, 4467 CrossRef CAS PubMed; (b) I. A. P. Thompson, L. Zheng, M. Eisenstein and H. T. Soh, Nat. Commun., 2020, 11, 2946 CrossRef CAS PubMed.
- V. J. Périllat, C. Pezzato and C. Pezzato, Mater. Today Chem., 2022, 25, 100918 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. Containing experimental procedures details. See DOI: https://doi.org/10.1039/d2cc06525h |
| ‡ V. J. P. and E. D. G. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |