
Minisci reaction of heteroarenes and unactivated C(sp3)–H alkanes via a photogenerated chlorine radical†
Zi-Tong
Pan
,
Li-Miao
Shen
,
Fentahun Wondu
Dagnaw
,
Jian-Ji
Zhong
 ,
Jing-Xin
Jian
* and
Qing-Xiao
Tong
*
,
Jing-Xin
Jian
* and
Qing-Xiao
Tong
*
College of Chemistry and Chemical Engineering, Key Laboratory for Preparation and Application of Ordered Structural Material of Guangdong Province, and Guangdong Provincial Key Laboratory of Marine Disaster Prediction and Prevention, Shantou University, Shantou, Guangdong 515063, China. E-mail: jxjian@stu.edu.cn; qxtong@stu.edu.cn
First published on 13th January 2023
Abstract
Here, an efficient Minisci reaction of heteroarenes and unactivated C(sp3)–H alkanes was achieved using an inexpensive FeCl3 as a photocatalyst. The photogenerated chlorine radical contributed to the HAT of C–H and subsequently initiated this reaction. Surprisingly, salt water and even seawater can act as a chlorine radical source, which provided an enlightening idea for future organic synthesis methods.
N-heteroarenes, such as benzothiazole and quinoline, are key structural motifs of many natural products, bioactive compounds, and pharmaceuticals (Scheme 1a).1 The derivatizations of the functionalization of benzothiazoles and quinolines have attracted the attention of many researchers because of their wide existence. The Minisci reaction, which involves the addition of carbon-centered radicals to basic heteroarenes followed by formal hydrogen atom loss,2 has provided a robust tool for the late-stage alkylation of N-containing heteroarenes because of its high atomic economy.3 But the photocatalytic derivatization and functionalization of alkylating reagents to produce alkylated carbon radicals are confined to active C(sp3)–H compounds. To date, only a few inactive C(sp3)–H compounds have been used as alkyl reagents. Besides, equivalent oxidants including peroxides,4 iodine reagents,5 persulfate (S2O82−)6 and Selectfluor7 are required to promote this type of reaction by activating the substrate.
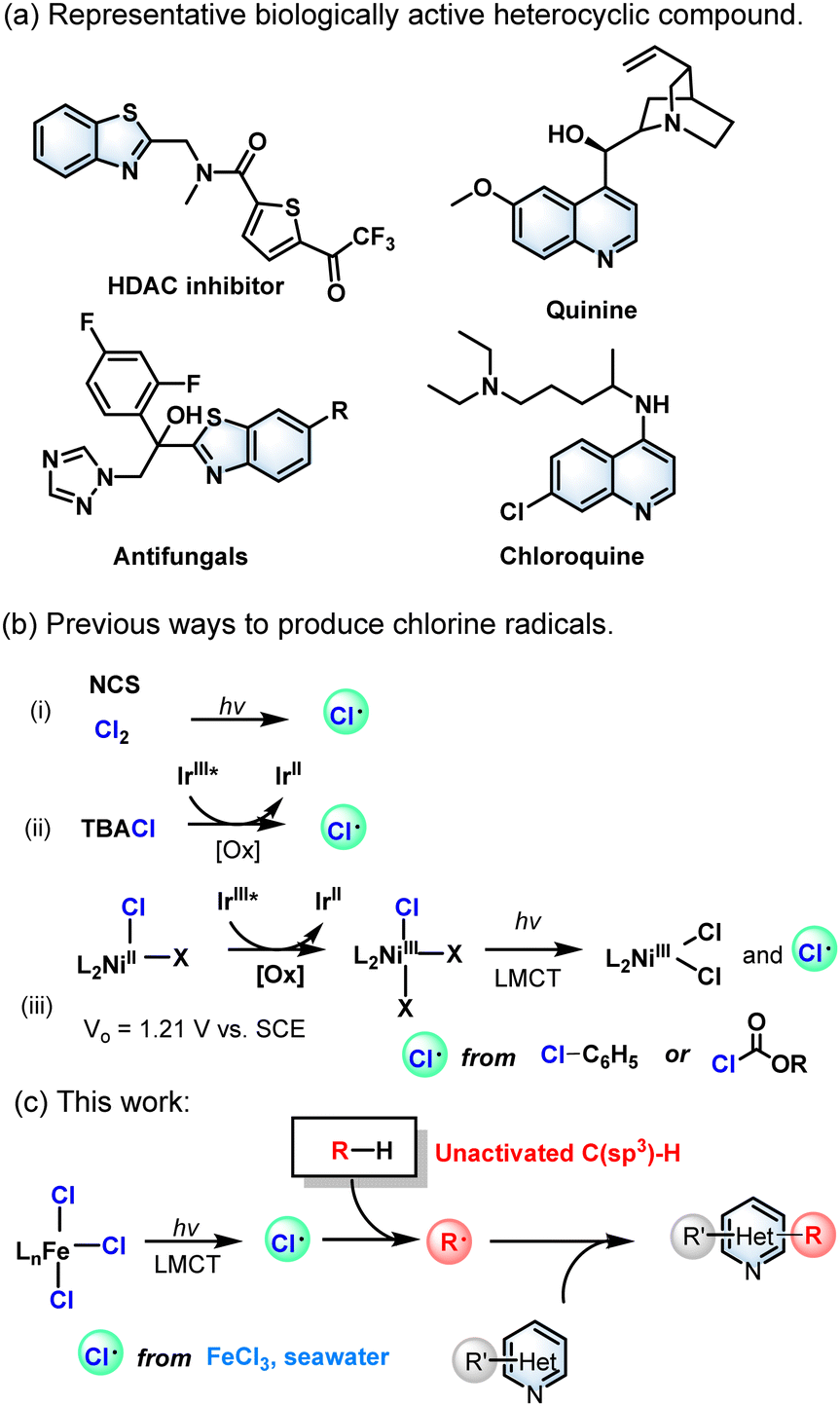 | ||
| Scheme 1 (a) Representative biologically active nitrogenous heterocyclic compound. (b) Previous ways to produce chlorine radicals. (c) This work. | ||
Alkanes, the major constituents of petroleum and natural gas, are widely distributed in nature and are mainly used as fuels, but few are utilized as economical chemical feedstocks.8 Unlike the C–H bonds activated by heteroatoms, aldehydes, benzylic and allylic groups,9 the unactivated C(sp3)–H bonds in samples of alkanes have great intrinsic inertness and uncontrollable chemoselectivity due to their high bond dissociation energies (BDEs) of >95 kcal mol−1.9b Therefore, the functionalization of non-activated C(sp3)–H bonds has always been a research hotspot in organic synthetic chemistry. In recent years, the photocatalytic system based on the intermolecular hydrogen atom transfer (HAT) process was developed,10 which could seize the hydrogen atoms of inactive C(sp3)–H. In addition, the selectivity of the HAT process depends not only on the activated C(sp3)–H bond of the substrate, but also on the electronic effects of the HAT reagent as well as on spatial effects. However, current strategies involving C(sp3)–H bond homolytic cleavage mainly rely on a HAT strategy with bromine, nitrogen, and oxygen as the central electrophilic heteroatom radicals, and strong reducing catalysts or oxidants are required under harsh experimental conditions.11
The chlorine radical (Cl˙) is an effective HAT reagent that can cleave various C(sp3)–H bonds.12 Chlorine (Cl2)13 and N-chlorosuccinimide (NCS) are the two conventional reagents that produce Cl˙, which in turn can afford alkyl radicals via the HAT process.14 Consequently, the chlorine radical (Cl˙) formed from chloride ions (Cl−) as the HAT reagents has a greater potential for C(sp3)–H bond functionalization (Scheme 1b). In previous reports, noble metal catalysts15 and strong chemical oxidants16 were usually required in this process. The direct oxidation of soluble chlorides to Cl˙ requires a high oxidation potential (+ 1.21 V vs. SCE),15c which is intolerant to some of the easily oxidized substrates. On the other hand, photoinduced ligand-to-metal charge transfer (LMCT) can well contribute to the generation of HAT reagents in photocatalytic systems.15a,17 However, a mild photocatalytic system avoiding the commonly used strong chemical oxidants and noble metal catalysts is still a serious challenge and is highly required for unactivated C(sp3)–H substrates. In recent years, there have been some reports about photoinduced iron-catalysed Minisci reactions.18 Herein, we describe a Minisci alkylation reaction of unactivated alkanes catalysed by inexpensive ferric chloride (FeCl3) under mild reaction conditions (Scheme 1c). Interestingly, salt water and even seawater can be used as chloride sources to promote this reaction, which ideally meets the concept of sustainable development and green chemistry.
For the outset, we began the optimization of the reaction conditions by choosing model substrates 4-methylquinoline (also called lepidine) (a) as a representative hetero compound and cyclohexane (b) as the model substrates (Scheme 2). After an extensive optimization study (Scheme 2, entries 1–10), the product c was obtained in a high isolated yield of 82% under the optimal conditions: lepidine (0.2 mmol, 1.0 equiv.), cyclohexane (0.5 mL, 20 equiv.), trifluoroacetic acid (TFA) (1.5 equiv.), 10% FeCl3 catalyst, 50% LiCl additive, in 2.0 mL CH3CN solvent, under irradiation of 385 nm light in an air atmosphere. The control experiments described in entries 2–3 showed that the reaction did not occur in the absence of light, and that there was a decreased yield under visible-light (>400 nm) illumination. The light source was indispensable for this photocatalytic cross-coupling reaction of a heterocyclic compounds with cyclohexane. In addition, the reaction did not proceed effectively without the strong acid additive of TFA (entries 1 and 4–5), indicating that the protonation of the N-atom can activate the heteroarenes, a typical feature of the Minisci reaction. Copper chloride could also be used as the catalyst rather than ferric chloride, but the yield was relatively low (entries 1 and 6–7). These results indicated that iron cations played a key role in the catalytic process. The isolated yields of the product after addition of LiCl have significantly increased from 25% to 82%, compared to that without LiCl (entries 1 and 8). Considering the abundance of the element chlorine in natural seawater, we assumed that it may also be feasible to use seawater as the source of chlorine ions. The evidence showed that it was also effective to replace the addition of LiCl with seawater, and the yield is up to 65% (entry 9). After replacing the air atmosphere with high purity argon, the yield of this reaction was dramatically decreased (entry 10), hence based on the optimized reaction conditions, oxygen is a crucial factor to promote the radical reaction.
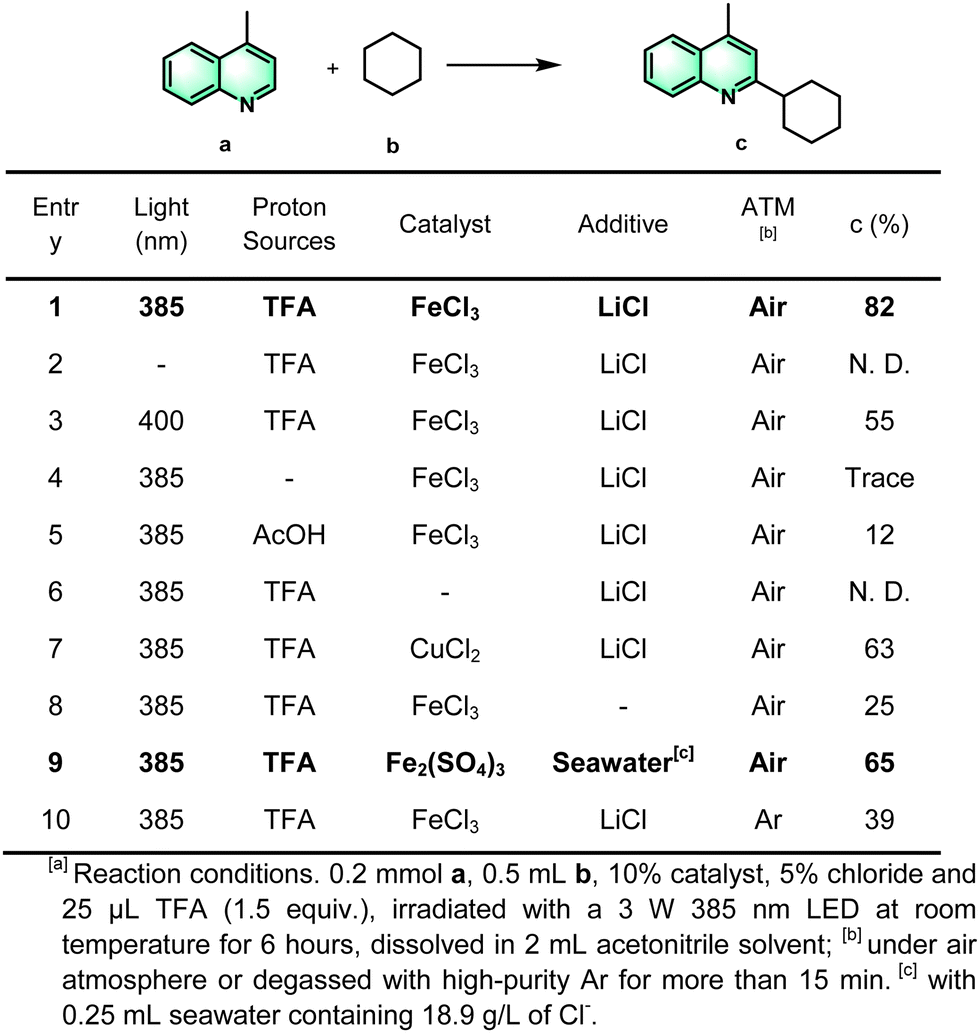 | ||
| Scheme 2 Optimization of the reaction conditions.a | ||
Under the optimal conditions, we expanded the range of different heterocyclic aromatic hydrocarbons. As shown in Scheme 3, quinolines substituted at the C4 or C2 positions reacted well to give the C2 or C4 coupling products (1c–2c), respectively, in good yields. In addition, for quinolines containing hydroxyl groups, the reaction was also tolerated, and the product (3c) was obtained in a medium yield. Moreover, for unsubstituted quinolines, the C2-substituted products were obtained in a moderate yield (4c). In order to study the applications of this type of reaction, bioactive intermediates, such as benzylation of 4-chloro-6,7-dimethoxyquinoline provided a fair yield (5c). Besides the quinoline, we also applied the optimized reaction conditions to heterocyclic compounds with two N atoms. To our delight, p-hydroxyquinazoline and quinazolinone also demonstrated satisfactory reaction efficiencies (6c–7c). Heterocyclic compounds containing S atoms and their derivatives, such as benzothiazole, are also very important in organic synthesis. Our group previously reported the synthesis of alkylated benzothiazole derivatives from alcohols as alkylating agents,19 and here, we were surprised to find that the present reaction conditions were also applicable to the synthesis of alkylated benzothiazoles (8c). Many cross-coupling-useful functional groups, such as cyano (9c), chloro (10c–11c), fluoro (12c), methyl (13c), and ether (14c–15c) groups were all compatible and achieved moderate to good yields.
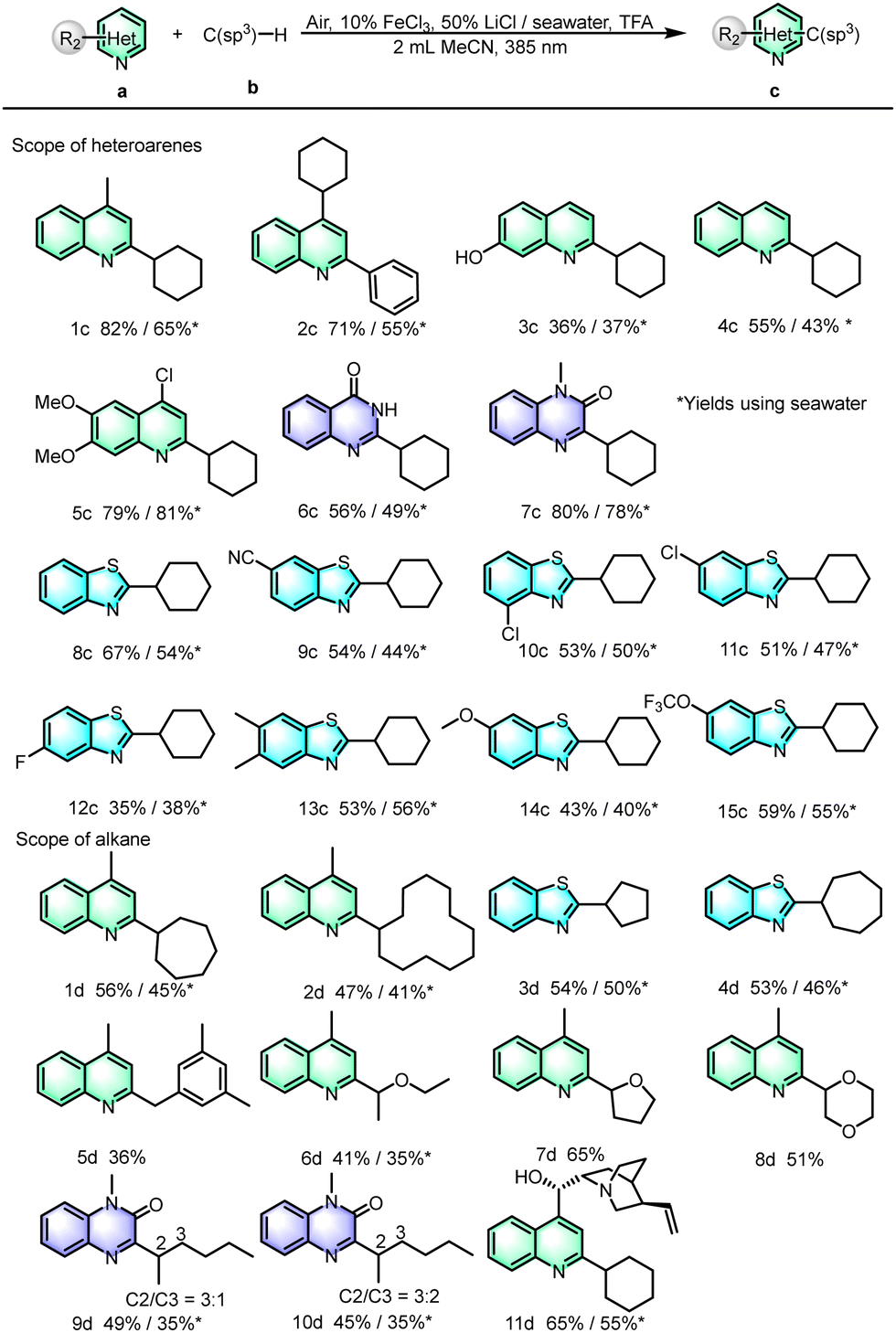 | ||
| Scheme 3 Scope of heteroarenes and alkanes. | ||
After investigating the reaction compatibility of heteroarenes, the substrate scope of unactivated alkanes was then examined. For instance, the corresponding products have been obtained in ordinary yields (1d–2d) for seven-membered cycloalkanes and even for cycloalkanes with up to twelve-membered rings, indicating that the chlorine radical is effective for the activation of non-activated hydrocarbon bonds with large positional resistance. Similarly, cyclopentane and cycloheptane can be coupled with benzothiazole in moderate yields to obtain coupling products (3d–4d). Furthermore, the C(sp3)–H substrates m-trimethylbenzene and ethers consisting of diethyl ether, tetrahydrofuran and 1,4-dioxane have been well reacted with quinoline to afford the C-2 substituted products in moderate to good yields (5d–8d). The linear alkanes n-pentane and n-hexane were tolerated for this reaction with moderate yields (9d–10d). In addition, the reactions between cyclohexane and the drug molecule cinchonine can proceed, giving the corresponding product (11d) with isolated yields of 65% and 55% in the absence and presence of seawater, respectively. All the results demonstrated the good functional group tolerance and utilization potential of the reaction.
To have a better understanding of the reaction mechanism, 1,1-diphenylethylene (DPE) was added and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was added as a suitable radical scavenger (Scheme 4a). The experiment result shows that the reaction process was significantly inhibited, indicating that the transformation may undergo a radical pathway. The formation of adducts (1c′) and (1c′′) indicates the generation of cyclohexyl and chlorine radicals.17c A kinetic isotope effect (KIE) experiment was carried out to determine whether the hydrogen atom transfer (HAT) is the rate-determining step of the whole reaction process (Scheme 4b). The preparation of the products 1c-d11 followed the general procedure using heteroarene 1a and deuterated cyclohexane 2b-d12 as the starting materials. The isolated products of 1c and 1c-d11 were obtained by column chromatography. The prominent isotope effect (kH/kD = 1.8) indicated that C–H homocleavage of cyclohexane was the rate-determine-step (Fig. S1, ESI†). The reaction quantum yield (Φ) was determined to be 23.6% (ESI,† Fig. S2), indicating that there was no significant radical chain propagation process in this reaction (Scheme 5).
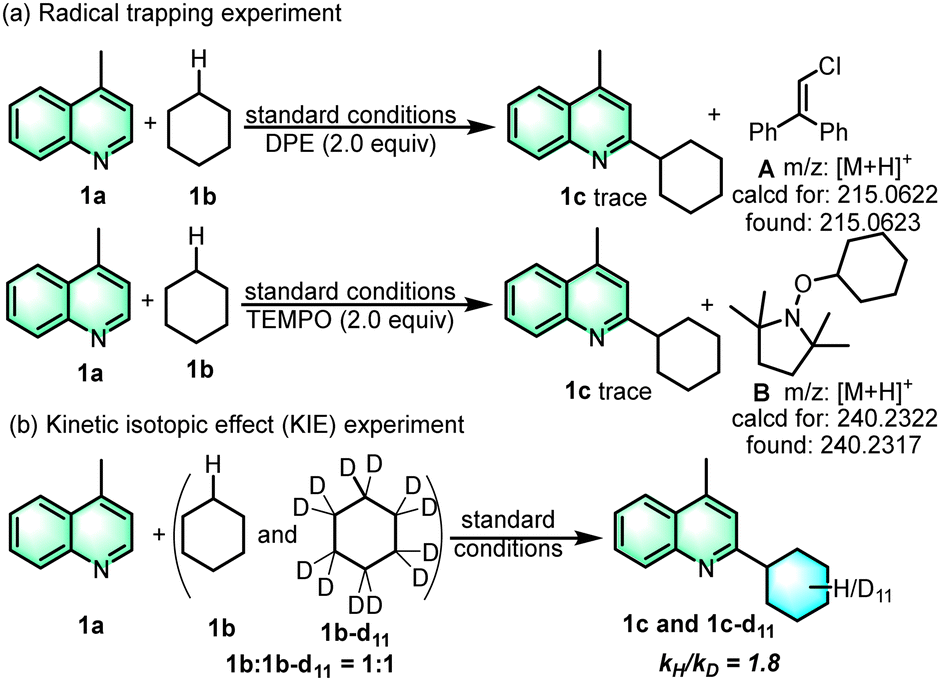 | ||
| Scheme 4 Mechanistic investigation experiments. | ||
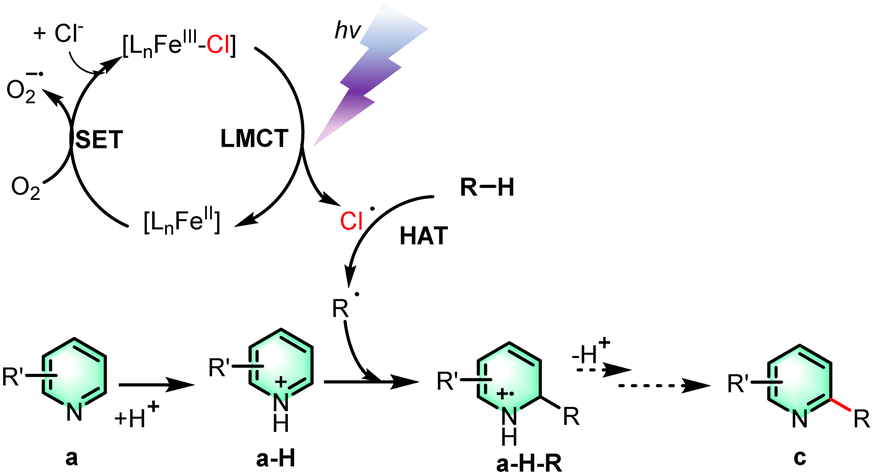 | ||
| Scheme 5 Proposed mechanism. | ||
Based on the results of the mechanistic investigation experiments, a feasible reaction mechanism has been postulated. Firstly, the trivalent iron complex is excited by light and undergoes the process of LMCT to form a chlorine radical and a molecule of the divalent iron complex. The divalent iron complex then undergoes a single electron transfer process with oxygen to re-form the iron complex. The resulting chlorine radical can seize the hydrogen atom from the alkane and undergo a hydrogen atom transfer process to form an alkyl radical, which subsequently attacks quinoline at the C-2 position that was previously protonated by proton abstraction from TFA. The intermediate a-H-R is deprotonated and oxidized to form the product c. Moreover, verification experiments showed that a-H-R went through the elimination process to get to the final product (see the ESI†). We speculated that the halogen atom at the C-2 position is readily departed under the reaction conditions to add to the alkane substrate.
In summary, we reported a highly efficient, green and mild method for the unactivated C(sp3)–H functionalisation of alkanes by using ferric chloride as the catalyst. Heterocyclic aromatics including quinoline, benzothiazole and quinazoline, as well as compounds with diverse functional groups can effectively react. The most critical part of this reaction is the generation of chlorine radicals from Fe(III)–Cl LMCT processes. The method has the potential to become an important tool for the functionalization of advanced synthetic intermediates, which have many potential applications in medicinal chemistry. Combined with its simplicity and sustainability, it will help to simplify the synthesis of complex heterocyclic compounds in academia and industry.
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 52273187 and 51973107), the Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme 2019 (GDUPS 2019), the start-up fund from Shantou University (NTF20033), the Natural Science Foundation of Guangdong Province (2022A1515110372) and the Department of Education of Guangdong Province (2021KCXTD032).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. G. Mortimer, G. Wells, J.-P. Crochard, E. L. Stone, T. D. Bradshaw, M. F. G. Stevens and A. D. Westwell, J. Med. Chem., 2005, 49, 179 CrossRef PubMed; (b) K. Komatsu, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 13447 CrossRef CAS PubMed; (c) C. Huang, J. H. Wang, J. Qiao, X. W. Fan, B. Chen, C. H. Tung and L. Z. Wu, J. Org. Chem., 2019, 84, 12904 CrossRef CAS PubMed.
- Y. H. Wu, N. X. Wang, T. Zhang, L. Y. Zhang, X. W. Gao, B. C. Xu, Y. Xing and J. Y. Chi, Org. Lett., 2019, 21, 7450 CrossRef CAS PubMed.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666 CrossRef CAS PubMed.
- (a) H. Zhao, Z. Li and J. Jin, New J. Chem., 2019, 43, 12533 RSC; (b) L. Zhou and H. Togo, Eur. J. Org. Chem., 2019, 1627 CrossRef CAS.
- (a) X. Li, C. Liu, S. Guo, W. Wang and Y. Zhang, Eur. J. Org. Chem., 2020, 411 Search PubMed; (b) X. Shao, X. Wu, S. Wu and C. Zhu, Org. Lett., 2020, 22, 7450 CrossRef CAS PubMed; (c) Y. Wang, L. Yang, S. Liu, L. Huang and Z. Q. Liu, Adv. Synth. Catal., 2019, 361, 4568 CrossRef CAS.
- H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709 CrossRef CAS PubMed.
- H. Zhao and J. Jin, Org. Lett., 2019, 21, 6179 CrossRef CAS PubMed.
- K. I. Goldberg and A. S. Goldman, Acc. Chem. Res., 2017, 50, 620 CrossRef CAS PubMed.
- (a) L. J. J. Laarhoven and P. Mulder, Acc. Chem. Res., 1999, 32, 342 CrossRef CAS; (b) S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 252 CrossRef.
- (a) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem. Catal., 2021, 1, 523 CrossRef; (b) C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714 CrossRef CAS PubMed; (c) C. M. Zhu, R. B. Liang, Y. Xiao, W. Zhou, Q. X. Tong and J. J. Zhong, Green Chem., 2023 10.1039/d2gc03347j; (d) X. K. Qi, H. Zhang, Z. T. Pan, R. B. Liang, C. M. Zhu, J. H. Li, Q. X. Tong, X. W. Gao, L. Z. Wu and J. J. Zhong, Chem. Commun., 2019, 55, 10848 RSC.
- (a) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875 CrossRef CAS PubMed; (b) Z. Ye, Y. M. Lin and L. Gong, Eur. J. Org. Chem., 2021, 5545 CrossRef CAS.
- (a) P. Xu, P. Y. Chen and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275 CrossRef CAS PubMed; (b) G. B. Panetti, Q. Yang, M. R. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Catal., 2022, 2, 853 CrossRef; (c) K. Niu, X. Shi, L. Ding, Y. Liu, H. Song and Q. Wang, ChemSusChem, 2022, 15, e202102326 CrossRef CAS PubMed; (d) X. Zhang and R. Zeng, Org. Chem. Front., 2022, 9, 4955 RSC; (e) Q. Zhang, S. Liu, J. Lei, Y. Zhang, C. Meng, C. Duan and Y. Jin, Org. Lett., 2022, 24, 1901 CrossRef CAS PubMed; (f) P. Lian, R. Li, L. Wang, X. Wan, Z. Xiang and X. Wan, Org. Chem. Front., 2022, 9, 4924 RSC.
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1593 CrossRef CAS PubMed.
- Z. Y. Dai, S. Q. Zhang, X. Hong, P. S. Wang and L. Z. Gong, Chem. Catal., 2022, 2, 1211 CrossRef.
- (a) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719 CrossRef CAS PubMed; (b) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191 CrossRef CAS PubMed; (c) S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664 CrossRef CAS PubMed; (d) M. Bhakat, B. Khatua and J. Guin, Org. Lett., 2022, 24, 5276 CrossRef CAS PubMed.
- (a) A. J. Cresswell, S. T. Eey and S. E. Denmark, Angew. Chem., Int. Ed., 2015, 54, 15642 CrossRef CAS PubMed; (b) J. C. Sarie, J. Neufeld, C. G. Daniliuc and R. Gilmour, ACS Catal., 2019, 9, 7232 CrossRef CAS.
- (a) S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729 CrossRef CAS PubMed; (b) D. Shao, Y. Wu, S. Hu, W. Gao, Y. Du, X. Jia, S. Liu, M. Zhou and J. Chen, ACS Sustainable Chem. Eng., 2022, 10, 10294 CrossRef CAS; (c) J. Xu, H. Cai, J. Shen, C. Shen, J. Wu, P. Zhang and X. Liu, J. Org. Chem., 2021, 86, 17816 CrossRef CAS PubMed.
- (a) Y. Ning, S. Wang, M. Li, J. Han, C. Zhu and J. Xie, Nat. Commun., 2021, 12, 4637 CrossRef CAS PubMed; (b) S. Wang, T. Li, C. Gu, J. Han, C. G. Zhao, C. Zhu, H. Tan and J. Xie, Nat. Commun., 2022, 13, 2432 CrossRef CAS PubMed; (c) S. Xia, K. Hu, C. Lei and J. Jin, Org. Lett., 2020, 22, 1385 CrossRef CAS PubMed; (d) N. Xiong, Y. Li and R. Zeng, Org. Lett., 2021, 23, 8968 CrossRef CAS PubMed.
- Z. T. Pan, X. K. Qi, Q. Xiao, X. W. Liang, J. J. Zhong, J. X. Jian and Q. X. Tong, Chem. Commun., 2022, 58, 8810 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06486c |
| This journal is © The Royal Society of Chemistry 2023 |