
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Converting pH probes into “turn-on” fluorescent receptors for anions
Evgeny A.
Kataev

Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Nikolaus-Fiebiger-Str. 10, 91058 Erlangen, Germany. E-mail: evgeny.kataev@fau.de
First published on 27th January 2023
Abstract
Recognition of anions by synthetic receptors is an integral part of supramolecular chemistry continuing to expand and find new application areas in our daily life. Many applications require visualization of anion recognition events, and the generated analytical signal is used to quantify anions in solution. Transferring a binding event to a measured signal is a challenging task. The design of a synthetic receptor must involve not only the perfectly positioned binding sites with complementary noncovalent interactions for a guest but should also realize the sensing mechanism that generates a strong analytical response upon guest binding. This feature article outlines the design concept for the construction of “turn-on” fluorescent receptors for anions involving fluorescent pH probes. Applications of this concept for the construction of synthetic fluorescent receptors for inorganic anions and nucleotides are described. Features of the obtained receptors and possible competing binding and sensing processes in solution are analyzed to understand the scope and limitations of the approach.
 Evgeny A. Kataev | Evgeny A. Kataev studied Chemistry at the M.V. Lomonosov Moscow State University (2003). He obtained his PhD in 2006 from the joint program of MSU (Prof. Y. A. Ustynyuk) and the University of Texas at Austin (Prof. J. L. Sessler). He was a postdoctoral fellow at EPFL Lausanne (Prof. Kay Severin) and the University of Regensburg (Prof. Burkhard König). In 2011 he joined TU Chemnitz as a junior professor. In 2019 he was an interim professor at FAU Erlangen-Nürnberg, and since April 2020 continued at FAU as a Heisenberg Fellow. His research interests are in the areas of organic synthesis, supramolecular recognition and sensing in water. |
Introduction
The challenge of supramolecular recognition of anionic species was postulated several decades ago.1 However, the significance of this area has only been realized during the last three decades, mainly due to the identification of the critical role of anion binding and transport in biological systems, catalysis and analytical applications.2 Moreover, nucleic acids are polyanions, and similar recognition and sensing principles could be applied.3The design of synthetic receptors for anions is a difficult task for several reasons. Being larger than the isoelectronic cations, anions have a smaller charge-to-radius ratio, which reduces the contribution of electrostatic interaction to their binding. The high ability to form hydrogen bonds with proton-donor solvents increases their solvation energies, so the receptor must have a very high affinity for the anion to compete effectively with solvent molecules in such environments. Therefore, it is especially challenging to design those synthetic hosts for anions that work in an aqueous solution. On the one hand, good hydrogen bond donors like NH, CH or OH-binding sites are ideal for binding anions, but on the other hand, these sites are strongly solvated by water.3a,4 The approaches how to override this problem have been discussed recently in several review articles.4,5
Another problem in the anion recognition area is the functionalization of a designed receptor with dyes to visualize the binding event. This problem can be easily recognized by comparison of the number of commercially available probes for cations and anions. There are more probes available for cations than for anions. In most cases, anionic species are detected by enzymatic assays.6 Fluorescent probes are highly attractive for many applications due to the successful precedents in cellular investigations7 and online monitoring of anions in the environment.8 Thus, there is a cogent need to develop general approaches to convert binding events into fluorescent signals.8b
Until now, several approaches have been proven to be versatile in many instances, such as indicator displacement assay,9 aggregation/disaggregation10 induced emission or quenching,11 monomer–excimer equilibrium,12 excited-state intramolecular proton transfer-based probes,13 metal-based molecular sensors14 and photoinduced electron transfer (PET)-based probes.15 Many fluorescent probes with receptor-fluorophore design, show quenching upon anion addition.15c,16
However, the “turn-on response” would be preferable in application to achieve high signal-to-noise ratio. PET was among the first approaches used to construct “turn-on” fluorescent probes for anions working in an aqueous solution. The working principle of PET sensors was demonstrated by Czarnik in 1989 with receptor 1.17 Further development of this approach resulted in a pyrophosphate probe, which was prepared by functionalization of anthracene at 1st and 8th positions with tetraamines.18 The coordination of an anion to 1 resulted in the protonation of the benzylic amine leading to the inhibition of PET and subsequent fluorescence enhancement (Fig. 1).19 It is suggested that the OH-group of the phosphate anion protonates the free benzylic amine. However, in the original paper, it is also mentioned that other anions, such as sulfate, citrate and ATP induce the similar effect. Although it can be valid for phosphate, sulfate does not bear OH-group but nevertheless induces a strong fluorescent enhancement. These results indicate that supramolecular pKa shift could be the reason for protonation. Since sulfate is a dianion at pH 6, additional protons should come from water (Fig. 1). Several research groups used this approach to detect anions and nucleotides in solution.20 These fluorescent probes rest on PET sensors and molecular logic gates developed by de Silva and others.15a,15b,15e,21 PET principle is applicable for detection of Zn(II) and other cations with the help of polyamine receptors. Consequently, these complexes can also be used to detect anions.10,22 Great contributions to the development of PET probes for anions were made by Lehn,23 Gunnlaugsson15c,15d,24 and Yoon,25 who explored receptors in both organic and in aqueous solutions. Pioneering examples of nucleotide detection come from the work of Lehn (1997)26 and Garćia-Espańa (1999).27 In these two publications, the authors found that protonated polyammonium receptors are responsible for the fluorescence enhancement with ATP. In 2006 Garćia-Espańa observed the fluorescence enhancement with macrocyclic receptor 2 and citrate at pH 6.28 The macrocycle design is similar to one reported by Czarnik. In both cases, the fluorescence of receptors are pH- and anion-sensitive, which precludes the supramolecular shift of the pKas of amine groups in the presence of anions. Based on observations of anion-induced pKa shifts, we have designed and explored several new families of receptors for anions in recent years and thus advanced the field of fluorescent probes.
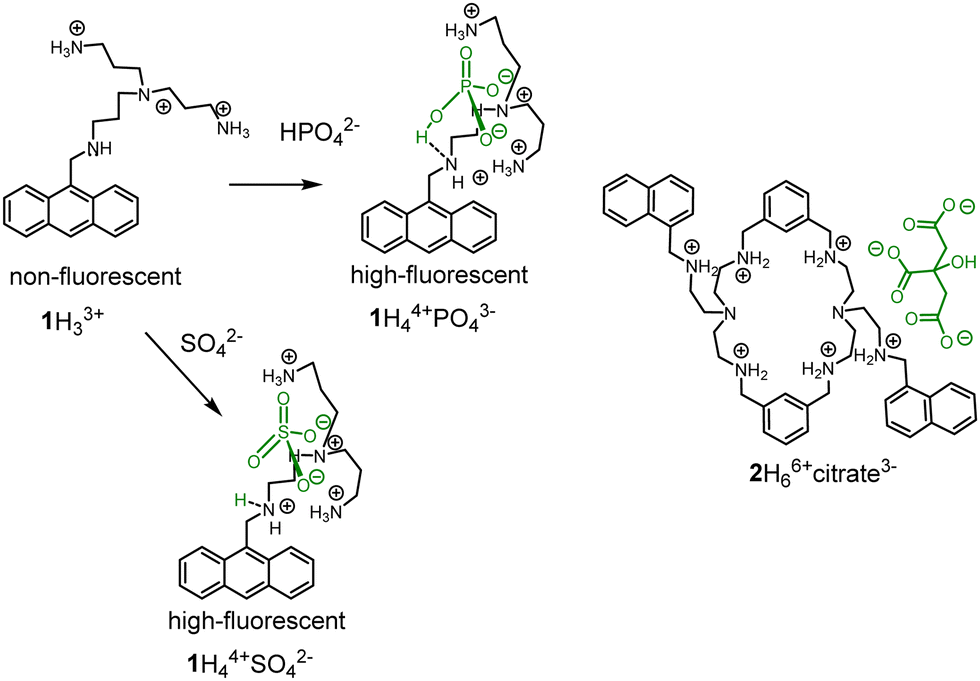 | ||
| Fig. 1 PET fluorescent probes for anions based on the protonation of a dye in an aqueous solution. | ||
In this feature article, we describe the design concept of fluorescent “turn-on” probes for anions involving pH-sensitive subunits in the framework of the receptor structure. We discuss in detail possible photoinduced processes and methods to determine optimal conditions for sensing in an aqueous buffered solution. Analysis of the receptor behavior at different conditions allows one to predict the response of receptors at chosen conditions and modulate the fluorescence answer. The application of the concept is demonstrated within three groups of anionic species: inorganic anions, nucleotides, and oligonucleotides. Finally, the scope and limitations of the systems are discussed.
Host design and supramolecular pKa shift
During recent years we have investigated a design concept for fluorescent receptors for anions with a “turn-on” response, which is schematically depicted in Fig. 2a. The critical element is a pH fluorescent probe attached to an anion-binding motif. The resulting structure is a pH-sensitive molecule, which switches on the fluorescence upon protonation. The presence of an anion in the anion-binding motif is expected to stabilize a positive charge of the amine group, similar to the mechanism described in Fig. 1.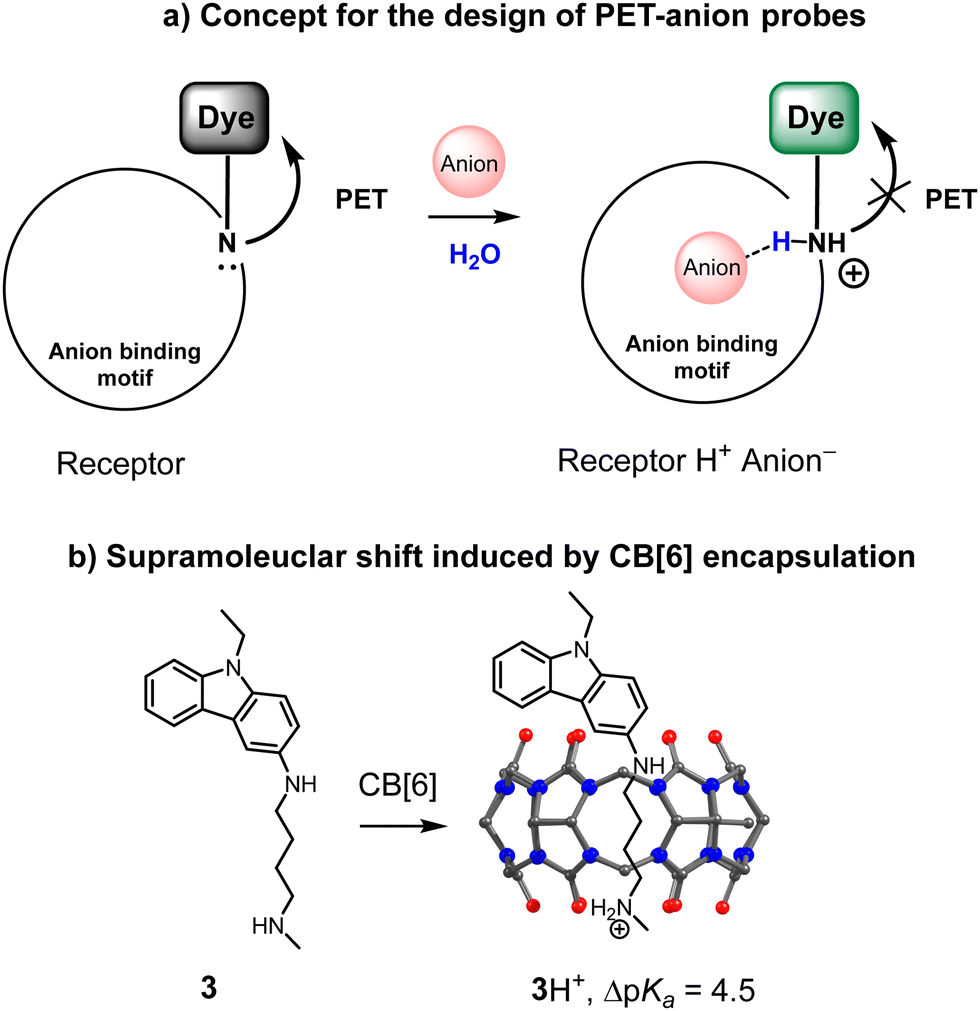 | ||
| Fig. 2 (a) Design concept of a fluorescent receptor for anions with a “turn-on” response. (b) Example of the supramolecular complexation of the pH probe in the cavity of cucurbit[6]uril. | ||
Consequently, the coordination of an anion should induce a supramolecular pKa shift. The conditions can be selected such that the receptor remains unprotonated until the anion is bound.
It was recently shown that supramolecular pKa shifts in the synthetic host–guest complexes could range from 1 to 5 pH units.29 Thus, the appropriate conditions for significant fluorescence changes upon anion recognition should be the pH region between the pKa values of the uncomplexed and complexed receptor. For instance, Nau and coworkers reported that ΔpKa for the complexation of an amine in the curucbituril cavity reaches 4.5 pH units (Fig. 2b).30 Encapsulation of the dye-functionalized amine can be considered as an inverted analogy of the sensing mechanism described in Fig. 2a. The amine is now not in the receptor structure but in a confined space. However, both systems are similar in terms of the supramolecular shift.
An important prerequisite to generate a strong “turn-on” response is the significant difference in fluorescence intensity of the dye in the non-protonated and protonated states. This value can be assessed as a ratio I(protonated)/I0(nonprotonated). The higher this ratio, the stronger the response of future receptors for anions. For instance, the Czarnik's compound generates nearly 2-fold enhancement upon protonation. Therefore, we began exploring naphthalimide-based fluorescent probes to achieve higher I/I0 ratios. Naphthalimides are unique dyes that can be functionalized stepwise at different positions and introduce different substituents. Such functionalization is convenient for receptor design.24,31
We started our studies with compound 4, which demonstrated 8-fold enhancement upon protonation in an aqueous solution.32 The acylation with dipyrrolylmethane diacid yielded 5. Since one amino group is now blocked with the amide bond, the pKa of the free amines shifted from 4.5 to 2.8. To adjust the condition for the anion detection, we used acetate buffer at pH 3.6. Under these conditions, a selective but small fluorescence enhancement (1.25-fold) was observed in the presence of sulfate. Most of the anions did not show any changes in fluorescence. Only iodide and bromide quench the fluorescence of the receptor because they have a low reduction potential and facilitate the PET from the dye to the anion.
The studies with 5 served as a starting point to explore various naphthalimide derivatives in terms of their pH-response and the ways to incorporate them into the receptor structure. The structures of the explored systems 6–11 are shown in Fig. 3 and discussed in detail below.
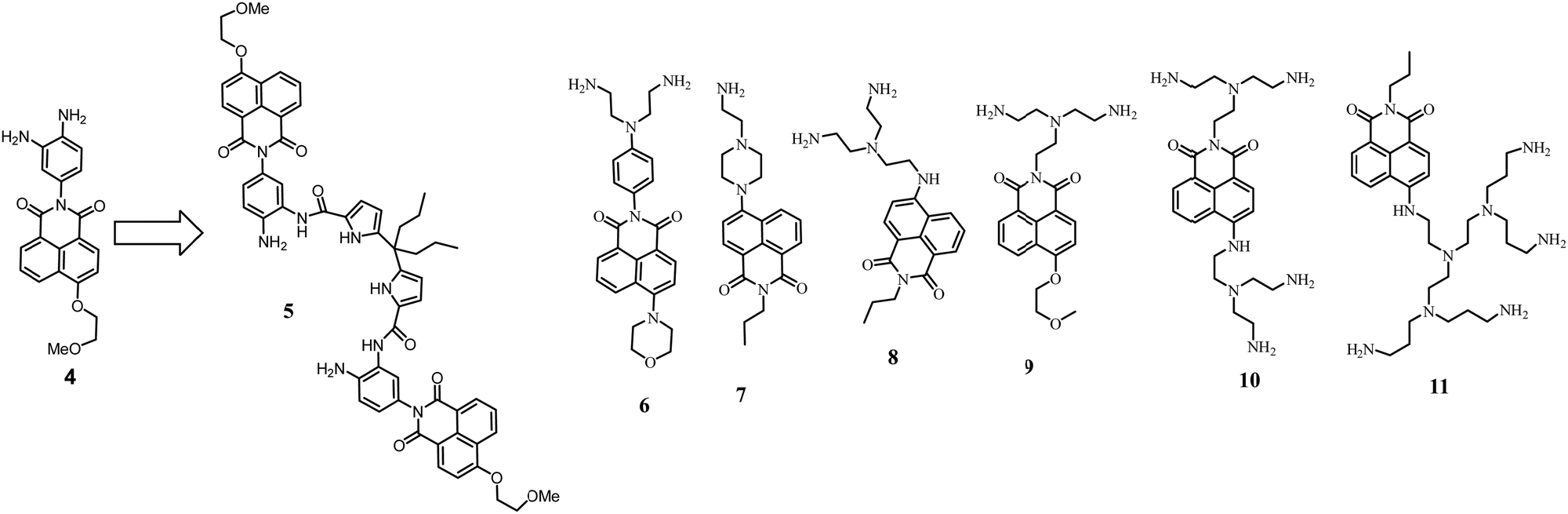 | ||
| Fig. 3 Functionalization of 4 with dipyrrolylmethane subunits to form 5. Naphthalimide dyes with appended amines explored in our work. | ||
Receptors for anions
Systematic studies of anion-binding and sensing properties were carried out with naphthalimide derivatives 6–11 bearing amine groups. These compounds also work as fluorescent pH probes in an aqueous solution. The PET from amine groups to naphthalimide is responsible for pH sensitivity. First, we tested how these dyes behave in the presence of highly charged anions, such as pyrophosphate.33 Since pyrophosphate carries multiple negative charges at neutral and basic pHs, the affinity of fully protonated polyamine receptors for pyrophosphate reaches high values, e.g., log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 8.31 for receptor 8. Binding constants with anions were determined with the help of potentiometric titrations. Although the structures of the dyes in Fig. 3 are very similar, they indeed showed different fluorescent properties and anion-binding behavior. For instance, 6 and 9 have very low I/I0 values upon protonation, which hinders further application in anion sensing. According to potentiometric titrations and fluorescence studies, 8 and 10 behave very similarly; namely, pKa values of the primary amines are around 8–12, and they are responsible for the PET process with naphthalimide. In the case of 11, tertiary nitrogen atoms with pKa values less than 6 induce PET. Therefore, “turn-on” responses e.g., for 10 and 11 are observed in different pH regions, 8–10 and 4–7, respectively.
K = 8.31 for receptor 8. Binding constants with anions were determined with the help of potentiometric titrations. Although the structures of the dyes in Fig. 3 are very similar, they indeed showed different fluorescent properties and anion-binding behavior. For instance, 6 and 9 have very low I/I0 values upon protonation, which hinders further application in anion sensing. According to potentiometric titrations and fluorescence studies, 8 and 10 behave very similarly; namely, pKa values of the primary amines are around 8–12, and they are responsible for the PET process with naphthalimide. In the case of 11, tertiary nitrogen atoms with pKa values less than 6 induce PET. Therefore, “turn-on” responses e.g., for 10 and 11 are observed in different pH regions, 8–10 and 4–7, respectively.
We found three different sensing mechanisms for anion detection that are realized depending on the pH of the solution. The first mechanism is represented by 8 and is based on the supramolecular pKa shift described above. A simple way to determine the pH range at which the receptor can detect anions is to measure the fluorescence in the absence and the presence of an excess (100 equiv.) of the anion to be detected. As can be inferred from Fig. 4a, pyrophosphate induces a stronger supramolecular pKa shift than sulfate. For both anions, an enhancement of fluorescence upon adding anions at pH 4.5–8 can be expected. A different situation is observed for 10, where enhancement and quenching are in different pH regions. Detailed studies revealed that aggregation of the dyes with anions leads to self-quenching, which we assigned to the second mechanism. Thus, 10 showed fluorescence enhancement at pH 8, while already at pH 9, quenching was detected (Fig. 4b). The third sensing mechanism was found at low pH values with fully protonated dyes. Excess sulfate and pyrophosphate were able to deprotonate the ammonium groups leading to quenching. Overall, these three mechanisms can be expected while designing new fluorescent receptors for anions. The pH of the solution and the self-aggregation behavior of the receptor have a dramatic effect on the resulting fluorescence response.
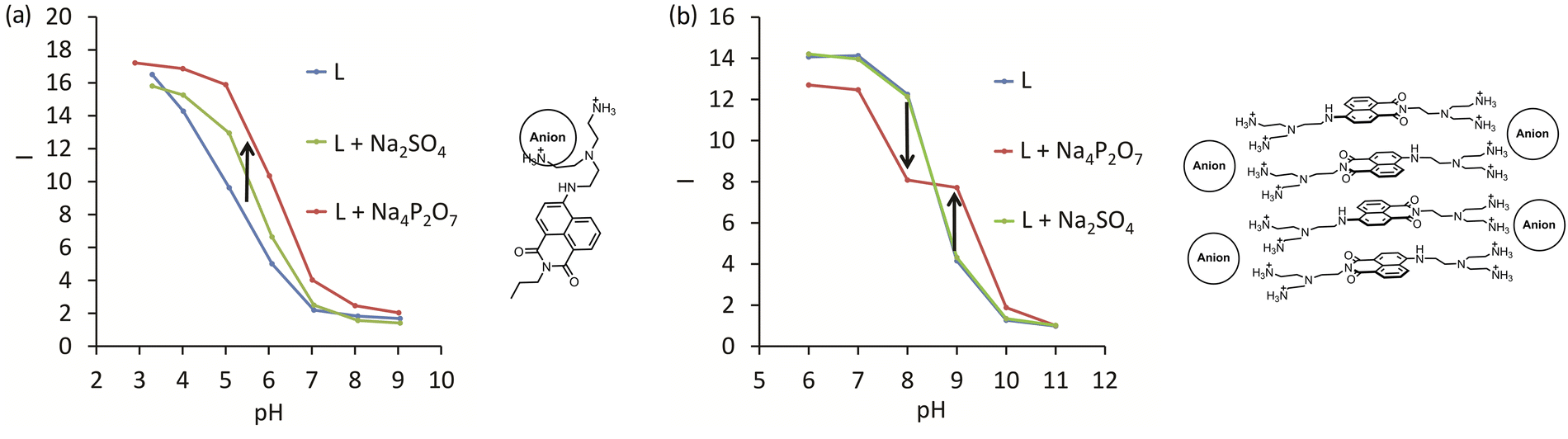 | ||
| Fig. 4 Structures and fluorescence response (I = Iobs/I11, where Iobs is observed intensity and I11 is the intensity at pH 11) for anion complexes with (a) 8 and (b) 10. L – is the intensity of the corresponding receptor depending on pH. Green and red lines are the intensities in the presence of sulfate and pyrophosphate, respectively. Adapted with permission from ref. 33. Copyright 2017 Wiley-VCH. | ||
The attachment of a neutral anion-binding motif to the amine-functionalized naphthalimides was found to be an efficient approach for achieving better selectivity for sulfate. Naphthalimide with an attached piperazine-ring was reported previously as an excellent pH probe with I/I0 > 50.34 This was the reason to use 7 in the construction of new receptor 12 for sulfate detection in water.35 The design of 12 involves a classical anion-binding motif, 2,5-pyridine-dicarboxamide, connected to two arms containing 7. The pH-depended fluorescence measurements (Fig. 5a) showed that sulfate could be detected between pH 3.5 and 4.5. The saturation with sulfate results in a pKa shift of pipeprazine of one pH unit. To show that the binding constant for sulfate and the fluorescence enhancement strongly depends on the pH of the solution, we conducted fluorescence titrations at seven different pH values 4.1–4.7 (Fig. 5b). With a difference of only 0.7 pH units, the affinity for sulfate drops one order of magnitude from logKa 3.30 (4.1) to 2.19 (pH 4.7). A decrease of fluorescence enhancement was also observed at saturation point: from 7-fold to 5-fold, respectively.
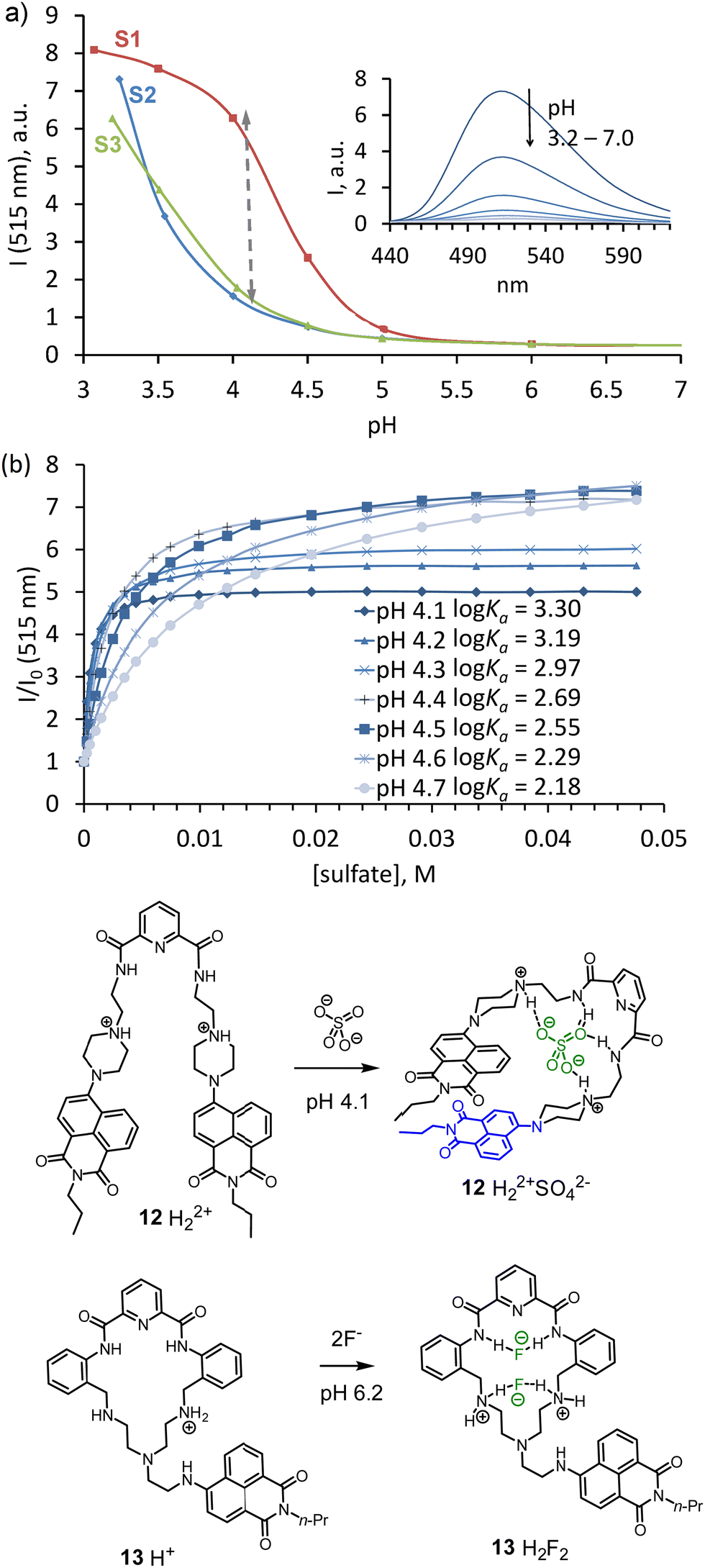 | ||
| Fig. 5 (a) Fluorescence vs pH ralationship for 12 (ex. 380 nm); spectra were recorded in a 1:1 water–THF mixture, containing 50 mM of acetate buffer; S2 – without any additives, S3 – in the presence of 10 mM NaH2PO4, S1 – in the presence of 10 mM Na2SO4. (b) Binding isotherms obtained by fluorescence titration of 12 in a 1:1 buffer–THF (pH 4.1) with anions as their sodium salts. Adapted with permission from ref. 35. Copyright 2018 Wiley-VCH. Proposed structures of complexes 12 and 13 with anions. | ||
Several exciting features of receptor 12 were discovered during the studies. For instance, ROESY NMR experiments revealed a dramatic change in the receptor's conformation upon sulfate coordination. Sulfate binding forces naphthalimides to form π–π interactions and contribute to the stability of the host–guest complex. These stacking interactions probably contribute to the host's rigidity in the complex. In addition, experiments with blocking hydrogen bond donating amide NHs by methylation uncovered their essential role in the selectivity and affinity of the host. Regarding the binding constant, two amide hydrogen bonds contribute one order of magnitude to the affinity for sulfate relative to the affinity provided only by two electrostatic interactions from the protonated piperazines.
As the previous example shows, preorganization is essential for highly selective binding. Thus, we explored the use of amine 8 in the construction of macrocycle 13 bearing pyridine-2,5-dicarboxamide binding site and rigid phenylene spacers. The macrocycle was designed to bind halides within a suitable cavity size. The pKa values of the free amine groups are close to neutral pH. Therefore, anion-binding measurements were carried out in a 10 mM MES buffer (pH 6.2). Binding studies revealed that fluoride best fitted the cavity, showing 105 M−1 affinity. The macrocycle can also bind chloride, however, with less affinity. Interestingly, adding an excess chloride led to the formation of receptor-anion aggregates that are held through electrostatic interactions. In the case of fluoride recognition, the receptor can perfectly accommodate one or two anions in the cavity, which prevents aggregation and induces protonation. The latter process resulted in a “turn-on” fluorescence enhancement, which agrees with the expected behavior.
Azacryptands are highly efficient receptors for anions and contain secondary amine groups.36 They are protonated in water and function as binding sites for anions. Several approaches how to visualize the binding events have already been reported. For example, the preparation of metal complexes, the use of indicator displacement assays,37 and the introduction of fluorescent dyes in the receptor structure. If azacryptands contain fluorescent dyes, they quench each other via stacking interactions. The coordination of anions (carboxylic acids) destroys these interactions and the fluorescence recovers.38 Amendola and coworkers utilized anthracene-containing azacryptands to detect TcO4− in an acidic aqueous solution. In this work, the heavy-atom containing anion quenches the fluorescence of azacryptands due to the selective encapsulation.39
In 2017, we adapted our approach to visualize anion binding with the known azacryptands and also developed new systems with new selectivities. Two strategies were investigated: (1) attachment of a dye to available secondary nitrogen in cryptands (as in 14–17) and (2) incorporation of a dye in the receptor's structure to be able to interact with a guest directly (as in 18). Anthracene and naphthalimide derivatives were investigated as fluorescent dyes introduced in cryptands. We proposed a simple approach to use halogen derivatives of dyes to alkylate secondary amines of azacryptands (Fig. 6). 1,3-Benzene and 2,5-pyridine azacryptands were converted to the corresponding products 14–17.40 Receptor 15 showed two pH units of supramolecular pKa shift with sulfate (Fig. 6a). We confirmed that it is also possible to detect the pH shift using potentiometric titrations, as shown in Fig. 6b. A comparison of the properties of anthracene and naphthalimide-containing cryptands suggests that the rigidity of azacryptands improves selectivity for some anions. Bulky anthracene connected to the azacryptands via only one CH2-group creates a steric hindrance and introduces rigidity in the receptor structure. Contrary, naphthalimide is smaller in size and is connected via two CH2-groups. Thus, it does not change the original selectivity of azacryptands considerably. For instance, anthracene-containing receptors 14 and 15, showed good selectivity towards sulfate and pyrophosphate at pH 3.6 and 6.2, respectively. The binding is strong enough to detect these anions in the presence of excess other anions. We found that fluorescence enhancements and selectivity for anions were changed upon shifting the pH of the solution from pH 3.6 to 6.2. 15 has a preference to bind oxalate and sulfate at pH 3.6 (Fig. 6c), while it is selective for pyrophosphate in terms of binding strength and fluorescence enhancement at pH 6.2 (Fig. 6d). This observation is consistent with stronger electrostatic interactions at pH 6.2. While oxalate and sulfate carry two positive charges over a broad range of pH, pyrophosphate has three negative charges at close to neutral pH.
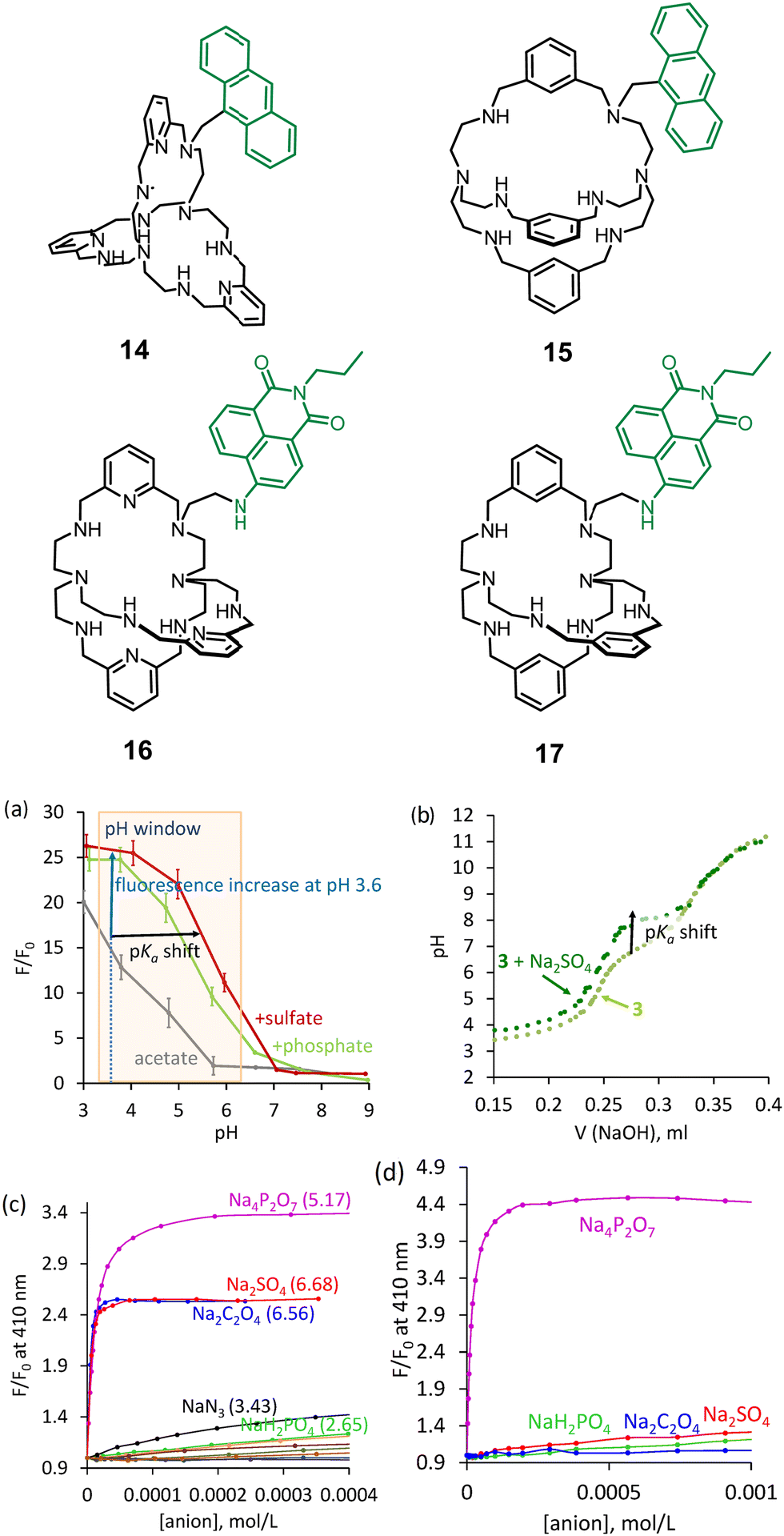 | ||
| Fig. 6 Structures of azacryptands functionalized with dyes. (a) Relationship between fluorescence and pH determined for 15. (b) Potentiometric titration of 15 in the absence and the presence of sulfate. Fluorescence titration of 15 with different anions at (c) pH 3.6 and d) pH 6.2. Reproduced from ref. 40 with permission from the Royal Society of Chemistry. | ||
We achieved the best selectivity for oxalate over sulfate with 18 – an anthracene-bridged azacyclophane, in which anthracene can directly interact with an anion.41 While previous receptors changed their selectivity at various pH values, 18 showed two orders of magnitude selectivity over other anions in the pH range 3.6–6.2. The best fluorescence discrimination was observed at pH 6.2 (50 mM MOPSO buffer), which is accomplished with 10-fold fluorescence enhancement. The effect was visible even by the naked eye. Strong fluorescence enhancement rests on the double protonation of two secondary NH-sites connected through CH2-groups to the anthracene (Fig. 7). Since the anthracene ring is located in close proximity to the coordinated anion, we observed that anions with a low reduction potential efficiently quenched the anthracene emission. As can be inferred from Fig. 7a, there is an almost linear correlation between the Stern–Volmer quenching constants and the reduction potential of anions. The structure of the oxalate complex was determined by the X-ray analysis and revealed a perfect geometry match between the receptor and the anion (Fig. 7b).
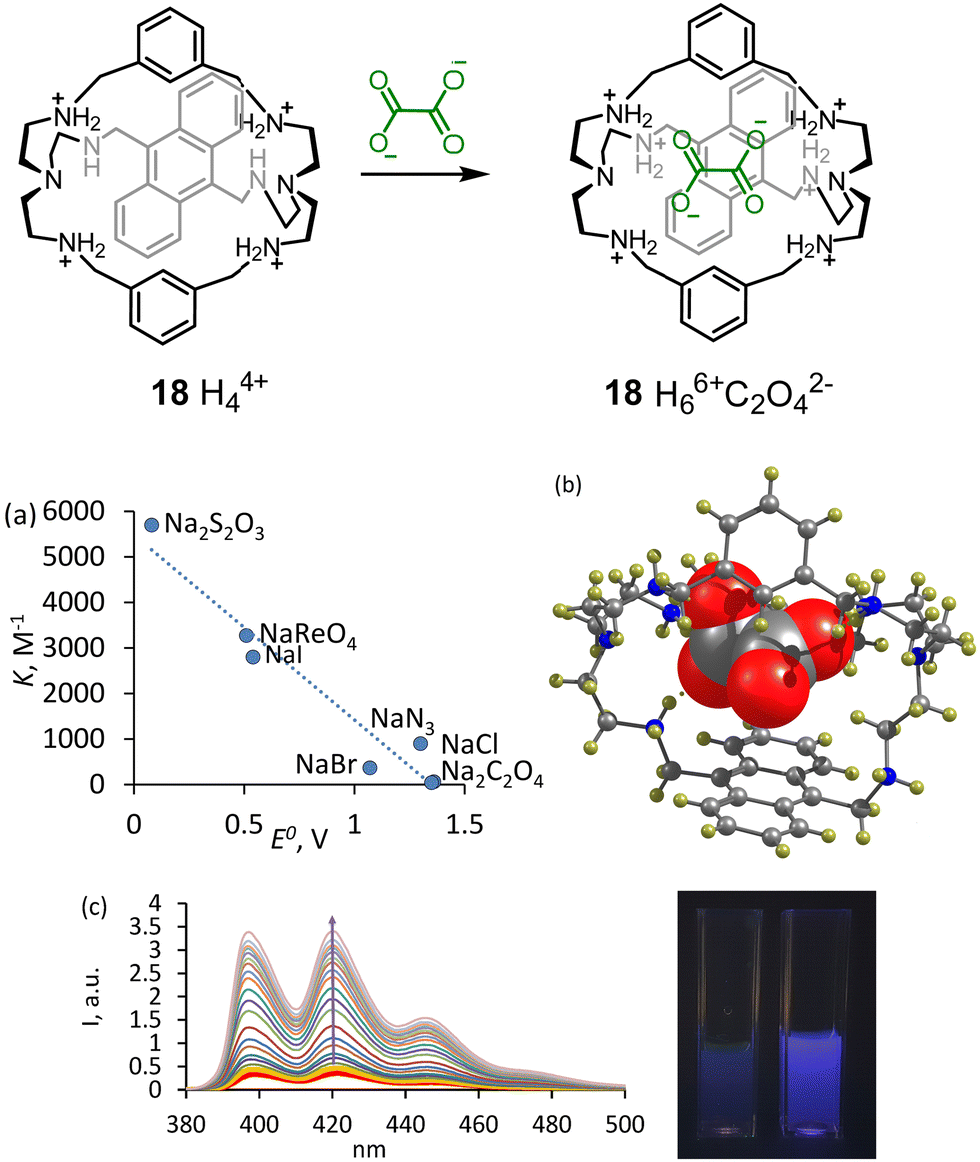 | ||
| Fig. 7 Complexation of 18 with oxalate. (a) Correlation between Stern–Volmer quenching and reduction potentials of the anions. (b) Molecular structure of the oxalate complex according to the single X-ray crystal analysis. Reproduced from ref. 41 with permission from the Royal Society of Chemistry. (c) Fluorescence changes observed upon gradual addition of oxalate to 18. The cuvettes show the fluorescence of 18 under UV-lamp without oxalate and with 10 equiv. of oxalate. | ||
Further studies with cryptands bearing two and three anthracene rings revealed a different sensing mechanism that appeared to be characteristic for chaotropic anions – those anions that in water solution disrupt the hydrogen bonding network inside the cavity.42 Thus, receptors 19 and 20 in the fully protonated state show higher fluorescence intensity in the presence of such anions as NO3−,ClO4−, PF6− and BF4− (Fig. 8).
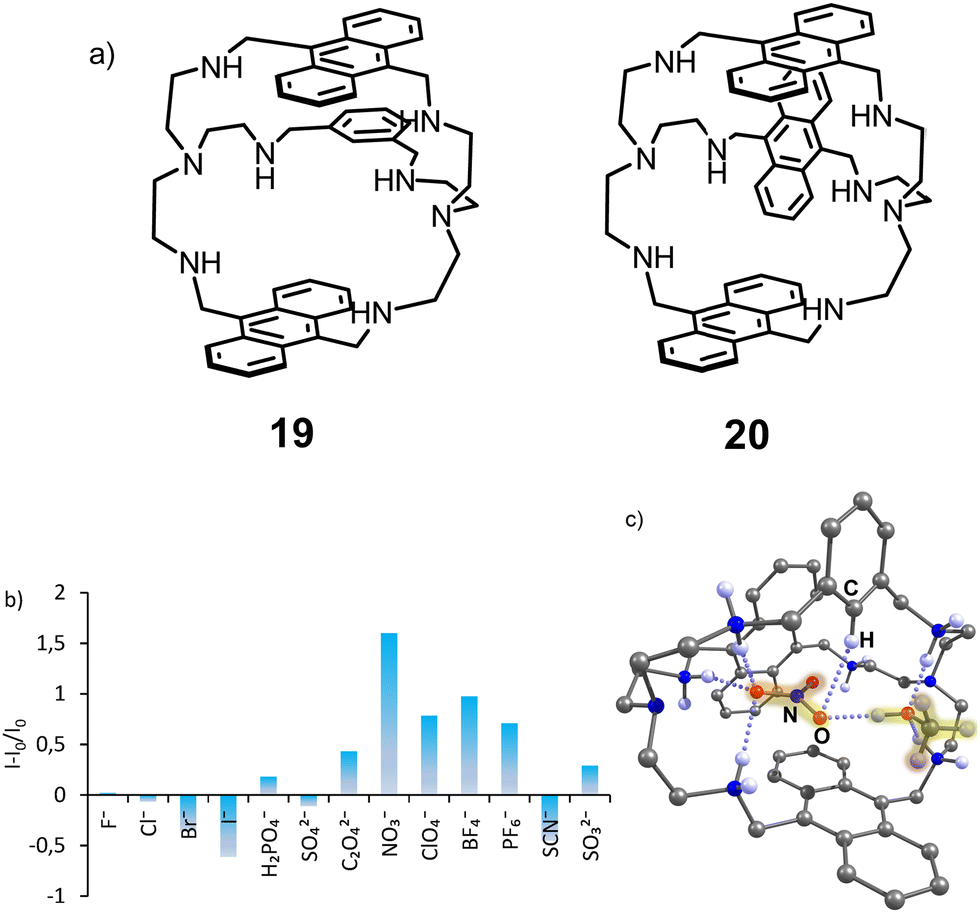 | ||
| Fig. 8 (a) Structures of receptors 19 and 20. (b) Fluorescence response of 19 measured in the presence of 200 equiv. of anions (50 mM acetate buffer, 5% DMSO, pH 3.6. (c) Molecular structure of the nitrate complex 19H66+(NO3−)(CH3OH) according to the X-ray analysis. Adapted with permission from ref. 42. Copyright 2019 American Chemical Society. | ||
The sensing mechanism, in this case, is different from that discussed previously. We suggested that the coordination of water inside and outside the hydrophobic cavity of the azacryptands quenches the anthracene fluorescence, while the coordination of nitrate and displacement of the water molecules leads to the fluorescence recovery. Interestingly, even though sulfate has a much higher affinity, it induces fluorescence quenching. According to the crystal structure obtained with 19(HNO3)6 and 20(HClO4)3, nitrate is encapsulated and forms π–π interactions with anthracene, while perchlorates are located outside the cavity. This encapsulation might contribute to the strongest “turn-on” response of the receptor for nitrate.
Fluorescent receptors for nucleotides
Nucleoside phosphates are also anions at neutral pH. However, they have more complex structures than inorganic anions and include variations in the sugar, phosphate, and nucleobase fragments. For instance, triphosphates have a higher negative charge than di- and monophosphates. Moreover, nucleobases have their own acid-base equilibrium in an aqueous solution, which should be considered when designing host–guest complexes. For molecular recognition and sensing of nucleotides, we utilized a simple approach in which we combined the anion recognition by azamacrocycles and the nucleobase recognition by π–π stacking with fluorescent dyes. Two possible designs have been explored. The first design we named the phosphate binding macrocycle-dye concept (PBM-dye), which relies on the attachment of a dye to the macrocycle. A possible coordination mode of a nucleotide is shown in Fig. 9. The second design stems from the Lehn's original cyclophanes, which were shown to bind nucleotides in water.23 A nucleobase can be encapsulated between two dyes, while positively charged amine-containing spacers can coordinate phosphate. In both design approaches, there is a possibility to realize the PET sensing mechanism investigated by us for inorganic anions and discussed in the previous section. However, more complex behavior is expected because fluorescence can be affected by many other interactions, such as π–π interactions between dyes in both ground and excited states and dye-nucleobase interactions.43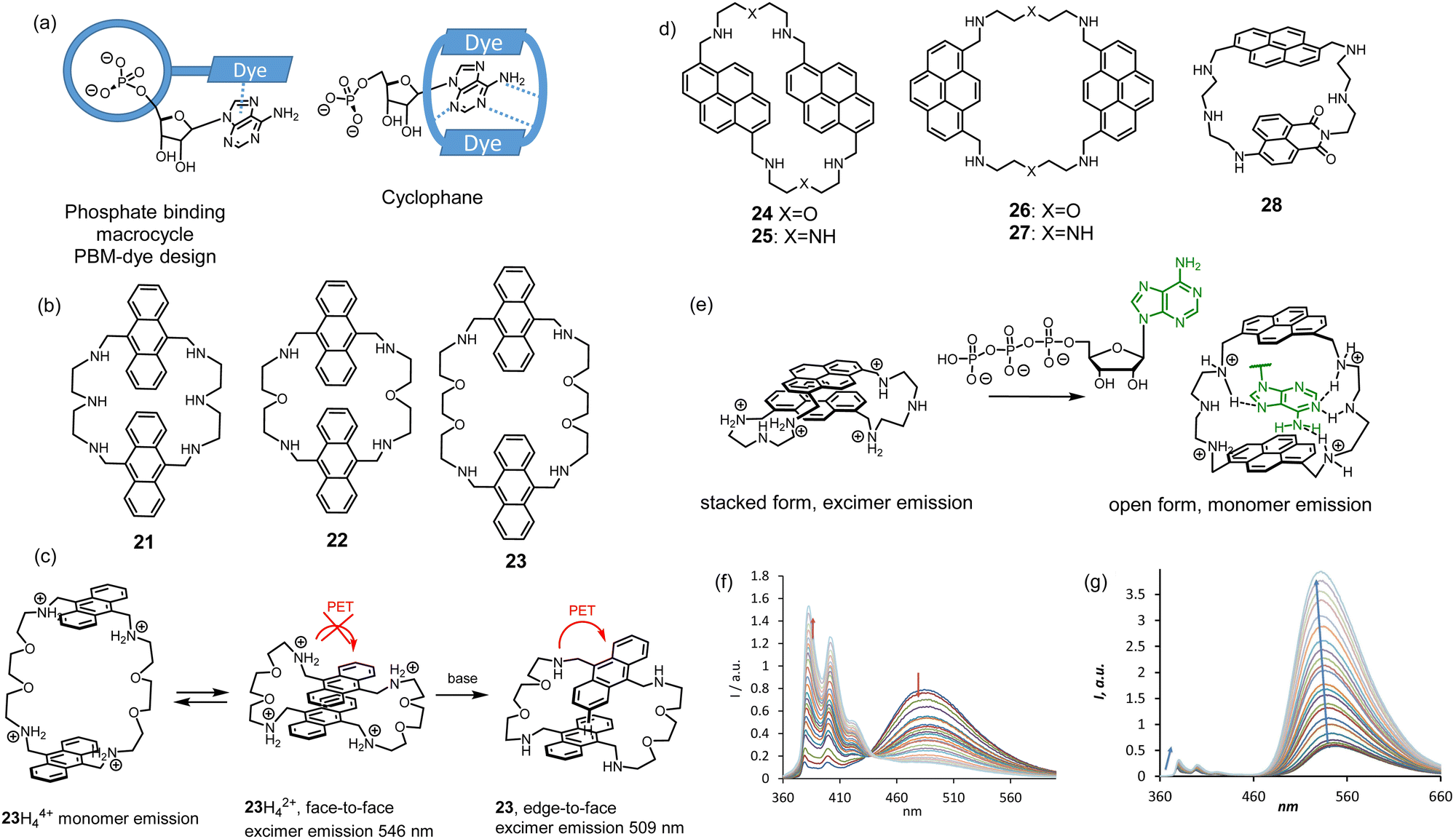 | ||
| Fig. 9 (a) Design approaches for nucleotide receptors. (b) Structures of anthracene-based cyclophanes. (c) Proposed interaction of anthracene rings depending on pH. (d) Structures of pyrene-based cylclophanes. (e) “Bellows-type” sensing mechanism of ATP by 27 and (f) the observed changes in the emission spectrum. (g) Fluorescence changes of 28 upon the addition of ATP or CTP. Conditions: 10 mM TRIS buffer (2 vol% DMSO, 100 mM NaCl, pH 7.4, ex.: 350 nm). Adapted with permission from ref. 45. Copyright 2020 Wiley-VCH. | ||
Anthracene-containing macrocycles were first studied in the context of the cyclophane design. The spacers between dyes were varied, namely the length and the number of amine groups (receptors 21–23).44 The PET process between anthracenes and amines was detected by varying the pH of the solution. However, more pronounced effects in fluorescence spectra were observed in the monomer–excimer emission equilibrium, which depended on the pH of the solution and the presence of nucleotides. Interestingly, the excimer emission peak shifted upon changing the pH. We proposed that this shift depends on the orientation of the anthracene rings to each other. For example, 23 shows different emission bands, which we assigned to different orientations of two anthracenes to each other (Fig. 9c). Binding studies in 50 mM MES buffer (pH 6.2, 2% DMSO) revealed the preference of anthracene-based cyclophanes for GTP. For all receptors, quenching of fluorescence was observed because of PET occurring between the anthracene and guanine. This quenching pathway appeared to be much stronger than the hindering of PET induced by protonation. However, other nucleobases that cannot quench anthracene in the excited state showed strong enhancement in the monomer emission.
Studies with anthracene systems showed that it was difficult to shift the monomer–excimer equilibrium with the help of nucleotides entirely in one or the other direction. Therefore, we explored a new family of pyrene-based cyclophanes 24–27 in an aqueous solution.45 The oxygen-containing spacer appeared to favor stacking between pyrene rings and led again to too strong interactions to be shifted by the coordination of a nucleobase. However, the triamine spacer was very suitable for this purpose. The best selectivity for ATP in terms of fluorescence detection showed receptor 27. As can be inferred from Fig. 9e, 27 works as a good PET probe for ATP with the sensing mechanism, which we called “bellows-type”. We proved that protonation of the receptor and the coordination of ATP leads to the same fluorescence response, namely the disappearance of the excimer emission and a strong increase in the monomer emission (Fig. 9f). DFT calculations suggested that the spacers and their spacial position provide high selectivity for ATP. The amine sites in the spacer are protonated and form hydrogen bonding interactions with adenine. The fact that 1,8-substituted pyrene has better selectivity than 1,6-substituted analog suggests that selectivity is highly sensitive to the spatial arrangements of spacers. We successfully applied 27 as a probe in real-time monitoring of the enzymatic activity of creatine kinase.
Interesting properties were found for cyclophane 28 containing different dyes: pyrene, and naphthalimide, connected with diethylenetriamine.46 We expected that donor–acceptor interactions between two dyes could prevail in an aqueous solution over electrostatic repulsion of the charged amine sites, like in the case of bis-pyrene cyclophane 28. Such interaction may result in the observation of Förster resonance energy transfer (FRET), as it is expected for the couple pyrene-naphthalimide. Indeed, we observed FRET in pure DMSO solution but not in water. The electrostatic repulsion was much stronger, and the stacked form was not detected. As can be seen in Fig. 9g, 28 shows excellent sensitivity towards nucleotides. Remarkably, the overall fluorescence enhancement of naphthalimide is much stronger than that of pyrene. The binding studies revealed a “turn-on” response for ATP and CTP with affinities logK = 4.80 and 4.53 (10 mM TRIS buffer, 100 mM NaCl, pH 7.4), respectively. The corresponding response and binding constants for GTP and UTP were one order of magnitude lower. Overall, these studies revealed that highly efficient PET observed for the naphthalimide derivatives is critical to generate a strong response for nucleotide detection.
The PBM-dye design depicted in Fig. 9a was realized with receptor 29 featuring two tertiary amine sites connected to naphthalimide.47 As we found previously for 8 and 10, this nitrogen is responsible for the PET quenching of naphthalimide, while upon protonation, the PET becomes repressed.
The first generation of such receptors was obtained by the reaction of 8 with the activated pyridine-2,5-dicarboxylic acid yielding 29 as an [2 + 2] acylation product. 29 has a unique feature to form a folded conformation upon anion recognition. Spectroscopic investigations suggest that this folded conformation of 29 found in the solid state (Fig. 10a) also exists in an aqueous solution. All the binding studies were performed at pH 3.6 for the sufficient solubility of the receptor in water. At this pH, the two tertiary nitrogens are protonated, and the receptor has overall two positive charges. Fluorescence titrations revealed that sulfate-induced the strongest quenching among other inorganic anions. Based on NMR investigations, we proposed that this quenching is due to the formation of π-stacks as it was found in the X-ray crystal structure. Thus, no additional PET processes can be expected in the presence of anions. Studies with nucleoside monophosphates identified the selectivity of 29 for guanine-containing nucleotides. The addition of cGMP resulted in the strongest fluorescence quenching among other nucleotides (Fig. 10b). Thus, we proposed that the formation of the host–guest complex with cGMP is accomplished by folding the receptor to the same conformation as we observed with sulfate. Since the quenching effect for cGMP was stronger than that for sulfate, π–π interaction between naphthalimide and guanine was proposed. The preference to bind guanines was additionally confirmed in binding studies with DNA tetranucleotides A4 (5′-AAAA-3′), G4, C4, and T4. As can be seen in Fig. 10d, 29 showed the highest affinity and quenching with G4 (logK > 8). In this study, we found interesting behavior of our receptor. It formed insoluble complexes with nucleoside di- and triphosphate, but the complexes with monophosphates and even with tetranucleotides were soluble. This property opens up an opportunity to explore the interaction of our receptors with longer oligonucleotide sequences at neutral pH.
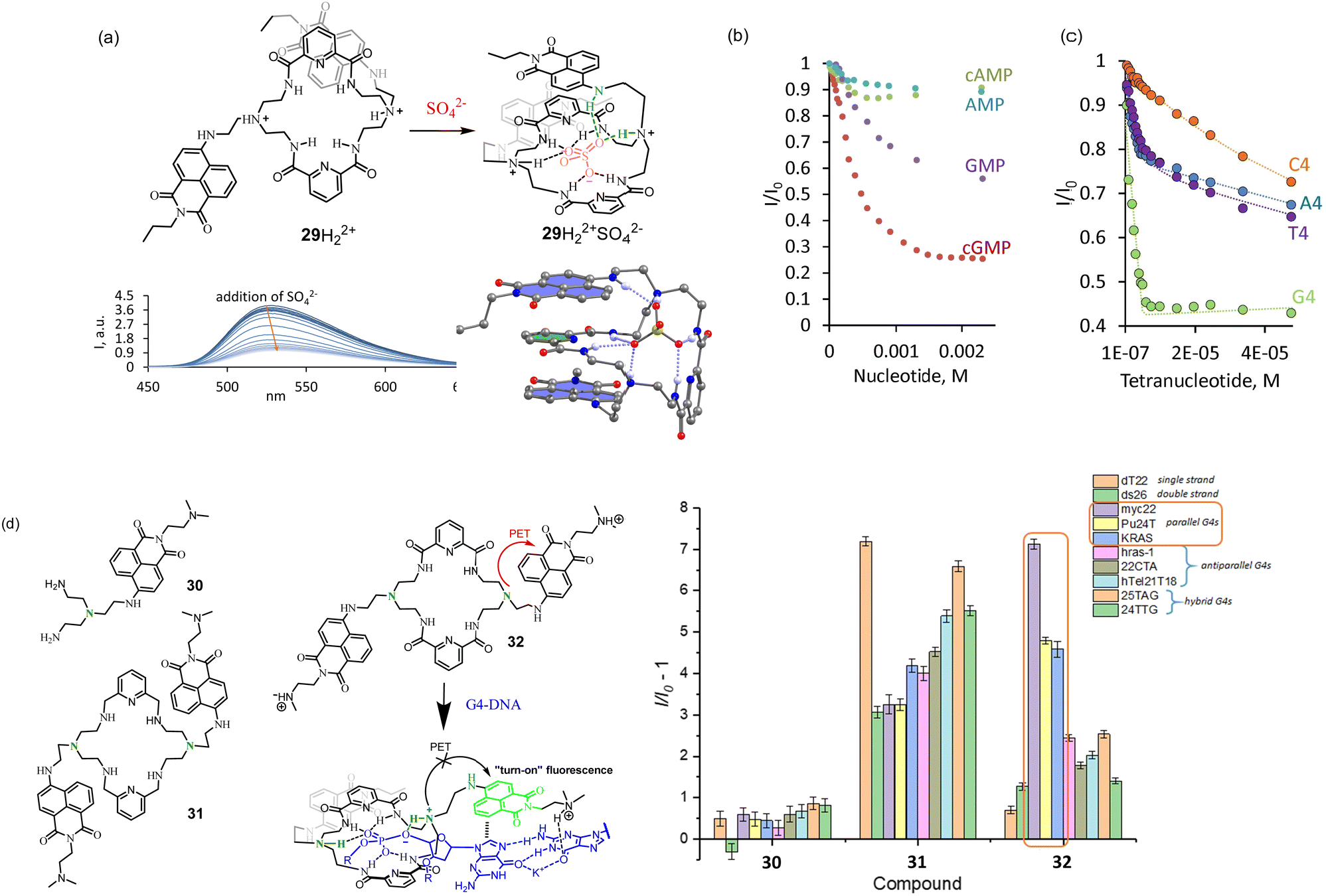 | ||
| Fig. 10 (a) Structure of the receptor with PBM-dye design and its fluorescence response to (b) nucleoside monophosphates and (c) tetranucleotides. Adapted with permission from ref. 47. Copyright 2019 American Chemical Society. (d) Second generation of macrocycles. Proposed coordination of 32 to G-quadruplex and fluorescence changes observed upon the interaction of 30–32 with DNA G-quadruplexes of different topologies. Conditions: 10 mM NaAsO2Me2 buffer (5% DMSO), 100 mM KCl, pH 7.2. Reproduced from ref. 48 with permission from the Royal Society of Chemistry. | ||
If we analyze the fluorescence data obtained for 29 depending on the pH of the solution, a possible fluorescence enhancement upon coordination of an anionic guest can be expected only at pH less than 5. Thus, at first glance, it is impossible to observe fluorescence enhancement at neutral pH. However, we postulated that the high negative charge of the oligonucleotides should induce a much stronger pKa shift of the tertiary amine group.
In order to increase the solubility of the receptor and increase the efficiency of PET, we prepared a new family of receptors 30–32 bearing protonable dimethylamino group.48 We investigated the properties of 29–32 in terms of their interaction with simple nucleotides and DNA G-quadruplexes of different topologies.
According to CD measurements, the sequence 5′-GGGG -3′ (G4) is able to form a quadruplex structure in the aqueous solution with potassium cations. The experiments at pH 7.4 demonstrated a slight enhancement of 32 fluorescence with G4 sequence. This experiment suggests that the PET process from the amine group to naphthalimide was completely blocked, and the PET between guanine and naphthalimide is less effective, likely because guanine is involved in supramolecular interactions. To our expectations, the fluorescence enhancement with longer oligonucleotides, able to form G-quadruplexes, was much stronger. A selective response was observed with sequences forming parallel G-quadruplexes, such as myc22 (Fig. 10d). According to the binding studies, 32 was bound to myc22 in a 1:2 (myc22:32) stoichiometry with dissociation constants K11 = K12 = 1.09 μM. Triamine 30 showed almost no response, while 31 bearing only amine groups in the structure of the macrocycle demonstrated a good response but no selectivity. Likely, it binds and responds to any G-quadruplex topologies due to the strong electrostatic interactions.
Outlook and conclusions
We have summarized in this article the approach of utilizing pH probes in the design of fluorescent receptors for anions explored by us during the last six years. We show many advantages of this approach to detect not only simple inorganic anions but also more complex anionic species, such as nucleotides. Furthermore, the design is simple and versatile. It can be applied to any polyamine cyclic or acyclic synthetic receptor by attaching a pH-sensitive dye. The presence of protonable amine groups often leads to good solubility of a synthetic receptor in water. As we showed with dye-functionalized azacryptands, the selectivity of the core receptor can be controlled by the introduction of a dye. This fact gives additional room for the optimization of binding properties. The systematic studies of more than 30 receptors show that anion recognition can be efficient in water and occurs even in the presence of more than 5000 equivalents of the buffer molecules (50 mM buffer).For optimization of the degree of the “turn-on” fluorescence response, it is important to use the dye possessing high “turn-on” response for pH. However, increasing the number of dyes in the receptor structure not always leads to a better fluorescence response. Several dye molecules present in the receptor structure can quench each other or form complexes in the excited state. As we saw with receptor 29, other mechanisms of fluorescence changes can be expected for fully protonated systems. For instance, special cases have been observed for the detection of nitrate by receptor 19, and sulfate by receptor 29. Interestingly, with a higher charge of a guest, like oligonucleotides, the supramolecular pKa shift can reach high values, 3–4 pH units. This fact allowed us to detect G-quadruplexes even at neutral pH, given that pKa of the receptor's amino group is around 4–5.
Indeed, there are several limitations of the design approach. The dependence of fluorescence response on pH values, the need for adjustments to appropriate pH values for maximum response, and optimization of a buffer system. Often multiple positive charges of the receptor preclude strong affinity for highly charged anionic species. Therefore it is difficult to achieve high binding constants for, e.g., mono- or dianions.
Even though there are some limitations, there is still much room for new developments in the area of fluorescent receptors for anions. For instance, there are many dyes with different sensing mechanisms to detect pH changes and the ability to form hydrogen bonds. The variation of the number of amine groups and their pKas will allow one to adjust the affinity of the systems to the desired targets. There are also many possibilities to use our approach for designing new ion-pair receptors, receptors for polyanions, detergents, phospholipids, anionic biologically active compounds, and molecular switches. We believe that our studies will attract more attention from the supramolecular community to use the approach in the visualization of anion binding and broadening their applications in the environment and biology.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DAAD and Deutsche Forschungsgemeinschaft (DFG), projects KA 3444/16-1 and SFB 953.Notes and references
- J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, ed. J. F. Stoddart, Royal Society of Chemistry, Monographs in Supramolecular Chemistry, 2006 Search PubMed.
- (a) P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed; (b) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- (a) A. M. Agafontsev, A. Ravi, T. A. Shumilova, A. S. Oshchepkov and E. A. Kataev, Chem. – Eur. J., 2019, 25, 2684–2694 CrossRef CAS PubMed; (b) E. García-España, R. Belda, J. González, J. Pitarch and A. Bianchi, Supramolecular Chemistry, John Wiley & Sons, Ltd, 2012 DOI:10.1002/9780470661345.smc066; (c) Y. Zhou, Z. Xu and J. Yoon, Chem. Soc. Rev., 2011, 40, 2222–2235 RSC; (d) D. Ramaiah, P. P. Neelakandan, A. K. Nair and R. R. Avirah, Chem. Soc. Rev., 2010, 39, 4158–4168 RSC.
- E. A. Kataev and C. Müller, Tetrahedron, 2014, 70, 137–167 CrossRef CAS.
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed.
- A. Amine and G. Palleschi, Anal. Lett., 2004, 37, 1–19 CrossRef CAS.
- (a) T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC; (b) M. H. Lee, J. S. Kim and J. L. Sessler, Chem. Soc. Rev., 2015, 44, 4185–4191 RSC.
- (a) C. Warwick, A. Guerreiro and A. Soares, Biosens. Bioelectron., 2013, 41, 1–11 CrossRef CAS PubMed; (b) H. M. Tay and P. Beer, Org. Biomol. Chem., 2021, 19, 4652–4677 RSC; (c) P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- A. C. Sedgwick, J. T. Brewster, T. Wu, X. Feng, S. D. Bull, X. Qian, J. L. Sessler, T. D. James, E. V. Anslyn and X. Sun, Chem. Soc. Rev., 2021, 50, 9–38 RSC.
- J. L. Sessler, J. M. Davis, V. Král, T. Kimbrough and V. Lynch, Org. Biomol. Chem., 2003, 1, 4113–4123 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) T. Noguchi, T. Shiraki, A. Dawn, Y. Tsuchiya, L. T. Ngoc Lien, T. Yamamoto and S. Shinkai, Chem. Commun., 2012, 48, 8090–8092 RSC.
- (a) S. Nishizawa, Y. Kato and N. Teramae, J. Am. Chem. Soc., 1999, 121, 9463–9464 CrossRef CAS; (b) F. Zapata, P. Sabater, A. Caballero and P. Molina, Org. Biomol. Chem., 2015, 13, 1339–1346 RSC.
- H. Tong, G. Zhou, L. Wang, X. Jing, F. Wang and J. Zhang, Tetrahedron Lett., 2003, 44, 131–134 CrossRef CAS.
- (a) T. Sakamoto, A. Ojida and I. Hamachi, Chem. Commun., 2009, 141–152 RSC; (b) X. Huang, Z. Guo, W. Zhu, Y. Xie and H. Tian, Chem. Commun., 2008, 5143–5145 RSC.
- (a) A. P. deSilva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (b) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS; (c) T. Gunnlaugsson, H. D. P. Ali, M. Glynn, P. E. Kruger, G. M. Hussey, F. M. Pfeffer, C. M. G. dos Santos and J. Tierney, J. Fluoresc., 2005, 15, 287–299 CrossRef CAS PubMed; (d) T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS; (e) B. Daly, J. Ling and A. P. de Silva, Chem. Soc. Rev., 2015, 44, 4203–4211 RSC.
- (a) P. Anzenbacher, K. Jursikova and J. L. Sessler, J. Am. Chem. Soc., 2000, 122, 9350–9351 CrossRef CAS; (b) H. Miyaji, P. Anzenbacher, J. L. Sessler, E. R. Bleasdale and P. A. Gale, Chem. Commun., 1999, 1723–1724, 10.1039/a905054j; (c) T. Gunnlaugsson, A. P. Davis and M. Glynn, Chem. Commun., 2001, 2556–2557, 10.1039/B107608F; (d) E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2008, 73, 8073–8076 CrossRef CAS PubMed.
- A. W. Czarnik, Chem. Biol., 1995, 2, 423–428 CrossRef CAS PubMed.
- D. H. Vance and A. W. Czarnik, J. Am. Chem. Soc., 1994, 116, 9397–9398 CrossRef CAS.
- M. E. Huston, E. U. Akkaya and A. W. Czarnik, J. Am. Chem. Soc., 1989, 111, 8735–8737 CrossRef CAS.
- (a) R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419–4476 CrossRef CAS PubMed; (b) M. Inclán, M. T. Albelda, E. Carbonell, S. Blasco, A. Bauzá, A. Frontera and E. García-España, Chem. – Eur. J., 2014, 20, 3730–3741 CrossRef PubMed.
- (a) P. A. de Silva, N. H. Q. Gunaratne and C. P. McCoy, Nature, 1993, 364, 42–44 CrossRef; (b) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS; (c) D. C. Magri, Coord. Chem. Rev., 2021, 426, 213598 CrossRef CAS; (d) S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Chem. Soc. Rev., 2018, 47, 2228–2248 RSC.
- (a) L. Fabbrizzi, M. Licchelli, P. Pallavicini and A. Taglietti, Inorg. Chem., 1996, 35, 1733–1736 CrossRef CAS PubMed; (b) L. Fabbrizzi, F. Gatti, P. Pallavicini and L. Parodi, New J. Chem., 1998, 22, 1403–1407 RSC.
- M. Dhaenens, J.-M. Lehn and J.-P. Vigneron, J. Chem. Soc., Perkin Trans. 2, 1993, 1379–1381, 10.1039/P29930001379.
- R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936–3953 RSC.
- S. K. Kim, H. N. Kim, Z. Xiaoru, H. N. Lee, H. N. Lee, J. H. Soh, K. M. K. Swamyand and J. Yoon, Supramol. Chem., 2007, 19, 221–227 CrossRef CAS.
- H. Fenniri, M. W. Hosseini and J.-M. Lehn, Helv. Chim. Acta, 1997, 80, 786–803 CrossRef CAS.
- M. T. Albelda, M. A. Bernardo, E. Garcia-España, M. L. Godino-Salido, S. V. Luis, M. João Melo, F. Pina and C. Soriano, J. Chem. Soc., Perkin Trans. 2, 1999, 2545–2549, 10.1039/A904894D.
- (a) M. P. Clares, C. Lodeiro, D. Fernández, A. J. Parola, F. Pina, E. García-España, C. Soriano and R. Tejero, Chem. Commun., 2006, 3824–3826, 10.1039/B607139B; (b) B. Verdejo, M. Inclán, M. P. Clares, I. Bonastre-Sabater, M. Ruiz-Gasent and E. García-España, Chemosensors, 2022, 10(1), 1, DOI:10.3390/chemosensors10010001.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132–5138 CrossRef CAS PubMed.
- A. Praetorius, D. M. Bailey, T. Schwarzlose and W. M. Nau, Org. Lett., 2008, 10, 4089–4092 CrossRef CAS PubMed.
- A. S. Oshchepkov, M. S. Oshchepkov, M. V. Oshchepkova, A. Al-Hamry, O. Kanoun and E. A. Kataev, Adv. Opt. Mater., 2021, 9, 2001913, DOI:10.1002/adom.202001913.
- A. M. Agafontsev, T. A. Shumilova, P. A. Panchenko, S. Janz, O. A. Fedorova and E. A. Kataev, Chem. – Eur. J., 2016, 22, 15069–15074 CrossRef CAS PubMed.
- A. S. Oshchepkov, R. R. Mittapalli, O. A. Fedorova and E. A. Kataev, Chem. – Eur. J., 2017, 23, 9657–9665 CrossRef CAS PubMed.
- M. H. Lee, N. Park, C. Yi, J. H. Han, J. H. Hong, K. P. Kim, D. H. Kang, J. L. Sessler, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 14136–14142 CrossRef CAS PubMed.
- T. A. Shumilova, T. Ruffer, H. Lang and E. A. Kataev, Chem. – Eur. J., 2018, 24, 1500–1504 CrossRef CAS PubMed.
- (a) G. Alibrandi, V. Amendola, G. Bergamaschi, L. Fabbrizzi and M. Licchelli, Org. Biomol. Chem., 2015, 13, 3510–3524 RSC; (b) V. McKee, J. Nelson and R. M. Town, Chem. Soc. Rev., 2003, 32, 309–325 RSC; (c) S. Kubik, in Chemistry of Nanocontainers, ed. M. Albrecht and E. Hahn, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–34 DOI:10.1007/128_2011_244; (d) Y. Han, Y. Jiang and C.-F. Chen, Tetrahedron, 2015, 71, 503–522 CrossRef; (e) V. Amendola, G. Bergamaschi and A. Miljkovic, Supramol. Chem., 2018, 30, 236–242 CrossRef CAS.
- M. A. Hortalá, L. Fabbrizzi, N. Marcotte, F. Stomeo and A. Taglietti, J. Am. Chem. Soc., 2003, 125, 20–21 CrossRef PubMed.
- M. P. Teulade-Fichou, J. P. Vigneron and J. M. Lehn, J. Chem. Soc., Perkin Trans. 2, 1996, 2169–2175, 10.1039/P29960002169.
- V. Amendola, G. Bergamaschi, M. Boiocchi, R. Alberto and H. Braband, Chem. Sci., 2014, 5, 1820–1826 RSC.
- R. R. Mittapalli, S. S. R. Namashivaya, A. S. Oshchepkov, E. Kuczynska and E. A. Kataev, Chem. Commun., 2017, 53, 4822–4825 RSC.
- R. R. Mittapalli, S. S. R. Namashivaya, A. S. Oshchepkov, T. A. Shumilova, T. Ruffer, H. Lang and E. A. Kataev, Chem. Commun., 2017, 53, 11345–11348 RSC.
- S. S. R. Namashivaya, A. S. Oshchepkov, H. Ding, S. Forster, V. N. Khrustalev and E. A. Kataev, Org. Lett., 2019, 21, 8746–8750 CrossRef CAS PubMed.
- I. Roy, A. H. G. David, P. J. Das, D. J. Pe and J. F. Stoddart, Chem. Soc. Rev., 2022, 51, 5557–5605 RSC.
- A. M. Agafontsev, T. A. Shumilova, T. Rüffer, H. Lang and E. A. Kataev, Chem. – Eur. J., 2019, 25, 3541–3549 CrossRef CAS PubMed.
- A. M. Agafontsev, T. A. Shumilova, A. S. Oshchepkov, F. Hampel and E. A. Kataev, Chem. – Eur. J., 2020, 26, 9991–9997 CrossRef CAS PubMed.
- A. M. Agafontsev, A. S. Oshchepkov, T. A. Shumilova and E. A. Kataev, Molecules, 2021, 26, 980 CrossRef CAS PubMed.
- A. S. Oshchepkov, T. A. Shumilova, M. Zerson, R. Magerle, V. N. Khrustalev and E. A. Kataev, J. Org. Chem., 2019, 84, 9034–9043 CrossRef CAS PubMed.
- A. S. Oshchepkov, O. Reznichenko, D. Xu, B. S. Morozov, A. Granzhan and E. A. Kataev, Chem. Commun., 2021, 57, 10632–10635 RSC.
| This journal is © The Royal Society of Chemistry 2023 |