
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overlooked potential of N,N-bidentate directing-groups in Ni-catalyzed C–H functionalization of benzamides†
Weirong
Wu
 a,
Xufang
Zhao
c,
Guang
Chen
c,
Lingjun
Liu
f,
Yulin
Li
b,
Tao
Chen
b,
Tony D.
James
*de and
Yuxia
Liu
*c
a,
Xufang
Zhao
c,
Guang
Chen
c,
Lingjun
Liu
f,
Yulin
Li
b,
Tao
Chen
b,
Tony D.
James
*de and
Yuxia
Liu
*c
aSchool of Environment and Chemical Engineering, Chongqing Three Gorges University, Chongqing, China
bKey Laboratory of Tibetan Medicine Research & Qinghai Key Laboratory of Qinghai-Tibet Plateau Biological Resuorces, Northwest Institute of Plateau Biology, Chinese Academy of Science, Xining 810001, Qinghai, P. R. China
cShaanxi Key Laboratory of Chemical Additives for Industry, College of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi’ an 710021, China. E-mail: liuyuxia2008@163.com
dDepartment of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: chstdj@bath.ac.uk
eSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, 453007, P. R. China
fSchool of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P. R. China
First published on 19th December 2022
Abstract
The Ni-catalyzed reactions of benzamides with bicyclic alkenes were explored using DFT calculations. An unprecedented “N–H deprotonation circumvented” catalytic mechanism was proposed, over the more common N–H/C–H activation mechanism, in which (i) the circumvention of N–H deprotonation ensures the presence of N–H⋯O hydrogen bond interaction, thereby stabilizing the critical ortho-C–H functionalization TS; and (ii) the N–H moiety retention results in a weak N⋯Ni σ-coordination, which is flexible to the configurational conversion during the key alkene insertion. These overlooked aspects of the functionalized N,N-bidentate directing groups will aid the design of new related catalytic reactions.
Transition-metal catalyzed C–H functionalization reactions have proven to be one of the most powerful and promising approaches for the construction of C–C and C–heteroatom bonds due to significant step- and atom-economy.1 The key challenge in exploring these reactions is how to discriminate multiple C–H bonds present in the reactant molecules. Considerable efforts have been devoted to incorporating directing groups (DGs) into the substrate2 to achieve site-selective C–H activation,3 in which, bidentate DGs are vital owning to their enhanced ability to achieve selective ortho-C–H functionalization, which is not possible using monodentate DGs.4 Since the seminal work by Daugulis et al.,5N,N-bidentate DGs have been widely used for C–H functionalization of aromatic amides catalyzed by various transition-metals complexes involving Ni, Co, Ru, Rh, Cu, and Pd.6 Among these C–H activation protocols, the coupling partners include alkynes, alkenes, and a wide range of halides etc.,7 significantly expanding the applications of these transformations.
Mechanistically, it is generally accepted that this type of reaction involves the programmed cleavages of the N–H and ortho-C–H bonds to form a metallacycle species as a key intermediate (Scheme 1A),8 in which, the N–H cleavage is an indispensable step to direct ortho-C–H activation. A series of computational studies have been successively established for these reactions with alkynes,9 arynes,10 halides11 and I2,12 where minimal divergence from the commonly accepted mechanism is observed.
 | ||
| Scheme 1 (A) Commonly reported mechanism for transition-metal catalyzed C–H activation of amides utilizing the N,N-bidentate directing group. (B) Ni-catalyzed reaction of N-(quinolin-8-yl)benzamide 1 with norbornene 2 reported by Chatani et al.13 | ||
Recently, using the 8-aminoquinoline(8-AQ) as a N,N-bidentate DG, Chatani et al. reported the Ni-catalyzed nonacidic C–H functionalization of aromatic amides with bicyclic alkenes13 to produce 1-indanone derivatives as potential pharmaceuticals for the treatment of Alzheimers,14 Parkinson's diseases15 and hepatitis C virus.16 A representative reaction is given in Scheme 1B, where N-(quinolin-8-yl)benzamide 1 and norbornene 2 in chlorobenzene at 140 °C produces the cyclic product P in high yield. However, a preliminary DFT examination (see the Computational details in the ESI†) for the reaction in Scheme 1B indicates that the generally accepted N–H/C–H activation mechanism is not operable under the given conditions due to high energy barrier (over 37 kcal mol−1 in Fig. 1). Thus, further theoretical explorations are required to unveil the mechanistic puzzle, and specially, the potential of the N,N-bidentate DG involved, which would be particularly useful for designing related catalytic reactions.
 | ||
| Fig. 1 Gibbs free energy profile in chlorobenzene solvent along the pathway forming the product P according to the commonly accepted route. The free energies are given in kcal mol−1. | ||
For the representative reaction in Scheme 1B, we first calculated the free-energy profile along the N–H/C–H activation pathway in Fig. 1. The reaction is initiated by the coordination of Ni(OTf)2 with BINAP ligand (denoted as L) to generate the complex IM1. Following participation of 1 and AgOAc, the N1–H deprotonation smoothly occurs via the cyclic transition state TS2-3 with a barrier of only 1.9 kcal mol−1, affording intermediate IM3. Upon LAgOAc addition to give IM4 after releasing LAgOTf and HOAc,17 a OAc-assisted concerted metalation-deprotonation (CMD) gives nickelacycle species IM5.18,19 Subsequently, the C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 bond of 2 inserts into the Ni–C1 bond forming IM6, a seven-membered energy nickelacycle species, which further evolves into product P.20 Note that the elementary step for alkene insertion viaTS5-6 has a barrier of 31.7 kcal mol−1 relative to IM5, which, results in an overall barrier of up to 37.6 kcal mol−1 (the difference between TS5-6 and IM3).9b Obviously, such a high energy barrier is impossible to accomplish under the given conditions. The main reason could be attributed to the instability of IM6, in which a seven-membered nickelacycle is involved. After N1–H deprotonation, compared with the previous weak Ni–N1 σ-coordination, a strong N1–Ni σ-bond forms, which results in a relatively rigid seven-membered nickelacycle in IM6. Thus, it is not surprising that a significant energy consumption occurs on formation of IM6. In this case, the commonly asserted pathway following a tandem N1–H deprotonation/C1–H-cleavage sequence is clearly not operative for the reaction under consideration and as such a more reasonable mechanism is required.
C5 bond of 2 inserts into the Ni–C1 bond forming IM6, a seven-membered energy nickelacycle species, which further evolves into product P.20 Note that the elementary step for alkene insertion viaTS5-6 has a barrier of 31.7 kcal mol−1 relative to IM5, which, results in an overall barrier of up to 37.6 kcal mol−1 (the difference between TS5-6 and IM3).9b Obviously, such a high energy barrier is impossible to accomplish under the given conditions. The main reason could be attributed to the instability of IM6, in which a seven-membered nickelacycle is involved. After N1–H deprotonation, compared with the previous weak Ni–N1 σ-coordination, a strong N1–Ni σ-bond forms, which results in a relatively rigid seven-membered nickelacycle in IM6. Thus, it is not surprising that a significant energy consumption occurs on formation of IM6. In this case, the commonly asserted pathway following a tandem N1–H deprotonation/C1–H-cleavage sequence is clearly not operative for the reaction under consideration and as such a more reasonable mechanism is required.
To circumvent the rigid seven-membered nickelacycle during the reaction, based on the theoretical investigation, we tentatively developed a N1–H deprotonation free mechanism, where an extra N–H⋯O hydrogen bond interaction is present in the OAc-assisted CMD process, as well as a weak Ni⋯N coordination in the crucial alkene insertion step. The proposed mechanism is initiated by C1–H activation. As shown in Fig. 2, from IM1 in Fig. 1, with the addition of 1 and AgOAc, the OAc-assisted CMD process proceeds to afford the five-membered cyclic complex IM8. The C1–H activated TS, TS7-8, includes a N1–H⋯O hydrogen bond, with a barrier of 11.8 kcal mol−1 relative to IM1. After the approach of 2 to IM8 with L release to give IM9,21 two elementary steps follow: the C4C5 coordination of 2 to the Ni centre (IM9 → IM10) and then C4C5 insertion into the Ni–C1 bond (IM10 → IM11), leading to the alkene inserted species IM11. The transformation from IM9 to IM11 is relatively facile with a barrier of 15.6 kcal mol−1 (the difference between TS10-11 and IM9).
 | ||
| Fig. 2 Gibbs free energy profiles in chlorobenzene solvent for P formation established in the present work. The free energies are given in kcal mol−1. | ||
After alkene insertion, according to Chatani's proposal,13 the product P would be obtained via an intramolecular nucleophilic cyclization followed by C3–N1 cleavage with the assistance of AgOAc. Our calculated results in Fig. 2 indicate that, from IM11, after the ligand exchange between L and HOAc,22 this intramolecular nucleophilic cyclization takes place viaTS12-13, in which, the (Ni-)OTf moiety is simultaneously transferring to the trans position from the cis position of the N(H) moiety to keep the planar configuration of the Ni center.23 This concerted step results in a barrier of 33.0 kcal mol−1 and thus is identified as rate-determining. The resultant complex IM13 then experiences a facile C3–N1 cleavage with a barrier of only 2.0 kcal mol−1 and gives rise to structure IM14, in which P has almost formed. Ultimately, IM14 combines with HOAc and 1 leading to product P and active catalyst IM2. The simultaneously obtained quinolin-8-amine 3, as a by-product, is further oxidized by AgOAc into N-(quinolin-8-yl)acetamide.13
To provide a better understanding on the pathway in Fig. 2, we performed comparative analyses of the transition states for two key steps involved in Fig. 1 and 2: the C1–H cleavage and alkene insertion, and the calculated results are given in Fig. 3. In the case of the C1–H cleavage process (left column), TS7-8 (including the N1–H⋯O hydrogen bond) is slightly higher in energy than TS4-5 (TS after N–H deprotonation) (−13.8 vs. −20.2 kcal mol−1). If no-hydrogen-bond is considered, the resultant C1–H activation TS, denoted as TS7-8′, is energetically less favorable by 10.1 kcal mol−1 than TS7-8. Thus, it is believed that the N–H⋯O hydrogen bond interaction involved facilitates the stability of TS7-8. From the performed noncovalent interaction analyses (NCIs),24 one can identify in Fig. 3 (left column) that a significant N1–H⋯O hydrogen bond interaction (blue isosurface) is present in TS7-8, but very weak dispersion interaction (green isosurface) is observed in TS7-8′. In general, the N1–H⋯O hydrogen bond plays a pivotal role on stabilizing TS7-8.
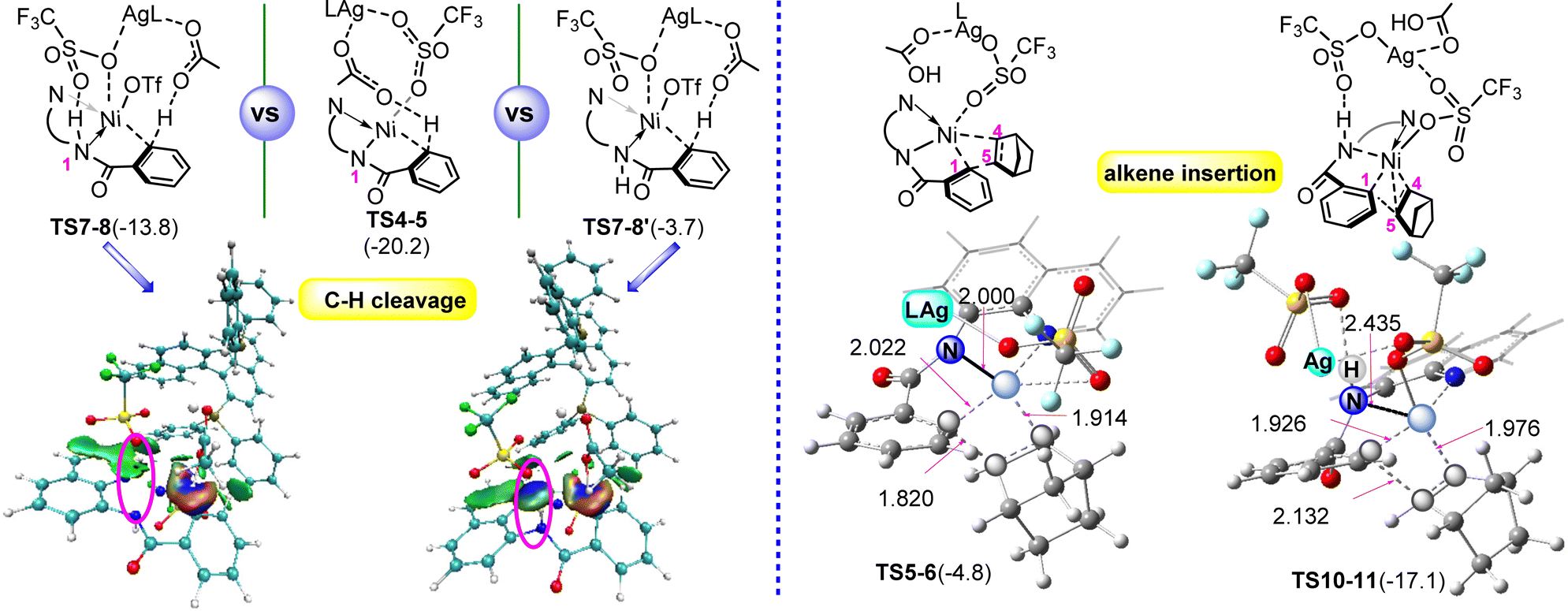 | ||
| Fig. 3 Noncovalent interaction analyses for the C–H cleavage TSs, TS7-8, TS4-5, TS7-8′ (left column, isosurface value = 0.01) as well as the geometries of two alkene insertion TSs, TS5-6 (HOAc is not shown for clarity) and TS10-11 (right column). The free energies are given in kcal mol−1. | ||
Next, we turned our attention to the alkene insertion step TSs (Fig. 3, right column). TS10-11 is found to have appreciably lower free energy than TS5-6, −17.1 vs. −4.8 kcal mol−1, which was supported by the calculated bond distances. In TS10-11, the Ni⋯C1 (1.926 Å) is much shorter than 2.022 Å in TS5-6, while the Ni⋯C4 and Ni⋯C5 (1.976 and 2.132 Å) are longer than the corresponding distances (1.914 and 1.820 Å) in TS5-6. Obviously, TS10-11 is easier to surmount than TS5-6. The origin might be derived from the discrepancy in the Ni–N1 interaction modes involved. It is noted that TS10-11 features weak Ni–N1 σ-coordination, which is flexible to adapt the configurational transformation resulting from the alkene insertion. In sharp contrast, the Ni–N1 σ-bond is included in TS5-6. Such a rigid bond brings about a large structural distortion and thus results in a significant energy penalty for TS5-6. Obviously, the N1–H retention, due to the lack of N1–H deprotonation, is intrinsically essential for facilitating the alkene insertion.
In summary, the detailed mechanisms for the Ni-catalyzed reaction of N-(quinolin-8-yl)benzamide 1 with norbornene 2 have been computationally evaluated. The commonly reported N–H/C–H activation mechanism was found to be kinetically inaccessible under the given conditions due to the high energy requirement. In this work, a unique “N–H deprotonation circumvented” catalytic mechanism is proposed, which highlights two properties of the N,N-bidentate group: (i) absence of N–H deprotonation leading to N–H⋯O hydrogen bond interaction, (ii) the N–H moiety retention resulting in N⋯Ni weak σ-coordination. It was found that the N–H⋯O hydrogen bond interaction facilitates the critical ortho-C–H functionalization, and N⋯Ni σ-coordination contributes significantly to the key alkene insertion, facilitating the reaction. These aspects have previously been overlooked, however we expect they may be important for other relevant catalytic reactions.
This work was jointly supported by the National Natural Science Foundation of China (No. 22174090), the Natural Science Foundation of Shaanxi Province (2022JM-089), Key Laboratory of Emergency and Trauma (Hainan Medical University), Ministry of Education (KLET-201903). TDJ wishes to thank the Royal Society for a Wolfson Research Merit Award and the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University for support (2020ZD01).
Conflicts of interest
There are no conflicts to declare.References
- (a) M. Albrecht, Chem. Rev., 2010, 110, 576 CrossRef CAS PubMed; (b) T. Brückl, R. D. Baxter, Y. Ishihara and P. S. Baran, Acc. Chem. Res., 2012, 45, 826 CrossRef PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- (a) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (b) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (c) K. M. Engle, T. S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. Daugulis, J. Org. Chem., 2013, 78, 9689 CrossRef CAS PubMed.
- (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (b) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705 CrossRef CAS; (c) M. S. Khan, A. Haque, M. K. Al-Suti and P. R. Raithby, J. Organomet. Chem., 2015, 793, 114 CrossRef CAS; (d) X. Yang, G. Shan, L. Wang and Y. Rao, Tetrahedron Lett., 2016, 57, 819 CrossRef CAS.
- (a) X.-F. Yang, X.-H. Hu and T.-P. Loh, Org. Lett., 2015, 17, 1481 CrossRef CAS PubMed; (b) P. Gandeepan, P. Rajamalli and C.-H. Cheng, Angew. Chem., Int. Ed., 2016, 55, 4308 CrossRef CAS PubMed; (c) Z. He and Y. Huang, ACS Catal., 2016, 6, 7814 CrossRef CAS; (d) B. Khan, R. Kant and D. Koley, Adv. Synth. Catal., 2016, 358, 2352 CrossRef CAS; (e) T. Uemura, M. Yamaguchi and N. Chatani, Angew. Chem., Int. Ed., 2016, 55, 3162 CrossRef CAS PubMed; (f) Y. Aihara and N. Chatani, ACS Catal., 2016, 6, 4323 CrossRef CAS; (g) Q. Zheng, C. Liu, J. Chen and G. Rao, Adv. Synth. Catal., 2020, 362, 1406 CrossRef CAS.
- (a) Y. Dang, X. Deng, J. Guo, C. Song, W. Hu and Z.-X. Wang, J. Am. Chem. Soc., 2016, 138, 2712 CrossRef CAS PubMed; (b) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed; (c) Y. Liu, K. Wang, B. Ling, G. Chen, Y. Li, L. Liu and S. Bi, Catal. Sci. Technol., 2020, 10, 4219 RSC; (d) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344 CrossRef CAS PubMed.
- (a) S. R. Neufeldt, G. Jiménez-Osés, J. R. Huckins, O. R. Thiel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 9843 CrossRef CAS PubMed; (b) K. Yamazaki, A. Obata, A. Sasagawa, Y. Ano and N. Chatani, Organometallics, 2019, 38, 248 CrossRef CAS; (c) H. M. Omer and P. Liu, ACS Omega, 2019, 4, 5209 CrossRef CAS PubMed.
- C. Chen, Y. Hao, T.-Y. Zhang, J.-L. Pan, J. Ding, H.-Y. Xiang, M. Wang, T.-M. Ding, A. Duan and S.-Y. Zhang, Chem. Commun., 2019, 55, 755 RSC.
- (a) L. Huang, Q. Li, C. Wang and C. Qi, J. Org. Chem., 2013, 78, 3030 CrossRef CAS PubMed; (b) A. L. Dewyer and P. M. Zimmerman, ACS Catal., 2017, 7, 5466 CrossRef CAS.
- B. E. Haines, J.-Q. Yu and D. G. Musaev, Chem. Sci., 2018, 9, 1144 RSC.
- A. Skhiri and N. Chatani, Org. Lett., 2019, 21, 1774 CrossRef CAS PubMed.
- L. Huang, H. Miao, Y. Sun, F. Meng and X. Li, Eur. J. Med. Chem., 2014, 87, 429 CrossRef CAS PubMed.
- A. Affini, S. Hagenow, A. Zivkovic, J. Marco-Contelles and H. Stark, Eur. J. Med. Chem., 2018, 148, 487 CrossRef CAS PubMed.
- L. Chan, O. Pereira, T. J. Reddy, S. K. Das, C. Poisson, M. Courchesne, M. Proulx, A. Siddiqui, C. G. Yannopoulos, N. Nguyen-Ba, C. Roy, D. Nasturica, C. Moinet, R. Bethell, M. Hamel, L. L’Heureux, M. David, O. Nicolas, P. Courtemanche-Asselin, S. Brunette, D. Bilimoria and J. Bédard, Bioorg. Med. Chem. Lett., 2004, 14, 797 CrossRef CAS PubMed.
- From IM3, the C–H cleavage followed by alkene insertion pathway is found to be less preferred than the one from IM4. Please see Fig. S1 for the details.
- The possible C–H oxidative additions from IM5 are also further considered. However, it is found that the resultant IM17 (NiIII intermediate) and IM18 (NiIV species) are even higher in energy than the alkene insertion transition state TS5-6. Therefore, the pathways leading to the nickel species with high oxidation state are not operative in the reaction under consideration. Please see Fig. S1 for the details.
- (a) Y. B. Dudkina, R. R. Fayzullin, K. A. Lyssenko, A. T. Gubaidullin, K. V. Kholin, A. I. Levitskaya, M. Yu. Balakina and Y. H. Budnikova, Organometallics, 2019, 38, 1254 CrossRef CAS; (b) S.-K. Zhang, R. C. Samanta, A. D. Vecchio and L. Ackermann, Chem. – Eur. J., 2020, 26, 10936 CrossRef CAS PubMed; (c) Y. H. Budnikova, Chem. Rec., 2021, 21, 2148 CrossRef CAS PubMed.
- The alkene insertion step viaTS5-6 is believed to be unachievable due to a high energy demand, and therefore, further calculations after IM6 were not performed.
- As compared to the L-involved case, the barrier for the no-L-involved alkene insertion process from IM8 is easier to overcome. Please see Fig. S3 for the details.
- Other possible nucleophilic cyclization pathways are collected in Fig. S3. It is found that these potential energy profiles are significantly higher than that starting from IM11.
- A diagram of the key bond length scans for TS12-13 along the IRC pathway is displayed in Fig. S4, which supports the concerted nucleophilic cyclization and the (Ni-)OTf migration.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed; (b) S. Manzetti and T. Lu, J. Phys. Org. Chem., 2013, 26, 473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06177e |
| This journal is © The Royal Society of Chemistry 2023 |