
Dual action of a tyrosinase–mesoporous silica nanoparticle complex for synergistic tissue adhesion†
Su-Hwan
Kim‡
ab,
Kwangsoo
Shin‡
cd,
Byung-Gee
Kim
 bef,
Nathaniel S.
Hwang
*bfg and
Taeghwan
Hyeon
*bc
bef,
Nathaniel S.
Hwang
*bfg and
Taeghwan
Hyeon
*bc
aDepartment of Chemical Engineering (BK21 FOUR), Dong-A University, Busan, 49315, Republic of Korea
bSchool of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul, 08826, Republic of Korea. E-mail: thyeon@snu.ac.kr; nshwang@snu.ac.kr
cCenter for Nanoparticle Research, Institute of Basic Science (IBS), Seoul, 08826, Republic of Korea
dDepartment of Biomedical Engineering, Yale University, New Haven, CT, USA
eInstitute of Molecular Biology and Genetics, Institute for Sustainable Development (ISD), Seoul National University, Seoul, 08826, Republic of Korea
fBio-MAX/N-Bio, Institute of BioEngineering, Seoul National University, Seoul, 08826, Republic of Korea
gInstitute of Engineering Research, Seoul National University, Seoul, 08826, Republic of Korea
First published on 28th November 2022
Abstract
Bridging biological tissues for immediate adhesion and long-term sustainability was accomplished using a combination of mesoporous silica nanoparticles (MSNs) and tyrosinase. Tyrosinase-loaded MSNs provided rapid physical adsorption, while tyrosinase within MSNs induced enzymatic chemical bond gluing of tissues. This synergistic strategy has robust potential in tissue adhesives for clinical settings.
Tissue adhesives have been widely used to immediately close a wound site to prevent loss of body fluids, minimize the chance of infection of injured tissues, and facilitate recovery of patients without any complications.1,2 Tissue adhesives have been used in various surgical situations in which staples and sutures cannot be used and have shown effectiveness with facile and complete sealing. Tissue adhesives meet the demands of long-term sustainability following immediate gluing under wet conditions.3 However, the moist, soft, and heterogenous surface of biological tissues makes the development of tissue adhesives with immediate and stable closure challenging. Cyanoacrylate-based tissue adhesives have been clinically translated and applied as they can chemically bond to neutrophilic biological substrates and are rapidly polymerized.4 However, due to toxicity and disparity between the physical properties of polymerized adherents and applied biological tissues, these adhesives cannot be applied to internal organs.5,6 Therefore, it is necessary to design a strategy to bridge biological tissues that can be adapted in clinical settings.
In this study, we combined two different actions of adhesion, i.e., the physisorption of mesoporous silica nanoparticles (MSNs) and the enzymatic reaction of tyrosinase, to develop a dual-action tissue adhesive to provide immediate and stable adhesion. MSNs have been suggested as adhesives for hydrogels and soft tissues.7 The MSN surface provides rapid adhesion and energy dissipation with biological substrates through physical adsorption, yielding an efficient and stable binding between biological tissues resistant to the large deformation of applied tissues. MSNs providing direct adhesion between biological tissues without blocking the molecular exchanges contribute to effective wound closures and regeneration of skin and liver.8,9 The versatility in the functioning of MSNs enables MSN adhesives to facilitate regeneration of the injured site, minimize inflammation and infection, and be applied via an image-guided procedure.10–12 Most importantly, the mechanism of adhesion by MSNs is based on physisorption, which provides immediate adhesion compared to chemical adhesives; however, further improvements with the stable and strong adhesion are necessary for physisorption-based MSN adhesives to be translated into clinics.
Meanwhile, tyrosinase-mediated tissue adhesives have been developed for biocompatible long-term tissue glue.13–15 Tyrosinase, an essential enzyme in skin pigmentation, recognizes specific phenolic moieties such as catechol, tyrosine, and gallol in melanocytes to synthesize a polydopamine called melanin. Substrate specificity and reactivity under physiological conditions make tyrosinase a desirable adhesive tissue suitable for clinical settings. The elucidated mechanism of tyrosinase-mediated tissue adhesion involves two consecutive oxidative reactions of tyrosine residues on biological tissues such as collagen. Ortho-quinone, the intermediate state of oxidized tyrosine, can react with primary amines, thiols, or imidazoles through non-enzymatic Michael-type addition or a Schiff-base reaction. This oxidative reaction is the main cascade to induce tissue gluing since primary amines and thiols are abundant in native tissues, including collagen. To combine both potentials providing rapid physisorption and enzymatic covalent binding, we successfully demonstrated tyrosinase–mesoporous silica nanoparticle (T–MSN)-based tissue adhesives by loading tyrosinase within MSNs with controlled pore size. When T-MSNs are intact with endogenous tissues such as collagen which has abundant phenolic residues, the MSN surface can be absorbed on collagen chains by physisorption. Then, loaded tyrosinase can oxidize phenolic moieties on collagen to bond tissues.
Since the hydrodynamic radius of tyrosinase calculated from the protein molecular weight is 2.75 nm, we decided to increase the pore size of MSNs to contain tyrosinase based on previous publications.9,11 Various amounts of mesitylene were introduced during MSN synthesis as a micelle swelling agent. Transmission electron microscopy (TEM) images showed that four MSNs having different pore sizes were successfully synthesized while maintaining the overall size of 70 nm without agglomeration (Fig. 1A). From the gas adsorption and desorption analysis, it was confirmed that the pore size, pore volume, and surface area of MSNs were increased by adding more mesitylene. The volume of tyrosinase-accessible-mesopores with their size between 5.5 and 10 nm starts to increase from MSN-3 and the volumes were larger in MSN-4 (Table 1 and Fig. S1, Table S1, ESI†). Various concentrations (6.25, 12.5, 25.0, and 50.0 μM) of tyrosinase were added for encapsulation of the prepared MSNs (Fig. 1B). We previously verified that up to 50 μM tyrosinase has no in vitro and in vivo toxicity whereas it has efficient tissue adhesion properties.13,14,16,17 The loadings of tyrosinase in MSNs depended on the amount of tyrosinase added and pore sizes. When tyrosinase was used at 6.25 and 12.5 μM, no significant difference was observed in the loading efficiency of different types of MSNs.
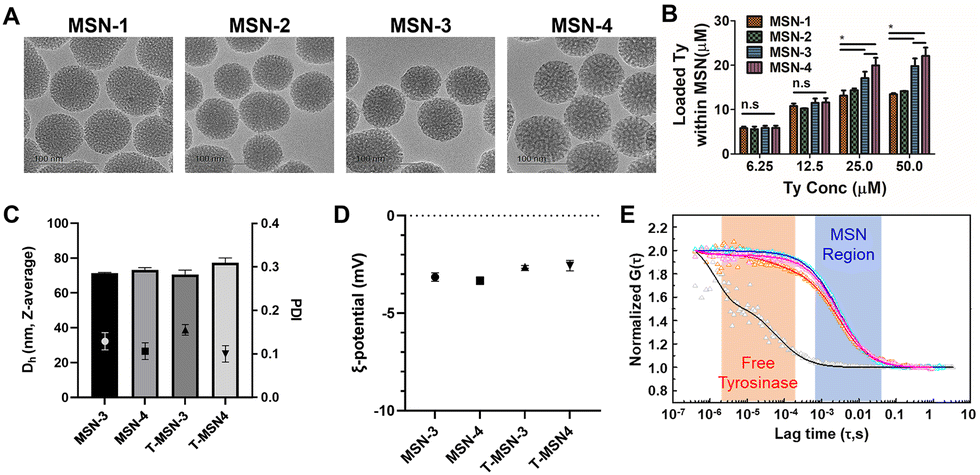 | ||
| Fig. 1 Characterization of T-MSNs. (A) TEM images of MSNs with various pore sizes (scale bar = 100 nm). (B) Quantification of loaded tyrosinase within MSNs. (C) Hydrodynamic size, polydispersity index, and (D) zeta-potentials of T-MSNs compared to those of MSNs. (E) Normalized correlation curves of tyrosinase (black), tyrosinase in T-MSNs (red), and T-MSNs (magenta) and cross-correlation curve of tyrosinase in T-MSNs and T-MSNs from fluorescence correlation spectroscopy. | ||
However, MSN-1 and MSN-2 with small pore sizes were saturated after the addition of 25 μM of tyrosinase. In contrast, MSN-3 and MSN-4 showed better loading, incorporating tyrosinase up to 22.1 ± 2.7 μM on 30 mg mL−1 of MSN, which could induce sufficient enzyme-mediated oxidation with native tissues. The loading of tyrosinase within MSNs can be attributed to both adsorption of tyrosinase on the MSN surface and encapsulation inside the enlarged pores. Tyrosinase loaded in MSN-1 and 2 would be mostly adsorbed on the NP surface, as their pore sizes are too small. Noticeable increases in tyrosinase loading of MSN-3 and 4 also confirm that enlarged pores contributed to tyrosinase being entrapped in pores and providing sufficient tyrosinase for enzymatic adhesion. Furthermore, the colloidal stability of MSNs was maintained after encapsulation of tyrosinase (Fig. 1C and D). The average hydrodynamic size of both MSNs and T-MSNs was 75 nm and their zeta-potentials were negative, confirming that MSNs were not agglomerated. Using fluorescence correlation spectroscopy with fluorescent-tagged tyrosinase and MSNs, we confirmed that tyrosinase was incorporated in MSNs (Fig. 1E). As shown in Fig. 1E, free-tyrosinase has a short diffusion time of 41 ± 6 μs, and MSNs have a longer diffusion time (3890 ± 1180 μs). When tyrosinase was incorporated into MSNs, the correlation curves of tyrosinase matched well with those of MSNs, with the same diffusion time of MSNs (3880 ± 1210 μs). Cross-correlation curves also confirmed that most of the tyrosinase was successfully incorporated and behaved together before release.
We examined tyrosinase activity of T-MSNs with L-tyrosine, gelatin, and tyrosine-conjugated hyaluronic acid (HA_t) (Fig. 2A–D) and compared with that of pristine tyrosinase. Tyrosinase activity on L-tyrosine showed no significant difference across the groups (20 μM tyrosinase, T-MSN-3, and T-MSN-4) (Fig. 2A). However, when T-MSNs were mixed with gelatin and HA_t, biopolymers containing tyrosine, the enzyme activities were significantly enhanced compared with those of tyrosinase itself (Fig. 2B and C). Additionally, T-MSN-4 with larger pores exhibited a higher enzyme activity than both pristine tyrosinase and T-MSN-3. The facilitated enzymatic oxidation of tyrosine-containing biopolymers is likely because the polymeric chain absorbed on the surface of MSNs can have more chance to be oxidized by tyrosinase on the surface and inside pores of MSNs (Fig. 2D).
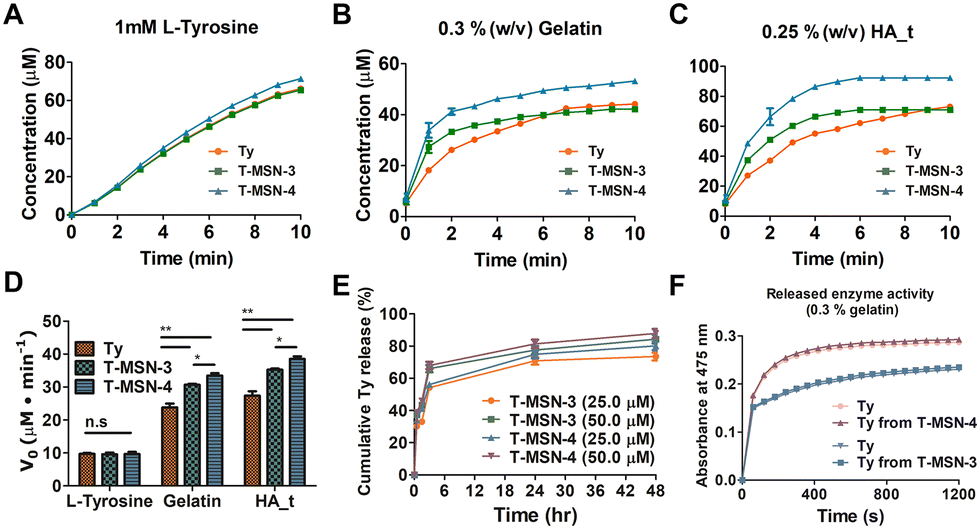 | ||
| Fig. 2 Analysis of tyrosinase (Ty) activity. (A–D) Monitoring of phenolic oxidation of L-tyrosine, gelatin, and HA_t and by Ty, T-MSN-3, and T-MSN-4. (E) Kinetics of Ty release from MSNs. (F) Reaction activity of released Ty from MSNs. | ||
We quantified the tyrosinase concentration released from MSNs at specific time points (0.5, 2, 3, 6, 24, and 28 h) (Fig. 2E). Both T-MSN-3 and -4 exhibited burst-releasing profiles, showing that approximately 60% of the encapsulated tyrosinase was released in 3 h. As there are no additional functional or structural modifications on MSNs to arrest tyrosinase in the mesopores, tyrosinase is expected to have burst-release profiles under diluted aqueous conditions. For the clinical translation in which T-MSNs are injected into the target tissue, released tyrosinase would be localized near the applied area since T-MSNs would be entrapped within the collagen structure. The released tyrosinase can also rapidly oxidize the phenolic groups in the collagen without direct contact with T-MSN, bridging two different tissues. The released tyrosinase had a similar enzyme activity compared to the same concentration of pristine tyrosinase (Fig. 2F).
As designed, T-MSNs have a dual mechanism of soft tissue adhesion. These two different mechanisms have complementary and synergistic strategies. MSNs have a high surface area to be quickly absorbed on tissue chains. Adhesion by physisorption of MSNs, however, can be weakened in vivo through an exchange between tissue chains and native tissue compounds such as blood constituents following hemolytic activity.18 Tyrosinase covalently glues tissue chains for prolonged and stronger tissue adhesion. Since MSNs were captured on tissue chains, tyrosinase inside and near MSNs can easily oxidize phenolic moieties (e.g., tyrosine groups) on tissue chains, such as collagen, forming a covalent bond with each tissue. The final product of T-MSN-based tissue adhesion will be permanently crosslinked to tissue chains in the adhesive layer, thereby stably gluing the tissue.
To demonstrate the proposed mechanism and evaluate the adhesion properties, we synthesized two types of hydrogels, a poly(dimethylacrylamide) (PDMA) hydrogel and a gelatin hydrogel (Fig. 3A, B and Fig. S2, S3, ESI†). The PDMA hydrogel is suitable to be a representative model hydrogel presenting MSN-based adhesion since it can drastically display MSN-based physisorption and charge interaction compared to other hydrogels (polyacrylamide, polyethylene oxide, and poly(N-isopropyl acrylamide)) with similar crosslinking density and swelling degree.7,19 However, it has no phenolic residues to react with tyrosinase. In contrast, the gelatin hydrogel, a denatured form of collagen, has abundant phenolic residues on polymeric chains. MSNs glued the PDMA pieces, but tyrosinase had a similar adhesive energy to the control group. The T-MSN group also provided adhesion between PDMA gels. T-MSNs on PDMA had a statistically significant decrease in adhesive energy compared to MSNs since partial tyrosinase was attached to the surface of MSNs (Fig. 3A). In contrast, synergistic adhesive effects of MSNs and tyrosinase were observed on the gelatin hydrogel (Fig. 3B). The gelatin hydrogel is negatively charged and has short free polymer chains where MSNs reversibly adsorb and desorb. Thus, the physisorption-based adhesion of a gelatin substrate was weaker than that of PDMA, but it also exhibited a statistically significant increase in adhesive energy compared to the control group (Fig. 3B). The adhesion energy of tyrosinase increased in a time-dependent manner since residual phenolic moieties in gelatin sequentially reacted via tyrosinase-mediated oxidation (Fig. 3C–F). In the case of MSN-mediated adhesion, the adhesive layer was immediately formed in a minute. However, there was no further gluing after the initial adhesion (Fig. 3D). T-MSNs on the gelatin hydrogel had synergistic effects on tissue adhesion since MSNs can be effectively absorbed on gelatin chains to crosslink two different gelatin pieces through tyrosinase-mediated oxidation, increasing the adhesion energy significantly by up to three times that of MSNs and tyrosinase (Fig. 3E). Hence, we confirmed that the combination of MSNs and tyrosinase can be a synergistic strategy for tissue adhesion, providing an immediate adhesive layer and permanent adhesion via chain crosslinking.
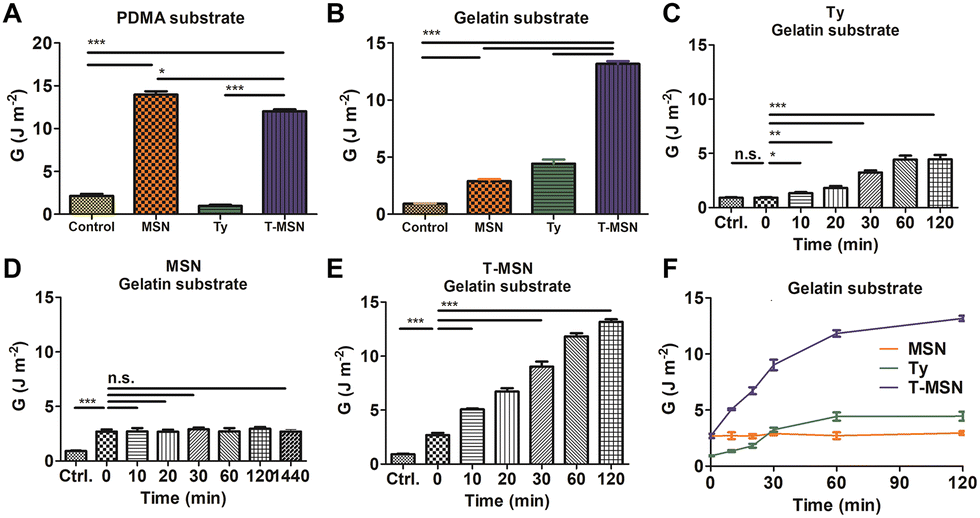 | ||
| Fig. 3 Adhesion properties of T-MSNs on two different substrates: (A) PDMA and (B) gelatin. (C–F) Time-dependent adhesion on the gelatin substrate (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
Along with the adhesion properties, we also evaluated the biodegradability and clearance of the T-MSN adhesive since it is important for the adhesive materials to be degraded at the rate of wound healing.5 The clinical translation of inorganic nanomaterial-based tissue adhesives can be concerning owing to their poor biodegradability. However, we employed large-pore MSNs that can degrade in vivo and whose by-product is known to be metabolically consumed and cleared.20,21 The clearance of MSNs and T-MSNs in vivo was examined after injecting adhesives subcutaneously. Both adhesive layers, MSNs and T-MSNs, were observed under the dermis after one day of administration; however, no adhesive layer was observed after 14 days (Fig. 4A and B). We also quantified the amount of silicon in the injected tissues and organs such as spleen, liver, and kidneys using inductively coupled plasma mass spectrometry (ICP-MS). Most of the silicon was cleared from the applied tissues within 14 days (Fig. 4C). Furthermore, significant levels of silicon were detected in spleen, liver, and kidneys on day 7, indicating their involvement in MSN clearance and by-product excretion (Fig. 4D). On day 28, most of the silicon was cleared both from the organs and applied tissues. Since exogenous proteins are not able to penetrate a lipid-based cellular membrane because of size limitation and their hydrophilicity,22 tyrosinase itself cannot diffuse into the melanocyte cytosol without any enhancers or carriers and induce tissue pigmentation. When tyrosinase is intact with native tissues, it might lead to unexpected tissue crosslinking, further increasing the stiffness of tissues. However, as tyrosinase is a nature-derived enzyme that is already involved in melanin synthesis, we hypothesize that tyrosinase has a relevant tolerance level that would not trigger side effects on tissues.
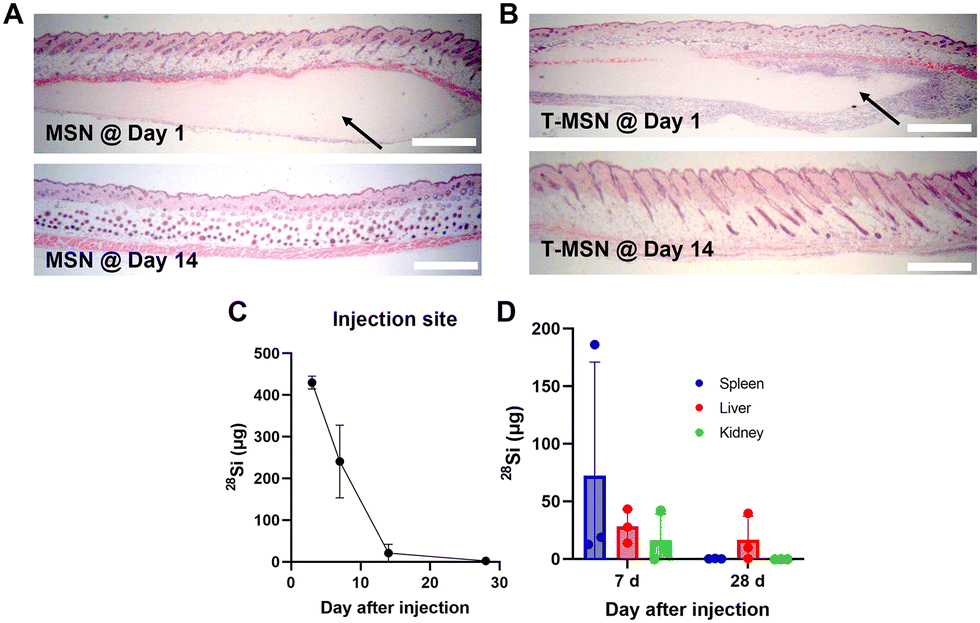 | ||
| Fig. 4 In vivo degradation of MSNs and T-MSNs. Histological analysis of subcutaneously injected (A) MSNs and (B) T-MSNs post-injection and after 14 days (scale bar = 500 μm). (C and D) Amounts of silicon in applied tissues and major organs, analyzed via ICP-MS. | ||
In conclusion, T-MSNs demonstrated the dual action of enzymatic- and physisorption-mediated tissue adhesion. T-MSNs were able to be prepared to have enough tyrosinase loading by increasing the pore size over tyrosinase. T-MSNs exhibited the facilitated enzymatic oxidation of tyrosine residues on biopolymers, possibly due to the adsorption of the polymers on MSNs. T-MSNs complementarily and synergistically glued tissue-mimetic substrates, showing immediate and long-term sustainable adhesion and overcoming the limitation of poor adhesion of the physisorption-based MSN adhesive and the delayed gluing of enzymatic crosslinking. Additionally, T-MSNs successfully degraded in mouse subcutaneous tissues within 2 weeks, indicating long-term in vivo biocompatibility. Further study is needed on the relationship between the facilitated enzymatic activity of tyrosinase on the NP surface and resulting adhesion synergy effects. As for the synergistic effect of MSNs and tyrosinase, we will provide a robust and practical method to glue biological tissues for clinical settings and expect that the synergistic combination of mechanisms may expand the design concept for further biological tissue adhesive developments.
This work was supported by the Institute for Basic Science in Korea (IBS-R006-D1) and the National Research Foundation of Korea (NRF-2022R1F1A1066631, NRF-2021M3E5E5096460, and NRF-2021R1A2C2008821). The Institute of BioEngineering at Seoul National University provided support for this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Dolgin, Nat. Med., 2013, 19, 124–126 CrossRef CAS.
- W. D. Spotnitz, Am. Surg., 2012, 78, 1305–1321 CrossRef.
- L. Ge and S. Chen, Polymers, 2020, 12, 939 CrossRef CAS.
- A. J. Singer and H. C. Thode Jr, Am. J. Surg., 2004, 187, 238–248 CrossRef CAS.
- S. Nam and D. Mooney, Chem. Rev., 2021, 121, 11336–11384 CrossRef CAS.
- P. A. Leggat, D. R. Smith and U. Kedjarune, Aust. N. Z. J. Surg., 2007, 77, 209–213 CrossRef PubMed.
- S. Rose, A. Prevoteau, P. Elziere, D. Hourdet, A. Marcellan and L. Leibler, Nature, 2014, 505, 382 CrossRef CAS.
- A. Meddahi-Pellé, A. Legrand, A. Marcellan, L. Louedec, D. Letourneur and L. Leibler, Angew. Chem., Int. Ed., 2014, 53, 6369–6373 CrossRef PubMed.
- J.-H. Kim, H. Kim, Y. Choi, D. S. Lee, J. Kim and G.-R. Yi, ACS Appl. Mater. Interfaces, 2017, 9, 31469–31477 CrossRef CAS.
- H. Wu, F. Li, S. Wang, J. Lu, J. Li, Y. Du, X. Sun, X. Chen, J. Gao and D. Ling, Biomaterials, 2018, 151, 66–77 CrossRef CAS PubMed.
- J. Kim, H. Y. Kim, S. Y. Song, S.-h Go, H. S. Sohn, S. Baik, M. Soh, K. Kim, D. Kim, H.-C. Kim, N. Lee, B.-S. Kim and T. Hyeon, ACS Nano, 2019, 13, 3206–3217 CrossRef CAS PubMed.
- K. Shin, J. W. Choi, G. Ko, S. Baik, D. Kim, O. K. Park, K. Lee, H. R. Cho, S. I. Han and S. H. Lee, Nat. Commun., 2017, 8, 1–12 CrossRef CAS.
- S.-H. Kim, S.-H. Lee, J.-E. Lee, S. J. Park, K. Kim, I. S. Kim, Y.-S. Lee, N. S. Hwang and B.-G. Kim, Biomaterials, 2018, 178, 401–412 CrossRef CAS PubMed.
- S.-H. Kim, K. Kim, B. S. Kim, Y.-H. An, U.-J. Lee, S.-H. Lee, S. L. Kim, B.-G. Kim and N. S. Hwang, Biomaterials, 2020, 242, 119905 CrossRef CAS.
- S. Choi, H. Ahn and S.-H. Kim, J. Appl. Polym. Sci., 2022, 139, e51887 CrossRef.
- U. J. Lee, J. Ko, S. H. Kim, P. G. Lee, Y. H. An, H. Yun, D. T. Flood, P. E. Dawson, N. S. Hwang and B. G. Kim, Adv. Sci., 2022, 9, e2103503 CrossRef.
- B. S. Kim, S.-H. Kim, K. Kim, Y.-H. An, K.-H. So, B.-G. Kim and N. S. Hwang, Mater. Today Bio, 2020, 8, 100079 CrossRef CAS PubMed.
- A. Yildirim, E. Ozgur and M. Bayindir, J. Mater. Chem. B, 2013, 1, 1909–1920 RSC.
- D. Hourdet and L. Petit, Symposia, 2010, 1, 141–158 Search PubMed.
- J. G. Croissant, Y. Fatieiev and N. M. Khashab, Adv. Mater., 2017, 29, 1604634 CrossRef PubMed.
- K. R. Martin, J. Nutr., Health Aging, 2007, 11, 94 CAS.
- L. L. Sun, Y. J. Gao, Y. G. Wang, Q. Wei, J. Y. Shi, N. Chen, D. Li and C. H. Fan, Chem. Sci., 2018, 9, 5967–5975 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S3. See DOI: https://doi.org/10.1039/d2cc05678j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |