
Combining tetraphenylethene (TPE) derivative cations with Eu3+-β-diketone complex anions for tunable luminescence†
Jicao
Han
,
Zhengyu
Zhang
,
Dongdong
Liu
 and
Xi
Wang
*
and
Xi
Wang
*
Marine College, Shandong University, Weihai, Weihai 264209, P. R. China. E-mail: xi_wang@sdu.edu.cn
First published on 28th November 2022
Abstract
Tetraphenylethene (TPE) derivative cations (TPE+) and Eu3+-β-diketone complex anions (Eu(ABM)4−) were combined to construct a novel dual energy transfer system (TPE+ to Eu3+ and ABM to Eu3+). Our system exhibits tunable luminescence in DMF/water mixtures under different fw conditions owing to the AIE and ACQ properties of TPE+ and ABM, respectively. Its luminescence can be also regulated by adding P-containing oxysalts or polyacrylic acids.
Propeller-like tetraphenylethene (TPE) derivatives, which show unique aggregation induced emission (AIE) behavior, are undergoing rapid development due to their widespread applications in light emission, bio-imaging and chemo/bio-sensing.1–5 Most typically, the construction of an electron-donating/accepting system is an effective approach to change the fluorescence that is attributed to the twisted conformation of TPE derivatives. As a result, the TPE backbones are usually decorated with different electron withdrawing substituent groups.6–9 However, these molecules require tedious synthesis methods and their fluorescence can barely be regulated conveniently, because even a small change in substituent group means that the synthesis route needs to be redesigned. Inspired by molecular assemblies via non-covalent interaction, the electron donating and accepting parts have been separated into cationic TPE derivatives and counteranions, respectively, resulting in the establishment of a novel flexible TPE structure with isolated donor–acceptor (D–A) motifs. There is no doubt that the counteranions have a very significant effect on the cationic TPE molecular packing upon aggregation as well as on the fluorescence, where the counteranions normally induce a change in the solubility.10 Thus, according to previous research, there is no need to take into account the luminescence of the anions. Moreover, the luminescence of these ionic compounds has normally been limited to the emission that originates from the TPE motif. However, when exploring excellent fine-tuning luminescent systems, the luminescence of the counteranions should not be neglected. Therefore, looking for suitable counteranions to interact with the TPE fluorophore is exciting yet challenging, wherein the energy transfer between the TPE cations and counteranions is an essential prerequisite. To the best of our knowledge, very few studies have focused on such cationic TPE/anion materials that exhibit combined electronic, structural and luminescent properties. Encouragingly, the unusual negatively charged complex that is formed by lanthanide and β-diketone is selected as the counteranion in this work.11 4,4,4-Trifluoro-1-phenyl-1,3-butanedione (ABM) is a typical type of β-diketone with aggregation-caused quenching (ACQ) characteristics, which can transfer energy to Ln3+.12–14 In addition, the phenyl ring of the TPE-motif is locked through an aggregated state via electrostatic interaction with the negatively charged Ln3+–β-diketone complex, so the AIE-active TPE cation can also transfer energy to Ln3+.15,16 Thus, this dual-ligand–Ln3+ energy transfer system can exhibit a stepwise pathway to sensitize the luminescence of Ln3+, which provides a novel approach for designing tunable fluorescent molecules. More strikingly, the luminescence regulation that is related to the dual energy transfer utilizing AIE/ACQ properties in distinct mixed solvents was investigated. To gain insight into the dual energy transfer process, a controllable approach for fine-tuning the luminescence was also studied.
The parallelogram-like TPE-based onium salt 1-(4-(1,2,2-triphenylvinyl)benzyl)pyridin-1-ium bromide (denoted as TPE+-Br−) was synthesized first, which was reacted by adding 1-(bromomethyl)-4-(1,2,2-triphenylethenyl)benzene to pyridine in toluene at 45 °C (Fig. 1 and Fig. S1–S3, ESI†).17–19 When the TPE+-Br− salt was dissolved in a mixture of THF/n-hexane, it showed typical aggregation-induced emission enhancement (AIEE) activity before the volume fraction of n-hexane (fhex) reached 70% (Fig. S4, ESI†). It was almost non-emissive when fhex increased from 0% to 30%, then strong aggregated-induced emission at 435 nm was observed when fhex was increased from 40 to 70% with a maximum 362 times enhancement (fhex = 70%). However, it exhibited a drastic decline in emission intensity at a higher n-hexane fraction (>70%), revealing the formation of amorphous molecular aggregates. To combine the typical AIE fluorophore with a lanthanide-based complex, the bromine ion of the TPE derivative was removed using a negatively charged lanthanide complex through adding the lanthanide salt and ABM ligand (mole ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) with stirring at room temperature. As a result, the bar-shaped TPE+–EuL4− compound was formed (Fig. 1 and Fig. S5, ESI†). XPS results suggest that the material consists of C, N, O, F and Eu (Fig. S6, ESI†). The results of XRD patterns reveal that the as-synthesized sample contains both TPE+ and EuL4− parts (Fig. S7, ESI†). The vibrational frequency of v(C
:4) with stirring at room temperature. As a result, the bar-shaped TPE+–EuL4− compound was formed (Fig. 1 and Fig. S5, ESI†). XPS results suggest that the material consists of C, N, O, F and Eu (Fig. S6, ESI†). The results of XRD patterns reveal that the as-synthesized sample contains both TPE+ and EuL4− parts (Fig. S7, ESI†). The vibrational frequency of v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and v(CF3) shifts towards lower frequency compared with ABM in the IR spectrum, indicating that ABM has coordinated with the Eu3+ ions (Fig. S8 and Table S1, ESI†). In addition, the contents of C, H, O, N and Eu are consistent with the calculated results, which demonstrates the pure phase (Table S2, ESI†). The typical AIE emission of TPE+–EuL4− (about 467 nm) was observed, but TPE+-Br− was almost non-emissive when dissolved in the DMF/H2O (fw = 90%), demonstrating restriction of the intramolecular rotation of the TPE moiety owing to the electrostatic interaction between the TPE-motif and the anionic lanthanide complex (Fig. S9, ESI†). Most excitingly, besides the TPE-motif fluorescence, TPE+–EuL4− displayed clear sharp peaks from 550 nm to 650 nm, which belong to the 5D0 → 7F1 and 5D0 → 7F2 transitions of typical Eu3+ luminescence,20–23 indicating the energy transfer process from the dual ligands to Eu3+. To elucidate the mechanism (Fig. S10–S12, ESI†), the Gd-based compound was synthesized, in which Gd3+ ions could not accept any energy from the excited state of most organic ligands due to the higher 6P7/2 energy level (Gd3+: 32500 cm−1).24,25 The UV-vis absorption and phosphorescence of the Gd-based compound TPE+–GdL′4− (L′ = citric acid) without ABM were detected to determine the singlet state (S1) (32680 cm−1) and the triplet energy level (T1) (20202 cm−1) of the TPE ligand, respectively. Similarly, S1 (30864 cm−1) and T1 (21413 cm−1) for ABM were calculated according to the UV-vis absorption and phosphorescence of (C2H5)3HN+–GdL4− (L = ABM) in the absence of TPE+. The energy gap ΔE1 between S1 and T1 is 12478 cm−1 and 9451 cm−1 for TPE-based ligand and ABM, respectively, implying an effective intersystem crossing process. According to the antenna effect,26 ΔE2 between T1 of the ligand and the excited state of the Ln3+ ions must be higher than 3500 cm−1 to achieve the irreversible energy transfer. In this system, ΔE2 is 2916 cm−1 (TPE-motif to Eu3+) and 4127 cm−1 (ABM to Eu3+). These results indicate that both ligands can contribute to transferring energy to Ln3+, but ABM can sensitize the luminescence more effectively.
O) and v(CF3) shifts towards lower frequency compared with ABM in the IR spectrum, indicating that ABM has coordinated with the Eu3+ ions (Fig. S8 and Table S1, ESI†). In addition, the contents of C, H, O, N and Eu are consistent with the calculated results, which demonstrates the pure phase (Table S2, ESI†). The typical AIE emission of TPE+–EuL4− (about 467 nm) was observed, but TPE+-Br− was almost non-emissive when dissolved in the DMF/H2O (fw = 90%), demonstrating restriction of the intramolecular rotation of the TPE moiety owing to the electrostatic interaction between the TPE-motif and the anionic lanthanide complex (Fig. S9, ESI†). Most excitingly, besides the TPE-motif fluorescence, TPE+–EuL4− displayed clear sharp peaks from 550 nm to 650 nm, which belong to the 5D0 → 7F1 and 5D0 → 7F2 transitions of typical Eu3+ luminescence,20–23 indicating the energy transfer process from the dual ligands to Eu3+. To elucidate the mechanism (Fig. S10–S12, ESI†), the Gd-based compound was synthesized, in which Gd3+ ions could not accept any energy from the excited state of most organic ligands due to the higher 6P7/2 energy level (Gd3+: 32500 cm−1).24,25 The UV-vis absorption and phosphorescence of the Gd-based compound TPE+–GdL′4− (L′ = citric acid) without ABM were detected to determine the singlet state (S1) (32680 cm−1) and the triplet energy level (T1) (20202 cm−1) of the TPE ligand, respectively. Similarly, S1 (30864 cm−1) and T1 (21413 cm−1) for ABM were calculated according to the UV-vis absorption and phosphorescence of (C2H5)3HN+–GdL4− (L = ABM) in the absence of TPE+. The energy gap ΔE1 between S1 and T1 is 12478 cm−1 and 9451 cm−1 for TPE-based ligand and ABM, respectively, implying an effective intersystem crossing process. According to the antenna effect,26 ΔE2 between T1 of the ligand and the excited state of the Ln3+ ions must be higher than 3500 cm−1 to achieve the irreversible energy transfer. In this system, ΔE2 is 2916 cm−1 (TPE-motif to Eu3+) and 4127 cm−1 (ABM to Eu3+). These results indicate that both ligands can contribute to transferring energy to Ln3+, but ABM can sensitize the luminescence more effectively.
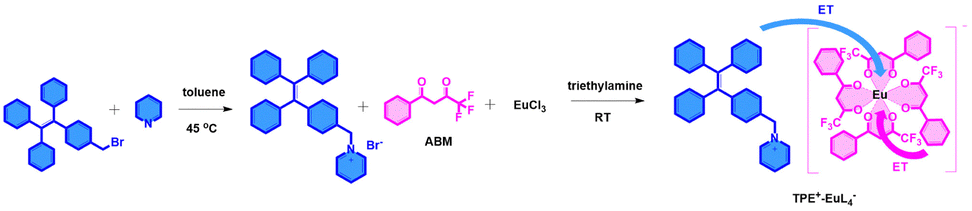 | ||
| Fig. 1 Synthetic strategy for TPE+–EuL4− materials. | ||
As ABM has been demonstrated to be one of the best sensitizers for Eu3+ emission in dilute DMF solutions and water is a poor solute for the organic ligand,13 the DMF/H2O system was selected and the fluorescence was investigated under different fw conditions (Fig. 2). Notably, the TPE-motif was almost non-emissive when fw increased from 0 to 60%, after which the fluorescence intensity at about 467 nm that is associated with the AIE properties enhances steadily when fw exceeded 70%. Meanwhile, the characteristic emission intensity of Eu3+ (5D0 → 7F2: 614 nm) declined gradually at first, reaching a minimum at 60%, and finally increased sharply when fw raised from 70 to 90%. When fw was below 60%, since the rotation of the TPE-based ligand was not totally restricted, the energy absorbed from the UV region was consumed by rotation of the benzene rings and an ineffective energy transfer process (TPE ligand to Eu3+) existed. Meanwhile, the energy transfer process from the ABM ligand to Eu3+ dominated,27,28 but the ACQ effect of ABM decreased its performance as a sensitizer, which was confirmed by the decline in luminescence intensity associated with Eu3+ when fw was raised from 0 to 60%. Interestingly, when fw exceeded 60%, the emission performance of Eu3+ was activated, in which the intensity of 614 nm soared to 888.20 (fw = 90%); this was because the restriction of the intramolecular motion of the TPE-motif blocked the non-radiative relaxation channels, and this restricted ligand began to transfer energy to Eu3+. However, ABM exhibited the ACQ effect under such a high-water ratio, so the TPE ligand acted as the main sensitizer for Eu3+. When fw reached 90%, the fluorescence lifetime detected at 467 nm associated with the TPE motif in TPE+–EuL4− (4.56 μs) declined compared with TPE+–GdL′4− (L′ = citric acid) (6.01 μs) (Fig. S13, ESI†), also implying that the TPE motif transferred energy to Eu3+. The energy-transfer efficiency of the TPE motif to Eu3+ could be calculated via E = 1 − τ/τ0, where τ0 and τ are the lifetime values detected at 467 nm in the absence and presence of the Eu3+ containing complex, respectively. The energy-transfer efficiency was lower (24.13%), which agrees with the lower ΔE2 of 2916 cm−1 (from the T1 of TPE+ to Eu3+). The lifetime detected at 614 nm (the 5D0 → 7F2 transition of Eu3+) for TPE+–EuL4− (503.5 μs) was longer compared with (C2H5)3HN+–EuL4− (249.8 μs) (Fig. S14, ESI†). Moreover, as shown in Fig. S15 (ESI†), the broad band located at 300–380 nm (attributed to organic ligands) was observed, indicating energy migration from the ligands to Eu3+. Notably, the intensity of the excitation peak that ranged from 300 to 350 nm was raised significantly (fw > 60%), which demonstrates that the TPE motif behaved as the main donor when the restriction of intramolecular rotation was enhanced (Fig. S15 and 16, ESI†). The quantum efficiency (QE) of TPE+–EuL4− detected in the DMF/water mixture (fw = 90%) was 10.95%, but the value of this solid climbed to 26.23%, implying that the restriction of intramolecular rotation in the TPE moiety can improve the QE of the material. In essence, the arrangement of the compound was the switch that controls the dual energy transfer process and luminescence when changing the ratio of solvents.
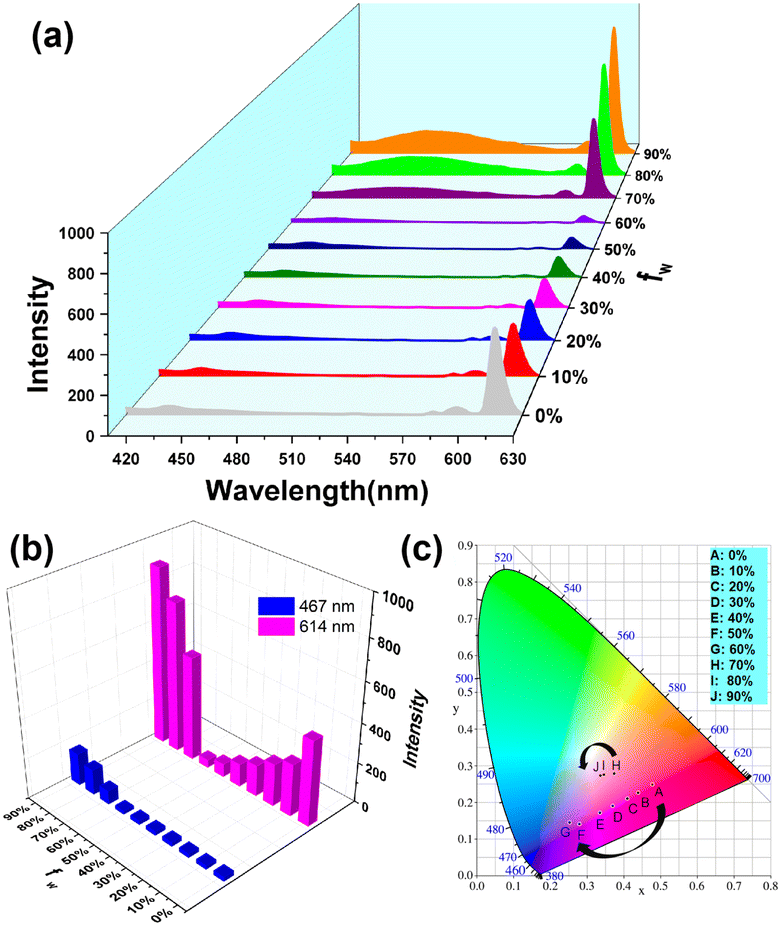 | ||
| Fig. 2 Emission spectra of TPE+–EuL4− materials dissolved in DMF/water mixtures with different water fractions (λex = 385 nm) (a); variation of I467nm and I614nm with different fw (b); and 1931 CIE chromaticity diagram (c). | ||
Adenosine 5'-triphosphate disodium salt (ATP disodium salt) is a central component of energy storage as well as an important endogenous signaling molecule in immunity and inflammation, so its detection strategy has been widely investigated.29 As shown in Fig. 3, the luminescence was quenched with an increasing concentration of ATP2−. Due to the characteristic three adjacent phosphate groups and the heterocyclic ring that contains both N and O coordinated atoms, ATP2− has a very strong coordinating ability with Eu3+, in which the adjacent phosphate groups can link to Eu3+ through several coordination modes. Since the excellent coordination ability of ATP2− helps to elevate the competitive capacity, ABM was substituted by ATP2− and dissolved in solution, which would further indicate that the new coordination interaction between Eu3+ and ATP2− existed. It was confirmed by the UV absorption spectrum of TPE+–EuL4− (1.0 × 10−4 mol L−1) in DMF/H2O (fw = 50%) upon addition of ATP2− (Fig. S17, ESI†). The intensity of the peak located at about 320 nm dropped, whereas the intensity of the relatively narrow peak (about 256 nm) was elevated. According to the UV absorption spectra of (C2H5)3HN+–EuL4− and TPE+–GdL′4− mentioned above (Fig. S10, ESI†), it can be deduced that both ABM and TPE+ contribute to the absorption peak at about 323 nm in TPE+–EuL4−. Notably, there is keto–enol tautomeric equilibrium in ABM, in which the absorption located at 225–265 nm denotes the keto-form, and the peak red-shifted to over 300 nm is the enol-form.30 In this system, the amount of TPE+ was not removed from the system, so the decline of the intensity was mainly induced by the decreasing enol-form of ABM. Although the intensity associated with the keto-form increased sharply, the absorption of ATP2− was also located around 256 nm (Fig. S18, ESI†). As a result, the absorption at >300 nm was mainly considered to measure ABM with the enol-form. The decline of the absorption at about 323 nm indicates that ABM (enol-form) dissociated from the surface of Eu3+ and then turned to the keto-form. Meanwhile, because the structure of Eu(ABM)4− was destroyed, TPE+ was no longer immobilized and it was attracted by the negative ATP2− instead (Fig. 3). The compound associated with ATP2− and TPE+ had a certain solubility in water owing to the hydrophilic groups of ATP2−, and TPE+ no longer displayed typical AIE behavior, which was confirmed by the non-emissive properties of the mixture of ATP2− and TPE+ (Fig. S19, ESI†). Moreover, when P2O74− and H2PO4− with decreasing numbers of coordinated sites were added, the luminescence of the peaks located at 467 nm and 614 nm also decreased, but were not quenched completely. However, H2PO2− and HPO32− cannot induce luminescence quenching due to their weak coordination ability (Fig. 4a and Fig. S20–S24, ESI†).
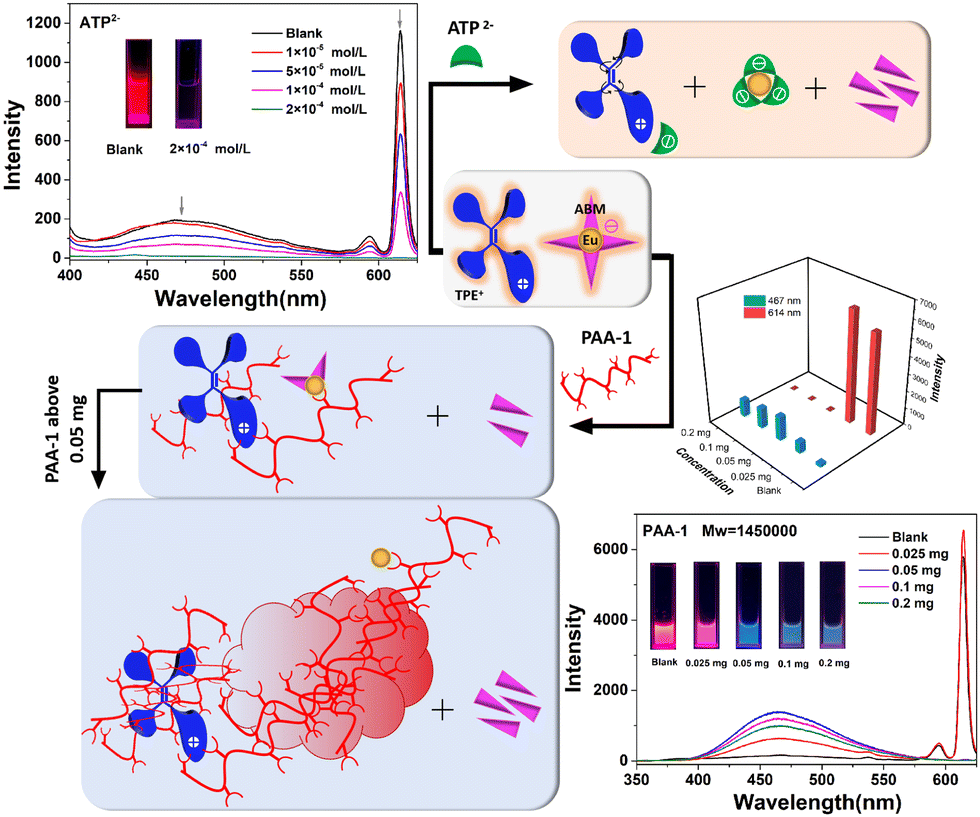 | ||
| Fig. 3 Emission spectra of TPE+–EuL4− (1.0 × 10−4 mol L−1) in DMF/H2O (fw = 90%) upon addition of increasing concentrations of ATP2− (λex = 385 nm) and PAA-1 (Mw = 1450000) (λex = 320 nm), respectively; emission intensity of the peaks (467 nm and 614 nm) varies with amount of PAA-1; possible mechanism of the different luminescence responses by adding ATP2− and PAA-1, respectively. | ||
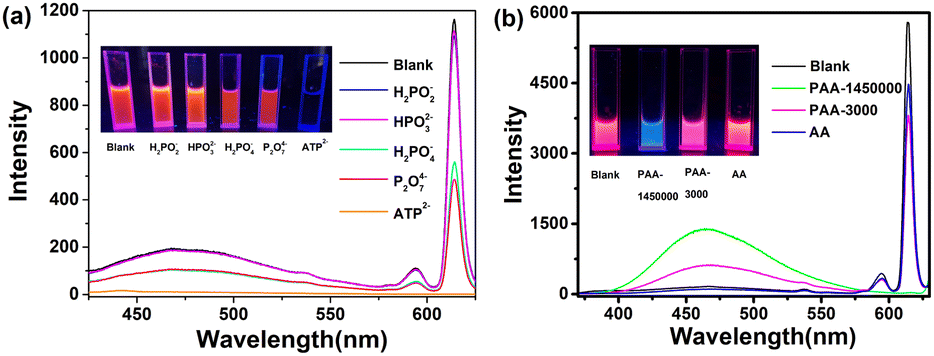 | ||
| Fig. 4 Emission spectra of TPE+–EuL4− (1.0 × 10−4 mol L−1) in DMF/H2O (fw = 90%) in the presence of aqueous solutions of different various salts (2.0 × 10−4 mol L−1) (λex = 385 nm) (a); emission spectra of TPE+–EuL4− (1.0 × 10−4 mol L−1) in DMF/H2O (fw = 90%) in the presence of PAA-1 (Mw = 1450000), PAA-2 (Mw = 3000), and AA (0.2 mg) (λex = 320 nm) (b). Insets in (a) and (b) show digital photos under 365 nm UV light. | ||
Based on the results above, the related negative phosphates do not have the ability to fix TPE+. To immobilize the TPE-motif, polyacrylic acid (PAA: Mw = 1450000, denoted as PAA-1) with long flexible carbon chains and multiple –COOH groups was introduced in the system (Fig. 3). The UV absorption peak located at 256 nm was only assigned to the keto-form of ABM, due to there being no PAA-1 absorption peak in this range (200–400 nm) (Fig. S18, ESI†). Similar to the condition with the addition of ATP2−, the dissociated ABM with the enol-form detached from the Eu–ABM complex and transformed to the keto-form, which was confirmed by the downward trend of I323nm/I256nm (Fig. S25, ESI†). However, a small amount of PAA-1 could only induce some ABM molecules to leave the complex, because the value of I323nm/I256nm only changed slightly when 0.025 mg of PAA-1 was added. As a result, PAA-1 behaved as a bridge to connect the donor (TPE+ and ABM) and acceptor (Eu3+). TPE+ was surrounded by polymer coils and the rotation was further restricted, leading to the promoted energy transfer process from TPE+ to Eu3+. This was confirmed by the tiny increase in the intensity of the peaks located at about 467 nm that belong to the TPE-motif and at 614 nm, which is associated with Eu3+ (Fig. 3). The insignificant extent of increase is consistent with the fact that TPE cannot sensitize the luminescence effectively as mentioned above. When more PAA-1 was added, nearly all the ABM dissolved, and the ACQ effects of ABM in the aqueous system prevented any further occurrence of energy transfer from ABM to Eu3+, even if the dissociative ABM existed. The intermolecular rotation of TPE+ was restricted owing to the twined carbon chains of PAA-1. However, when the carbon chains were intertwined with each other to a greater extent, the increased steric hindrance resulted in blocked energy transfer (TPE+ to Eu3+). Therefore, although the peaks assigned to the TPE-motif increased obviously, the luminescence intensity associated with Eu3+ fell rapidly and was quenched completely. When the amount of PAA-1 reached above 0.05 mg, both TPE+ and Eu3+ were wrapped around a large amount of PAA-1 with multiple –COOH groups, leading to increasing hydrophilicity and a decreased intensity of the peaks (467 nm). When 0.1 mg of PAA-1 was added, nearly all the PAA-1 substituted the ABM, because the value of I323nm/I256nm remained basically unchanged when more PAA-1 was added (0.2 mg) (Fig. S25, ESI†). To verify the fact that steric hindrance was unfavorable for energy transfer from TPE+ to Eu3+, the same weight (0.05 mg) of PAA (Mw = 3000, denoted as PAA-2), PAA-1 and acrylic acid (AA) were added, respectively (Fig. 4b and Fig. S26, S27, ESI†). Because they have the same amount of –COOH groups, the change in energy transfer caused by the disassociation of ABM in the acidic system was not considered. For the intensity associated with TPE+, the sequence I467nm-PAA-1 > I467nm-PAA-2 > I467nm-AA indicated that the ability to restrict the rotation of TPE+ was weakened from AA to PAA-1, which further demonstrated that the steric hindrance caused by the wrapped acids decreased from PAA-1, PAA-2 to AA. The intensity sequence located at about 614 nm (I614nm-PAA-1 < I614nm-PAA-2 < I614nm-AA) also verified that the stronger steric hindrance prohibited energy transfer from TPE+ to Eu3+ more seriously.
In summary, a novel lanthanide-based ionic compound was synthesized by combining TPE+ derivatives with AIE properties and the EuL4− (L = ABM) complex built using a ligand with ACQ behavior. The compound exhibited a stepwise energy transfer pathway from dual AIE/ACQ ligands to lanthanide in the DMF/H2O system. The addition of ATP2− and PAA had a significant effect on the dual energy transfer process and the tunable luminescence, respectively. This discovery provides new insight into the tunable fluorescence of the compounds combined with AIE ligands, ACQ ligands and lanthanide ions.
This work was financially supported by the National Natural Science Foundation of China (21601107) and the Shandong Provincial Natural Science Foundation, China (ZR2021MB052).
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. Erdemir, G. Suna, S. Gunduz, M. Şahin, S. Eğlence-Bakır and E. Karakuş, Anal. Chim. Acta, 2022, 1218, 340029 CrossRef CAS PubMed.
- L. Liu, Q. Wan, C. Gui, P. He, Z. Zhao, Z. Wang and B. Z. Tang, Chem. Commun., 2022, 58, 5769–5772 RSC.
- L. Zhang, Y. Li, G. Mu, L. Yang, C. Ren, Z. Wang, Q. Guo, J. Liu and C. Yang, Anal. Chem., 2022, 94, 2236–2243 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- Z. Li, L. Xu, H. Yuan and P. Zhang, Analyst, 2022, 147, 2930–2935 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- C. Liu, Y. Hang, T. Jiang, J. Yang, X. Zhang and J. Hua, Talanta, 2018, 178, 847–853 CrossRef CAS.
- Y. Liu, C. M. Deng, L. Tang, A. J. Qin, R. R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660–663 CrossRef CAS.
- V. G. Naik, S. D. Hiremath, A. Thakuri, V. Hemmadi, M. Biswas, M. Banerjee and A. Chatterjee, Analyst, 2022, 147, 2997–3006 RSC.
- G. Zhang, H. Zhu, M. Chen, M. Pietraszkiewicz, O. Pietraszkiewicz, H. Li and J. Hao, Chem. – Eur. J., 2018, 24, 15912–15920 CrossRef CAS PubMed.
- L. R. Melby, E. Abramson, J. C. Caris and N. J. Rose, J. Am. Chem. Soc., 1964, 86, 5117–5125 CrossRef CAS.
- Y. Ma and Y. Wang, Coord. Chem. Rev., 2010, 254, 972–990 CrossRef CAS.
- G. G. Condorelli, G. Malandrino and I. L. Fragala, Coord. Chem. Rev., 2007, 251, 1931–1950 CrossRef CAS.
- X. Zhang, Z. Zhang, Y. Liu, S. Shi, Y. Zhang, Y. Cao, L. Li, C. Geng, Y. Xia, J. Zhu and S. Xu, J. Phys. Chem. Lett., 2021, 12, 11710–11716 CrossRef CAS.
- Y. Y. Li, Y. Y. Zhou, Y. Yao, T. Gao, P. F. Yan and H. F. Li, New J. Chem., 2021, 45, 7196–7203 RSC.
- P. R. Su, L. J. Liang, T. Wang, P. P. Zhou, J. Cao, W. S. Liu and Y. Tang, Chem. Eng. J., 2021, 413, 127408 CrossRef CAS.
- X. Li, S. Wu, S. Chen, Z. Lai, H. B. Luo and C. Sheng, Org. Lett., 2018, 20, 1712–1715 CrossRef CAS PubMed.
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed.
- A. Zhang, J. Zhang, Q. Pan, S. Wang, H. Jia and B. Xu, J. Lumin., 2012, 132, 965–971 CrossRef CAS.
- A. Wada, M. Watanabe, Y. Yamanoi and H. Nishihara, Chem. Commun., 2008, 1671–1673 RSC.
- N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201–228 CrossRef CAS.
- B. R. Judd, Phys. Rev., 1962, 127, 750–761 CrossRef CAS.
- G. J. Grant, W. N. Chen, A. M. Goforth, C. L. Baucom, K. Patel, P. Repovic, D. G. VanDerveer and W. T. Pennington, Eur. J. Inorg. Chem., 2005, 479–485 CrossRef CAS.
- X. S. Ke, Y. Y. Ning, J. Tang, J. Y. Hu, H. Y. Yin, G. X. Wang, Z. S. Yang, J. L. Jie, K. H. Liu, Z. S. Meng, Z. Y. Zhang, H. M. Su, C. Y. Shu and J. L. Zhang, Chem. – Eur. J., 2016, 22, 9676–9686 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- H. Q. Yin, X. Y. Wang and X. B. Yin, J. Am. Chem. Soc., 2019, 141, 15166–15173 CrossRef CAS PubMed.
- S. S. Skourtis, C. R. Liu, P. Antoniou, A. M. Virshup and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8115–8120 CrossRef CAS PubMed.
- S. Speiser, Chem. Rev., 1996, 96, 1953–1976 CrossRef CAS PubMed.
- J. Deng and A. Walther, Adv. Mater., 2020, 32, 2002629 CrossRef CAS PubMed.
- J. C. Sloop, C. L. Bumgardner, G. Washington, W. D. Loehle, S. S. Sankar and A. B. Lewis, J. Fluor. Chem., 2006, 127, 780–786 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The detailed experimental procedure and related spectroscopic data. See DOI: https://doi.org/10.1039/d2cc03903f |
| This journal is © The Royal Society of Chemistry 2023 |