
Understanding cyclic(alkyl)(amino)carbene–copper complex catalysed N–H and O–H bond addition to electron deficient olefins†
Akshi
Tyagi
,
Sunita
Mondal‡
,
Anmol‡
 ,
Vikas
Tiwari‡
,
Tarak
Karmakar
* and
Subrata
Kundu
*
,
Vikas
Tiwari‡
,
Tarak
Karmakar
* and
Subrata
Kundu
*
Department of Chemistry, Indian Institute of Technology Delhi, Delhi 110016, India. E-mail: skundu@iitd.ac.in; tkarmakar@iitd.ac.in; Tel: +91-11 2654 8443
First published on 24th November 2022
Abstract
The hydroamination of electron-deficient olefins was carried out using the (CAAC)Cu–Cl (CAAC = cyclic (alkyl)(amino)carbene) catalyst with an excellent yield at room temperature and under an open atmosphere. Furthermore, the catalyst shows excellent efficiency in the hydroaryloxylation and hydroalkoxylation of alkenes under mild conditions. The efficiency of the catalyst was tested for a wide range of substrates with different electronic and steric functionalities. Detailed computational studies have been carried out to understand the mechanism of these Cu(I) catalyzed reactions, which revealed that the reaction proceeds via either a four-membered or a six-membered cyclic transition state containing the copper ion.
Since their discovery in 2005, cyclic (alkyl)(amino) carbenes (CAACs) have received much attention and are regarded as excellent donor ligands due to their stronger nucleophilicity and electrophilicity compared with NHC.1,2 Consequently, several powerful catalysts have been produced using CAACs.3,4 For instance, compared with their NHC analogues, e.g., Ru–H2IMes complexes, CAAC–Ru complexes exhibit an extraordinary performance in metathesis macrocyclization and ethenolysis reactions.3 In general, it has been observed that the reactions involving CAAC–metal complexes as a catalyst have a better efficiency than the corresponding NHC analogues.4c,5 These observations show the multiple perspectives of CAAC–metal complexes for catalytic applications; however, they have been much less explored than NHC–metal complexes. Inspired by the progressive development in catalytic reactions using CAAC–metal complexes,6 in this work we investigate the potential application of CAAC–CuCl complex towards hydroamination and hydroalkoxylation reactions.
Hydroamination reactions are among frequently performed C–N bond-forming reactions that are useful in the synthesis of various pharmaceuticals and biologically active compounds.7 The hydroamination of alkenes is more challenging compared with alkynes because of their lower reactivity.8 Related to this, the catalytic anti-Markovnikov hydroamination of olefins was named one of the “Ten Challenges for Catalysis”.9 During the past two decades, several metal-catalyzed hydroamination reactions have been performed using, but not limited, to transition metals, alkali metals, alkaline earth metals, and lanthanides.7,10–12 Nonetheless, many of these catalysts are air- and moisture-sensitive, comparatively expensive, and have limited substrate scope. Despite the major advances in recent years, reports on the activation of amines via Cu-complexes with electron-donating ligands are rare. Earlier, Gunnoe and co-workers employed the well-defined copper amido complex coordinated to an electron-rich NHC ligand to catalyze the conjugate addition of aniline to activated olefins, where β-amino carbonyls were obtained as the desired products under mild conditions.12a However, this methodology requires pre-functionalization of the catalyst before the reaction. Glovebox techniques are necessary for all synthesis and catalytic processes as the catalyst is extremely air- and moisture-sensitive. Recently Lee and co-workers reported Cu salt as a catalyst with NHC or phosphine ligands for the hydroamination of an alkene with heterocyclic amines.12d It is worth mentioning that CAAC–Au(I) and CAAC–Hg(II) complexes have previously been used for hydroamination reactions of allenes and alkynes.11
In this work, we applied an easily accessible, air- and moisture-stable catalyst, (CAAC)Cu–Cl (1),13 for Nu–H (N = N, O) bond addition reactions to electron deficient olefins. When we compared our results with the reported NHC-based Cu-catalyst, we found compound 1 to be a competitive catalyst, especially regarding alkene substrates bearing the ester group. Another goal of this work was to gain a clear understanding of the catalytic mechanism of the hydroamination reaction. As far as we know, there are no reports on the detailed mechanistic study of this reaction in the presence of a carbene-stabilized copper catalyst. This lack of mechanistic details motivated us to explore numerous possibilities and uncover the most probable pathway(s) of this reaction using ab initio DFT calculations. So far, in the literature, two catalytic routes have been conjectured: one in which there is the formation of a zwitterion intermediate, where product formation occurs without the direct involvement of the metal ion; the other reaction route involves a conventional organometallic pathway and goes via metal olefin complex formation.12b So far, none of these routes has been confirmed either by experiments or computational studies. Our study reveals that the reaction proceeds via either a four-membered or a six-membered transition state involving the copper ion. The other explored reaction pathways have high energy barriers and, thus, they are less likely to be followed during the catalytic reaction.
The catalytic utility of 1 was evaluated for N–H and O–H bond addition to alkenes. Initially, aniline and methyl acrylate substrates were chosen for the model reaction. Detailed optimization of the hydroamination reaction revealed that toluene is the optimal solvent, and tBuOK serves an effective base (Table S1, see ESI†). The substrate scope for the hydroamination reaction was explored using the optimized reaction conditions (Scheme 1). A variety of alkenes having ester, nitrile, and sulphone as the electron withdrawing group (EWG) were explored, along with a variety of amines. The reaction of methyl acrylate and aniline as well as its p-fluoro, p-chloro, and p-methoxy derivatives afforded the desired products in 98, 94, 92, and 96% yield, respectively (Scheme 1). It is worth mentioning that an improved yield (in a few cases up to 43% increased yield) was observed using catalyst 1 compared with the literature known NHCCu(I) catalyst.12a Subsequently, a secondary amine, N-methyl benzylamine, was also examined as a substrate with methyl acrylate, which resulted in 98% conversion to the hydroaminated product (4e, Scheme 1). With ethyl acrylate, both electron rich and electron deficient aryl amines gave excellent conversion (95–98%) (Scheme 1). Again, a variety of anilines was examined with vinyl-phenyl sulfone. As the data in Scheme 1 indicate, β-amino sulfones were produced efficiently from the corresponding amines in excellent yields (92–97%). Along with the desired mono-alkylated product, a small amount (up to 5%) of the double-alkylated product was also formed. With cyclohexylamine, the selectively monoalkylated product was obtained in excellent yield (4p, 98%). Furthermore, the applicability of catalyst 1 was examined for the hydroamination of acrylonitrile with aniline and its derivatives with an excellent conversion (85–98%) to the products. Internal alkene (ethyl crotonate), electron rich alkenes, and vinyl-ethyl ether were also evaluated for the hydroamination reaction; however, no desired product was formed. The results depicted in Scheme 1 indicate that catalyst 1 functions well in the presence of electron-withdrawing as well as electron-donating groups on the aromatic ring.
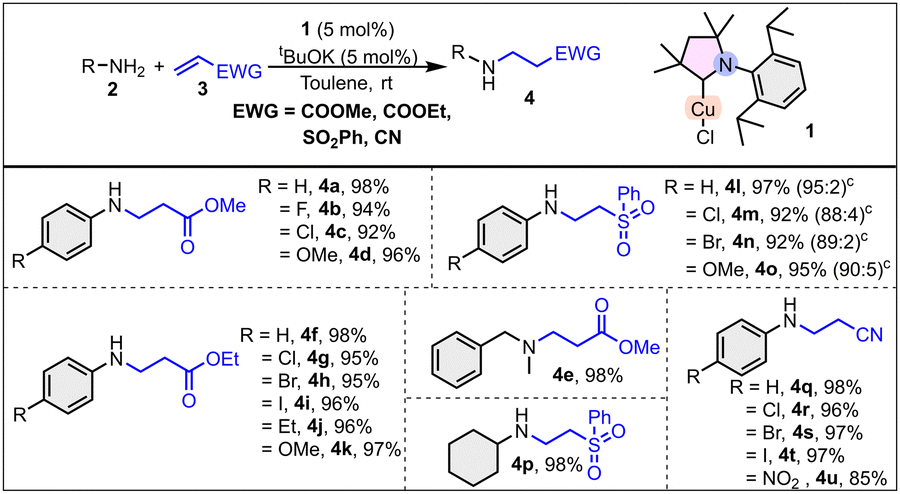 | ||
| Scheme 1 Scope of hydroamination reaction. Reaction conditions: aniline/alkene/base/catalyst = 1.1/1/0.05/0.05 mmol), toluene (2 mL) at RT, for 6 h. Yields were determined via GC-MS using mesitylene as an internal standard. cSmall amount of double alkylated product. | ||
Compared with the addition of amine N–H bonds to alkenes, the addition of O–H bonds of alcohols to alkenes is more challenging.14 We explored the possibility of using catalyst 1 for the hydroalkoxylation and hydroaryloxylation of electron deficient alkenes (Scheme 2). Catalyst 1 showed excellent efficiency towards O–H bond addition reactions. A product formation of up to 95% was observed when phenyl-vinyl sulphone was used as the substrate with MeOH and EtOH. An alcohol containing an electron withdrawing group, such as the trifluoroethyl moiety, gave a lower yield (52%, 6e) of product. However, phenol and phenyl-vinyl sulphone/acrylonitrile gave only 30% conversion to product 6c/6d at RT, which increased to 75% upon increasing the reaction temperature to 70 °C (Scheme 2). The low catalytic turnover and its increase upon the increase in the reaction temperature indicates a high energy barrier involved in the reaction. Moreover, we will see in a later section that this is indeed the case. Before delving into that specific case, first, let us understand the reaction mechanism.
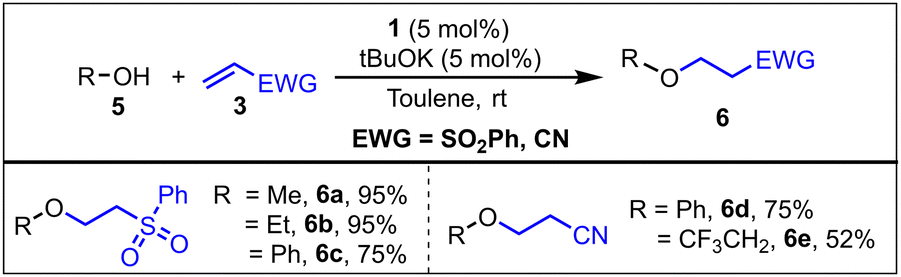 | ||
| Scheme 2 Scope of O–H bond addition. Reaction conditions: alcohol/alkene/base/catalyst = 1.1/1/0.05/0.05 mmol), toluene (2 mL) at RT. Yields were determined via GC-MS using mesitylene as an internal standard. Reactions with trifluoroethanol and phenol were performed at 60 and 70 °C, respectively, and a time of 12 h. | ||
We anticipated that the CAAC ligand in catalyst 1 plays two major roles: (i) facile electron donation from the ligand CAAC to the metal, which helps in the stabilization of Cu(I); and (ii) due to its bulky nature, the catalyst exerts steric selectivity. These intuitions encouraged us to explore the reaction mechanism using an exhaustive number of DFT calculations. As discussed earlier, the reaction is ineffective in the absence of a base, so, at first, the amine is activated by the abstraction of a proton by tBuOK base. This activated amine then attacks the copper (Cu) centre of the catalyst to form Int_1via the removal of a chloride ion. In the subsequent step, Int_1 acts as the active catalyst for this reaction. After analysing some initial benchmark calculations, we deciphered two major reaction pathways as (i) metal-assisted olefin activation (MAOA) and (ii) proton-assisted olefin activation (PAOA). Among these two pathways, MAOA was found to be the plausible one, which is discussed below, whereas the PAOA pathway is discussed in ESI† Section 6. The very first and most important step of this reaction is the olefin activation. In the MAOA pathway, Cu of the active catalyst (Int_1) assists the olefin activation, which can be achieved via the formation of four-membered TS_1A, or via six-membered TS_1A′ (Fig. 2). In TS_1A, the β-carbon of the olefin interacts with the nitrogen (cat-NH-Ph) atom of the active catalyst, and the Cu ion helps to activate the olefin by interacting with the α-carbon of the olefin. On the other hand, TS_1A′ is formed due to the interaction of the Cu ion with the hetero-atom (O, N) of the EWG of the olefin. In the case of the –CN group, due to its linear geometry, TS_1A′ is strained, which results in an ∼20 kcal mol−1 higher free energy barrier compared with the TS_1A. (Fig. 2) This implies that in the presence of –CN, olefin activation occurs via a favourable four-membered TS. By contrast, olefins containing methyl vinyl ketone (CH2CHCOMe), methyl acrylate (CH2CHCOOMe), and vinyl sulphone (CH2CHSOOPh) have a lower activation energy barrier for the six-membered TS due to their geometrical feasibility. (Fig. 1) As a result of this, the energy of the six-membered TS is similar to that of the four-membered TS. In the case of methyl vinyl ketone, the energy of the six-membered TS is in fact lower than that of the four-membered transition state. Hence, in this case, the reaction must occur via this six-membered TS. However, in the other two cases, i.e., methyl acrylate and vinyl sulphone, both TSs are equally feasible, and hence the reaction can proceed via either of these two transition states.
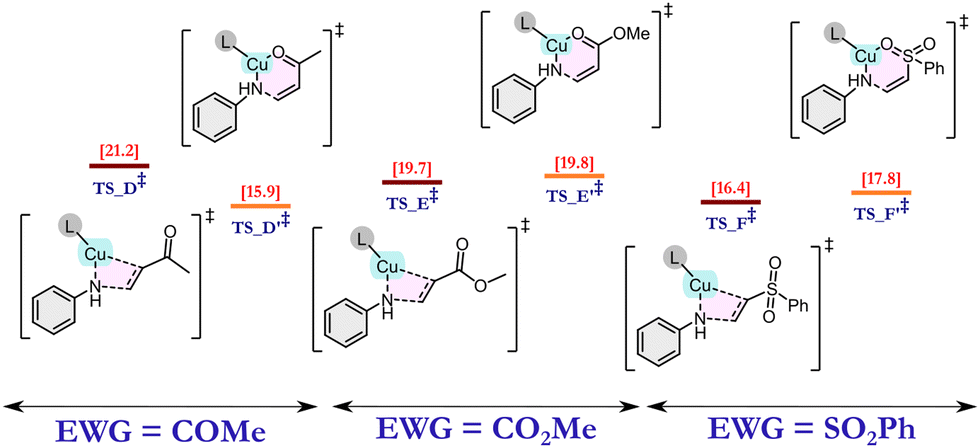 | ||
| Fig. 1 Comparison of Gibbs free energy of activation of 4- and 6-membered transition states formed in the MAOA mechanism for different electron withdrawing groups on the olefin, such as COMe, COOMe and SO2Ph. | ||
Experimentally it was observed that the catalyst is unable to activate the olefin in the absence of an EWG. This observation can be explained on the basis of NBO charge analysis (see ESI† Table S2). As a result of the olefin activation, a negative charge develops on the α-carbon of olefin in TS_1A, which is stabilized by the metal as well as the EWG of the olefin. TS_1A gives the intermediate Int_2A in which the copper is covalently bonded to the α-carbon atom. This intermediate can lead to the product via two different pathways (Fig. 2). The first pathway (rearrangement pathway) involves the rearrangement of the Cu–C bonded intermediate to the Cu–N (nitrogen of –CN group) bonded intermediate (Int_3A) viaTS_2A by crossing an energy barrier of 18.8 kcal mol−1. This nitrogen-bonded intermediate can give the product viaTS_3A, which requires less energy compared with TS_2A. Another pathway (direct pathway) involves the direct conversion of Int_2A to the product viaTS_3A′ but requires a higher energy. Both Int_2A and Int_3A lead to the product formation and regeneration of the active catalyst through the transfer of a proton from an incoming aniline molecule to the activated olefin. TS_3A and TS_3A′ differ in one of the Cu–N interactions. TS_3A has an almost planar configuration of bonds in which the Cu interacts with the aniline molecule's nitrogen and the CN nitrogen attached to the olefin's α-carbon. By contrast, TS_3A′ has an envelope-like arrangement (like the envelope conformation of cyclopentane), and the Cu interacts with the nitrogen attached to the β-carbon instead of the –CN. Since the TS in the rearrangement pathway needs less energy than that in the direct pathway, vinyl cyanide is hydroaminated through the rearrangement pathway.
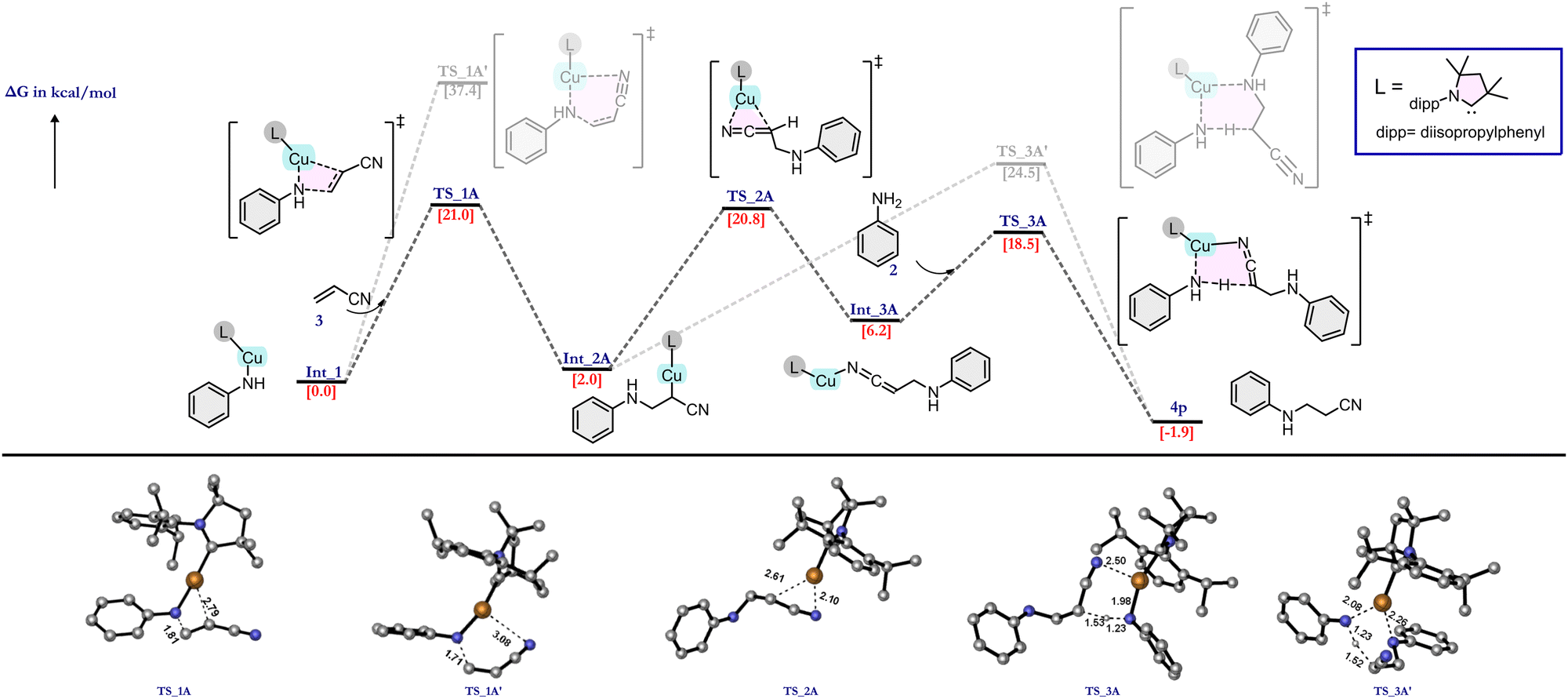 | ||
| Fig. 2 Energy profile diagram of the proposed mechanisms for the hydroamination reaction of vinyl cyanide showing the most probable pathway (the less probable pathway is shown in faded colours). | ||
In the case of the hydroalkoxylation reaction, a higher temperature is required and gives only moderate yields (Scheme 2). To gain a clear understanding of this phenomenon we investigated the reaction pathway for the hydroalkoxylation reaction of vinyl cyanide with phenol. Here, the calculated energy barrier for olefin activation is higher than for the hydroamination reaction, suggesting the need for an elevated temperature. Interestingly, this reaction was found to follow a different reaction pathway, which is discussed in detail in the ESI† Section 7.
In conclusion, we have developed an alternative route for the hydroamination and hydroalkoxylation of electron deficient olefins using a simple CAAC based Cu(I) catalyst under mild conditions. The CAAC–Cu catalyst showed a higher efficiency than the corresponding NHC-based catalyst reported previously. Due to the lack of mechanistic details related to this reaction, we carried out an extensive investigation of the mechanistic pathway using DFT calculations. In the literature, two pathways had been hypothesized: one proceeds via the formation of a zwitterion, and the other involves the formation of a metal olefin complex.12a No evidence – either experimental or computational – was provided to support any of these pathways. Later, some kinetic studies were performed to support the formation of a zwitterion but they did not provide convincing evidence to support that pathway.12b In this communication, we carried out ab initio DFT calculations to determine the mechanistic route, and we propose a distinct pathway (MAOA) that provides a better understanding of the reaction mechanism (Fig. 3). The mechanism proceeds via the olefin activation step, which has a significant dependence on the type of EWG present on the olefin, and it also plays a crucial role in stabilizing the TS. In addition, the hydroalkoxylation reaction proceeds via a slightly different pathway and consists of a higher energy TS compared with the hydroamination reaction.
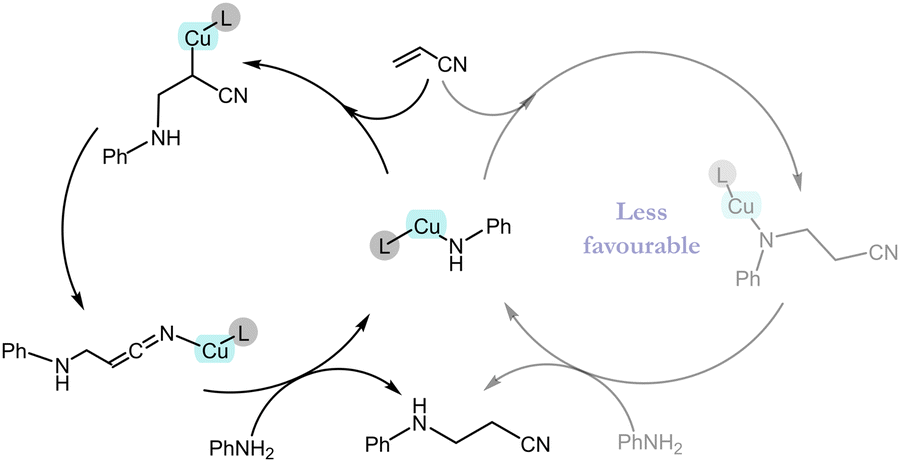 | ||
| Fig. 3 DFT computed pathway for the hydroamination reaction. | ||
This work was financially supported by the SERB, New Delhi, India (SRG/2020/000191). All the authors gratefully acknowledge CRF, IIT Delhi, for the instrumental and HPC facilities, for computational resources. TK acknowledges the IITD seed grant for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed; (b) H. V. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS.
- (a) V. M. Marx, A. H. Sullivan, M. Melaimi,S., C. Virgil,B., K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919 CrossRef CAS; (b) J. Zhang, S. Song, X. Wang, J. Jiao and M. Shi, Chem. Commun., 2013, 49, 9491 RSC; (c) R. Gawin, A. Kozakiewicz, P. A. Guńka, P. Dąbrowski and K. Skowerski, Angew. Chem., Int. Ed., 2017, 56, 981 CrossRef CAS PubMed; (d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed.
- (a) M. Bhunia, S. R. Sahoo, G. Vijaykumar, D. Adhikari and S. K. Mandal, Organometallics, 2016, 35, 3775 CrossRef CAS; (b) M. T. Proetto, K. Alexander, M. Melaimi, G. Bertrand and N. C. Gianneschi, Chem. – Eur. J., 2020, 27, 3772 CrossRef; (c) L. Zhao and X. Zeng, Chem, 2022, 8, 2082 CrossRef CAS.
- (a) A. M. du Vigné and N. Cramer, Organometallics, 2022, 41(19), 2731 CrossRef; (b) R. K. Singh, T. K. Khan, S. Misra and A. K. Singh, J. Organomet. Chem., 2021, 956, 122133 CrossRef CAS; (c) S. K. Thakur, M. Kaur, K. K. Manar, M. Adhikari, A. R. Choudhury and S. Singh, Chem. – Eur. J., 2022, e202202237 CAS.
- (a) S. Xu, X. Zhang, W. Xiong, P. Li, W. Ma, X. Hu and Y. Wu, Chem. Commun., 2022, 58, 2132 RSC; (b) Y. Gao, S. Yazdani, A. Kendrick IV, G. P. Junor, T. Kang, D. B. Grotjahn, G. Bertrand, R. Jazzar and K. M. Engle, Angew. Chem., Int. Ed., 2021, 60, 19871 CrossRef CAS; (c) W. Ma, G. Zhai, X. Hu and Y. Wu, Org. Biomol. Chem., 2020, 18, 4272 RSC; (d) E. A. Romero, R. Jazzar and G. Bertrand, J. Organomet. Chem., 2017, 829, 11 CrossRef CAS; (e) E. A. Romero, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 165 RSC; (f) L. Jin, E. A. Romero, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 15696 CrossRef CAS; (g) X. Hu, M. Soleilhavoup, M. Melaimi, J. Chu and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 6008 CrossRef CAS.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef.
- S. Borman, Chem. Eng. News, 2004, 82, 42 CrossRef.
- (a) J. Haggin, Chem. Eng. News, 1993, 71, 23 CrossRef; (b) A. Fischer, T. Mallat and A. Baiker, Catal. Today, 1997, 37, 167 CrossRef CAS.
- (a) M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125(19), 5608 CrossRef CAS; (b) S. Hong and T. J. Marks, J. Am. Chem. Soc., 2002, 124, 7886 CrossRef CAS PubMed; (c) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (d) Y. Xi, S. Ma and J. F. Hartwig, Nature, 2020, 588, 254 CrossRef CAS.
- (a) X. Zeng, G. D. Frey, S. Kousar and G. Bertrand, Chem. – Eur. J., 2009, 15, 3056 CrossRef CAS PubMed; (b) D. Bawari, B. Goswami, S. K. Thakur, R. V. Varun Tej, A. R. Choudhury and S. Singh, Dalton Trans., 2018, 47, 6274 RSC.
- (a) C. Munro-Leighton, E. D. Blue and T. B. Gunnoe, J. Am. Chem. Soc., 2006, 128, 1446 CrossRef CAS; (b) C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483 CrossRef CAS; (c) S. A. Delp, L. A. Goj, M. J. Pouy, C. Munro-Leighton, J. P. Lee, T. B. Gunnoe, T. R. Cundari and J. L. Petersen, Organometallics, 2011, 30, 55 CrossRef CAS; (d) S. Kim, S. Kang, G. Kim and Y. Lee, J. Org. Chem., 2016, 81, 4048 CrossRef CAS PubMed.
- A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Chem. – Eur. J., 2017, 23, 4625 CrossRef CAS PubMed.
- (a) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed; (b) Catalytic Heterofunctionalization, ed. A. Togni and H. Grützmacher, Wiley-VCH, Weinheim, 2001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05613e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |