
Selectfluor®-enabled photochemical selective C(sp3)–H(sulfonyl)amidation†
Yuehua
Chen
a,
Boxuan
Yang
a,
Qian-Yu
Li
a,
Yu-Mei
Lin
 a and
Lei
Gong
*ab
a and
Lei
Gong
*ab
aKey Laboratory of Chemical Biology of Fujian Province, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China. E-mail: gongl@xmu.edu.cn
bInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361005, China
First published on 29th November 2022
Abstract
Transition metal- and photosensitizer-free C(sp3)–H (sulfonyl)amidation reactions have been realized by employing Selectfluor® as a versatile reagent, functioning as a photoactive component, a HAT precursor and an oxidant. Various toluene derivatives, cycloalkanes, natural products and bioactive molecules can be converted into N-containing products under mild conditions in good yield and with high chemo- and site-selectivity.
The C–N bond is a basic building block in many biologically important molecules and natural products, and creating such a bond is a fundamental concern in organic synthesis.1–3 Direct C–H amination and amidation reactions provide atom- and step-economic access to C–N bond formation, and have attracted significant attention from the synthetic community (Scheme 1a).4 It remains a challenging task however to convert relatively unreactive C(sp3)–H units, such as those in alkyl fragments, into the corresponding amine or amide moieties.5 These reactions face a number of challenges, including: (i) the intrinsic inertness of these strong, neutral C(sp3)–H bonds, (ii) the difficulty of achieving a balance between reactivity and selectivity, (iii) competitive side reactions such as β-H elimination and self-coupling, and (iv) the incompatibility of some N-containing functionalization reagents with transition metal or strong redox systems.6 In this field, the groups of Chang, Muñiz, Du Bois and others have reported significant progesses.7,8 Nevertheless, there remains a pressing need for the design of new, effective reaction systems that can facilitate C(sp3)–H activation and enable subsequent selective C–N bond formation.
 | ||
| Scheme 1 Overview of this study. TM = transition metal, PC = photocatalyst. | ||
1-(Chloromethyl)-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis-tetra-fluoroborate, otherwise known as Selectfluor®, is widely used as a fluorination reagent, an oxidant and a Lewis acid in organic synthesis.9–11 Some recent studies have shown that under thermal or photochemical conditions, Selectfluor® can function as a C–H activation agent with specific substrates, additives or external photocatalysts.12,13 For example, Humbeck et al. reported a benzylic C–H fluorination of aromatic azaheterocycles, in which a single electron transfer (SET) from a heterocyclic substrate to Selectfluor® gives rise to a benzylic radical that is involved in further C–F bond formation.14 Baxter et al. found that simple pyridine substrates formed [N–F–N]+ halogen bonds with Selectfluor® thus facilitating single electron reduction by catalytic Ag(I), leading to a C–H fluorination reaction.15 Lei et al. reported that photochemical Minisci-type C(sp3)–H arylation of alcohols and cycloalkanes with heteroarenes are promoted by Selectfluor® via hydrogen atom transfer (HAT) pathways,16,17 and Chupakhin et al. developed a blue light-promoted C(sp2)–H azolation reaction of nonaromatic 2H-imidazole 1-oxides mediated by Selectfluor®.18
Inspired by these studies and our previous research on photochemical synthesis,19,20 we chose to investigate if employing Selectfluor® as a multifunctional reagent would enable the photochemical activation of strong C(sp3)–H bonds, further single-electron oxidation and combination with different N-centered nucleophiles. Such a process would allow us to develop a versatile and general platform for C–N bond formation without the requirement for transition metal catalysts or external photosensitizes (Scheme 1b).
We began our study of the sulfonylamidation of strong C(sp3)–H bonds with 4-ethyl-1,1′-biphenyl (1a) as a model substrate and p-toluenesulfonamide (2a) as the reaction partner (Table 1). Upon irradiation of a mixture of 1a, 2a and Selectfluor® with a LEDs lamp (λmax = 395 nm) for 48 h, the desired product (3) was obtained in 76% yield (entry 1). Screening of the solvents showed that dichloromethane was the best choice. Chloroform and 1,2-dichloroethane as solvents gave declined yields of 52% and 66%, respectively (entry 2), while N,N-dimethylformamide, dimethyl sulfoxide and acetonitrile failed to afford the product (entry 3). The replacement of Selectfluor® by other oxidative reagents such as N-fluorobenzenesulfonimide (NFSI) (entry 4) or MnO2 (entry 5) led to no product, and use of PhI(OAc)2 (entry 6) or m-chloroperoxybenzoic acid (m-CPBA) (entry 7) in place of Selectfluor® gave only 21% or 11% yield, respectively. Control experiments showed that Selectfluor® and light irradiation were both essential for the transformation (entries 8–11). When the reaction was performed at 60 °C or 120 °C in the dark, the product (3a) could not be detected (entries 10 and 11). The photochemical reaction could be conducted in air, which gave only a slightly reduced yield of 72% and meanwhile afforded 4-acetylbiphenyl as a side-product in 7% yield (entry 12). Addition of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), a radical quencher, completely inhibited the formation of 3, suggesting a plausible radical pathway (entry 13). The reduced reaction temperature at 25 °C led to a lower yield of 50% (entry 14).
|
|
||
|---|---|---|
| Entry | Variations from standard conditionsa | Yieldb (%) |
| a Standard conditions: 1a (0.30 mmol), 2a (0.20 mmol), Selectfluor® (0.60 mmol), CH2Cl2 (1.0 mL), 40 °C, a 50 W LED lamp, under argon, 48 h, see the ESI for more details concerning the screening of solvent and concentration. b Isolated yield. c 4-Acetylbiphenyl was isolated as a side-product in 7% yield. n.a. = not applicable. | ||
| 1 | None | 76 |
| 2 | CHCl3 or ClCH2CH2Cl as solvent | 52 or 66 |
| 3 | DMF, DMSO or CH3CN as solvent | n.a. |
| 4 | NFSI instead of Selectfluor® | n.a. |
| 5 | MnO2 instead of Selectfluor® | n.a. |
| 6 | PhI(OAc)2 instead of Selectfluor® | 21 |
| 7 | m-CPBA instead of Selectfluor® | 11 |
| 8 | In the absence of Selectfluor® | n.a. |
| 9 | In dark | n.a. |
| 10 | 60 °C in dark | n.a. |
| 11 | 120 °C in dark | Trace |
| 12 | In air | 72c |
| 13 | TEMPO (4.0 eq.) | n.a. |
| 14 | In 25 °C | 50 |
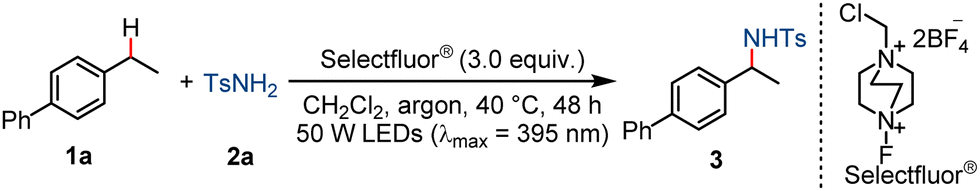
With the optimal conditions, we evaluated the substrate scope of the photochemical C–H sulfonylamidation reaction (Scheme 2). It was found that benzenesulfonamides containing a methyl (product 3), methoxy (5), trifluoromethoxy (6), fluoro (7), chloro (8) or trifluoromethyl group (9) at the para position, fluoro (10) at the meta position, and methyl (11), chloro (12), bromo (13) or trifluoromethyl (14) at the ortho position were all tolerated, delivering the corresponding products in 64–96% yields. Benzenesulfonamides with two or three substituents on the phenyl moiety (products 15–17), a fused ring (18), a heterocycle (19) and an N-methyl group (20) provided yields of 40–97%. Aliphatic sulfonamides also proved to be good substrates. For instance, the reaction of methanesulfonamide under standard conditions delivered the product (21) in 71% yield. It was found that sulfonyl diamide, a compound containing two sulfonamide moieties, reacted with two equivalents of 1a, forming a bis-sulfonamide product (22) in 62% yield and with 3.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. This reaction is compatible with pharmaceutically useful compounds with more complex structures. For example, Celecoxib, a non-steroidal anti-inflammatory drug, was found to react smoothly with 1a to furnish a benzyl-substituted Celecoxib (23) in 84% yield.
:1 d.r. This reaction is compatible with pharmaceutically useful compounds with more complex structures. For example, Celecoxib, a non-steroidal anti-inflammatory drug, was found to react smoothly with 1a to furnish a benzyl-substituted Celecoxib (23) in 84% yield.
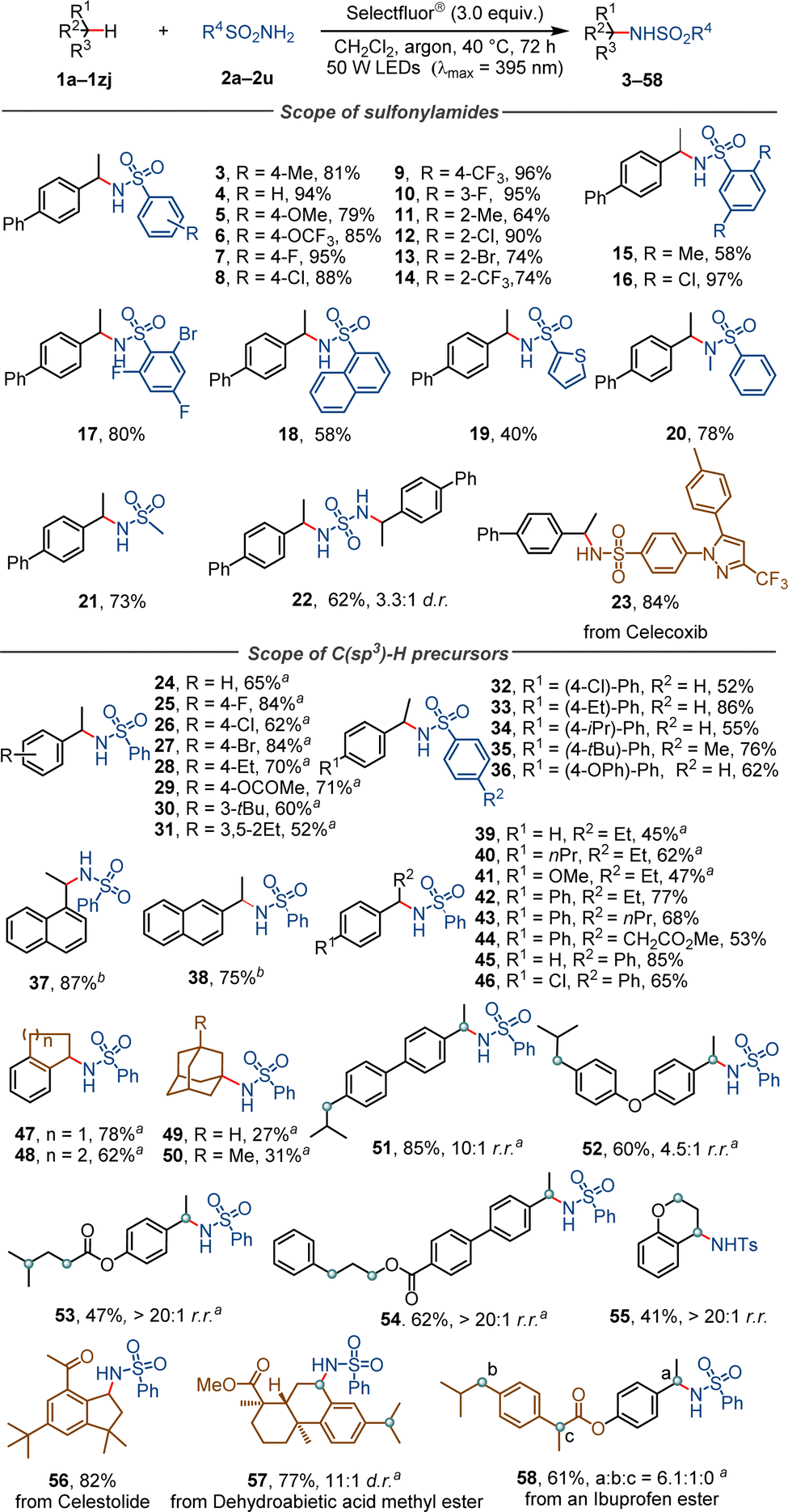 | ||
| Scheme 2 Substrate scope. aReaction upon irradiation with a purple LED lamp (λmax = 365 nm). | ||
The reactions of different precursors with C(sp3)–H bonds were examined. Ethylbenzene derivatives featuring diverse electronic and steric properties for example, were successfully converted into the target compounds (24–38) in 52–87% yield. Secondary benzylic substrates with a longer phenyl substituent (products 39–46) or a fused aliphatic ring (47, 48) also gave satisfactory yields. Adamantane is a cycloalkane which has a tertiary C–H bonds with a bond dissociation energy (BDE) of 99 kcal mol−1, higher than the 96 kcal mol−1 of its secondary C–H bonds.21 In this reaction, adamantane and its methyl-substituted derivative were exclusively functionalized at their tertiary positions, affording the corresponding products (49, 50) as single regioisomers. The site-selectivity of this photochemical process was further investigated by employing ethylbenzene derivatives containing distinct and competitive C(sp3)–H bonds as the substrates. For example, 4-ethyl-4′-isobutyl-1,1′-biphenyl with two different secondary benzylic C–H bonds was selectively functionalized and afforded the product (51) in 85% yield and with 10:1 r.r. 3-Phenylpropyl 4′-ethyl-[1,1′-biphenyl]-4-carboxylate, a compound bearing O-α C–H bonds and different secondary benzylic C–H bonds, was converted into the product (52) with >20:1 r.r. These outcomes demonstrate the precise site recognition of the Selectfluor®-enabled reaction towards specific C(sp3)–H bonds.22
This reaction was also applied to the late-stage modification of natural products and bioactive molecules. For example, Celestolide, a natural product with a musky, sweet and animal odor, was converted under standard conditions into its analog (56) in 82% yield. Dehydroabietic acid methyl ester was selectively functionalized at a secondary benzylic position, affording the product (57) in 77% yield and 11:1 d.r. A sulfonylamidated derivative (58) was obtained in 61% yield and with a site-selectivity of 6.1:1:0 (a:b:c), starting from an Ibuprofen ester with three closely similar benzylic C–H bonds.
A gram-scale reaction of 1a (0.82 g, 4.5 mmol) and 2u (1.14 g, 3.0 mmol) was carried out and gave the product (23) in a slightly reduced yield (67%, 1.12 g, 2.0 mmol) in 96 h (Scheme 3a). Other N-containing reagents including carbamates (products 59–61) and benzamide (62) can be used as alternative reagents in the reaction with 4-ethyl-1,1′-biphenyl (1a) (Scheme 3b). Interestingly, the employment of readily available nitriles as reaction partners led to the development of a highly effective C–H amidation reaction (Scheme 3c). In the presence of 0.5 equivalent of concentrated sulfuric acid under slightly modified conditions, the reaction of ethylbenzene or its derivatives with benzyl cyanide gave rise to the corresponding phenylacetamide derivatives (63–74) in 35–78% yield. Various alkyl nitriles and an aryl nitrile were examined and found to afford amide products (75–90) in satisfactory yields. A convenient three-step synthesis of Rasagiline, a drug used for the treatment of Parkinson's disease, was developed from readily available 2,3-dihydro-1H-indene (1y) on the basis of this photochemical C–H sulfonylamidation reaction (Scheme 3d). The sulfonylamidation of the benzylic compound (1y) to generate N-(2,3-dihydro-1H-inden-1-yl)benzenesulfonamide (47), followed by N-propargylation with propargyl bromide in the presence of sodium hydride, and reduction with LiAlH4, afforded Rasagiline (92) in a total yield of 71%.
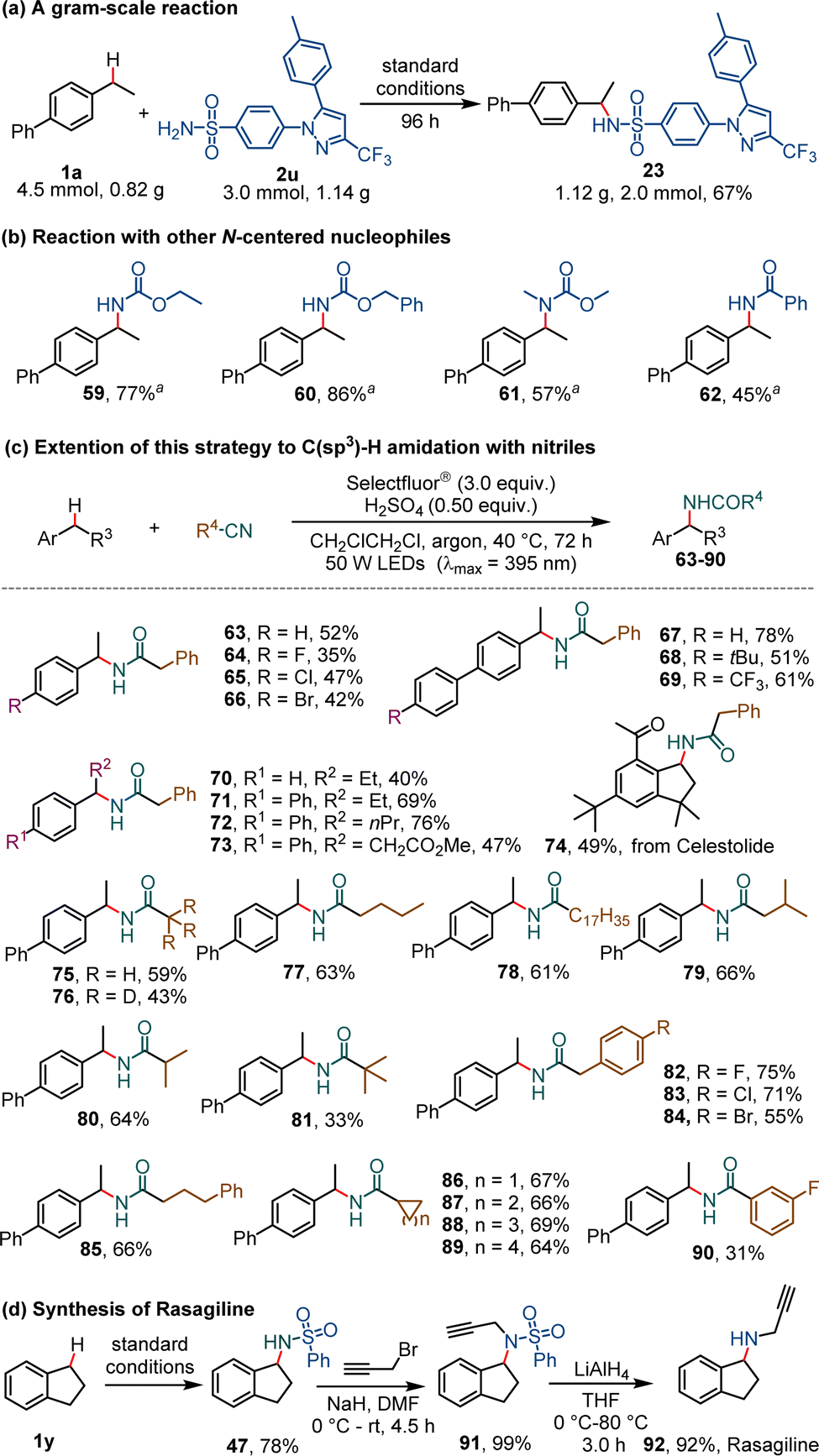 | ||
| Scheme 3 Synthetic utility of this method. | ||
Several control experiments were conducted to gain insight into the reaction mechanism. Addition of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy, 3.0 equiv.) to the reaction of 1a + 2a → 3 led to complete inhibition of the formation of 3. Two TEMPO-coupled products (93 and 94) were detected by high resolution mass spectrometry (HRMS), confirming the engagement of benzyl and fluorine radicals in the photochemical sulfonylamidation reaction (Scheme 4a). The incorporation of the nucleophilic sodium acetate into the reaction gave rise to a benzyl acetate (95) in 15% yield, suggesting a possible pathway via carbon cations (Scheme 4b). When 2.8 equiv. of H218O was added to the reaction of 4-ethyl-1,1′-biphenyl (1a) with benzyl cyanide, an isotopically labeled product (96) was detected by HRMS (Scheme 4c). This result suggests that the formation of amide moieties might derive from hydrolysis.
 | ||
| Scheme 4 Studies of the mechanism. | ||
On the basis of these experiments, a plausible reaction mechanism was proposed and is shown in Scheme 4d. Irradiation of Selectfluor® with a LED lamp (λmax = 395 nm) leads to cleavage of the N–F bond. The N-centered radical cation produced (I) undergoes hydrogen atom abstraction from a C(sp3)–H bond in the hydrocarbon substrate to generate an alkyl radical (II). Further oxidization of radical II by Selectfluor®, F radical or intermediate I furnishes a carbocation species (III).23 This electronic intermediate (III) is rapidly captured by an N-centered nucleophile to generate a new C–N bond. In a photochemical C–H amidation reaction, the nitrile serves as a nucleophile to react with the carbocation (III) to give a nitrilium ion, which is subsequently hydrolyzed in the presence of concentrated sulfuric acid to form the amide product.
In summary, we have developed a convenient strategy relying on multiple functions of commercially available Selectfluor® for the sulfonylamidation and amidation of strong C(sp3)–H bonds. Various benzylic compounds, cycloalkanes, natural products and bioactive molecules can effectively react with different N-centered nucleophiles, affording structurally diverse products in good yields and with high chemo- and site-selectivity. These reactions are scalable and can install different N-containing functionalities into C(sp3)–H moieties. We expect that they will provide a general platform for C–N bond formation without the requirement of transition metals and external photosensitizers.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Zhang and A. Lei, Synthesis, 2019, 83–96 Search PubMed.
- (a) J. R. Clarke, K. Feng, A. Sookezian and C. M. White, Nat. Chem., 2018, 10, 583–593 CrossRef PubMed; (b) X. Zhao, H. Xu, X. Huang and J. S. Zhou, Angew. Chem., Int. Ed., 2019, 58, 292–296 CrossRef CAS PubMed; (c) A. Nasrallah, Y. Lazib, V. Boquet, B. Darses and P. Dauban, Org. Process Res. Dev., 2020, 24, 724–728 CrossRef CAS.
- (a) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS; (b) D. Hazelard, P.-A. Nocquet and P. Compain, Org. Chem. Front., 2017, 4, 2500–2521 RSC; (c) J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS; (d) G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 124, 7498–7510 CrossRef; (e) S. Y. Hong, Y. Park, Y. Hwang, Y. B. Kim, M.-H. Baik and S. Chang, Science, 2018, 359, 1016–1021 CrossRef CAS.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS.
- H. Chen, S. Schlecht, T. C. Semple and J. F. Hartwig, Science, 2000, 287, 1995–1997 CrossRef CAS.
- (a) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS; (b) S.-W. Wu, J.-L. Liu and F. Liu, Org. Lett., 2016, 18, 1–3 CrossRef CAS PubMed; (c) S. Sakaguchi, T. Hirabayashi and Y. Ishii, Chem. Commun., 2002, 516–517 RSC; (d) G. Laudadio, Y. Deng, K. Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS.
- (a) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed; (b) D. Ramesh, U. Ramulu, K. Mukkanti and Y. Venkateswarlu, Tetrahedron Lett., 2012, 53, 2904–2908 CrossRef CAS; (c) A. Nasrallah, Y. Lazib, V. Boquet, B. Darses and P. Dauban, Org. Process Res. Dev., 2020, 24, 724–728 CrossRef CAS; (d) Y.-H. Ye, J. Zhang, G. Wang, S.-Y. Chen and X.-Q. Yu, Tetrahedron, 2021, 67, 4649–4654 CrossRef.
- (a) Y. Zhang, B. Feng and C. Zhu, Org. Biomol. Chem., 2012, 10, 9137–9141 RSC; (b) X. Lu, Y. Shi and F. Zhong, Green Chem., 2018, 20, 113–117 RSC; (c) F. Wu, J. P. Ariyarathna, N. Kaur, N.-E. Alom, M. L. Kennell, O. H. Bassiouni and W. Li, Org. Lett., 2020, 22, 2135–2140 CrossRef CAS PubMed.
- D. D. Bume, S. A. Harry, T. Lectka and C. R. Pitts, J. Org. Chem., 2018, 83, 8803–8814 CrossRef CAS PubMed.
- K. Chen and S. Zhu, Synlett, 2017, 640–653 CAS.
- K. Yang, M. Song, A. I. M. Ali, S. M. Mudassir and H. Ge, Chem. – Asian J., 2020, 15, 729–741 CrossRef CAS PubMed.
- R. Narobe, K. Murugesan, C. Haag, T. E. Schirmer and B. König, Chem. Commun., 2022, 58, 8778–8781 RSC.
- M. González-Esguevillas, J. Miró, J. L. Jeffrey and D. W. C. MacMillan, Tetrahedron, 2019, 75, 4222–4227 CrossRef.
- K. E. Danahy, J. C. Cooper and J. F. V. Humbeck, Angew. Chem., Int. Ed., 2018, 57, 5134–5138 CrossRef CAS.
- A. M. Hua, S. L. Bidwell, S. I. Baker, H. P. Hratchian and R. D. Baxter, ACS Catal., 2019, 9, 3322–3326 CrossRef CAS.
- L. Niu, J. Liu, X.-A. Liang, S. Wang and A. Lei, Nat. Commun., 2019, 10, 467 CrossRef PubMed.
- X.-A. Liang, L. Niu, S. Wang, J. Liu and A. Lei, Org. Lett., 2019, 21, 2441–2444 CrossRef CAS.
- A. A. Akulov, M. V. Varaksin, A. N. Tsmokalyuk, V. N. Charushin and O. N. Chupakhin, Green Chem., 2021, 23, 2049–2057 RSC.
- S. Cheng, Q. Li, X. Cheng, Y.-M. Lin and L. Gong, Chin. J. Chem., 2022, 40, 2825–2837 CrossRef CAS.
- (a) Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016–1026 CrossRef CAS; (b) Y. Li, K. Zhou, Z. Wen, S. Cao, X. Shen, M. Lei and L. Gong, J. Am. Chem. Soc., 2018, 140, 15850–15858 CrossRef CAS; (c) S. Cao, W. Hong, Z. Ye and L. Gong, Nat. Commun., 2021, 12, 2377 CrossRef CAS; (d) S. Zhang, S. Cao, Y.-M. Lin, L. Sha, C. Lu and L. Gong, Chin. J. Catal., 2022, 43, 564–570 CrossRef CAS; (e) S. Cao, Z. Ye, Y. Chen, Y.-M. Lin, J. Fang, Y. Wang, B. Yang and L. Gong, CCS Chem., 2022, 4, 3122–3133 CrossRef CAS.
- H.-B. Yang, A. Feceu and D. B. C. Martin, ACS Catal., 2019, 9, 5708–5715 CrossRef CAS.
- Z.-W. Hou, D.-J. Liu, P. Xiong, X.-L. Lai, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef CAS PubMed.
- L. Wang, X. Wang, G. Zhang, S. Yang, Y. Li and Q. Zhang, Org. Chem. Front., 2019, 6, 2934–2938 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05569d |
| This journal is © The Royal Society of Chemistry 2023 |