
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of a nanoscale Cu(II)31-oxo-carboxylate cluster, and effect of Cu–oxo cluster size on visible-light absorption†
Thomas J.
Barnes
 a,
Jack
Payne
b and
Sebastian D.
Pike
*a
a,
Jack
Payne
b and
Sebastian D.
Pike
*a
aDepartment of Chemistry, University of Warwick, Coventry, UK. E-mail: sebastian.pike@warwick.ac.uk
bDepartment of Chemistry, University of Cambridge, Cambridge, UK
First published on 24th November 2022
Abstract
Cu16–Cu31 Cu(II)-oxo-carboxylate clusters are reported, including those with condensed 1.5 nm Cu–O cores supported exclusively by O-donor ligands. A size–colour correlation is observed due to a red-shift of the charge transfer absorption band on increasing size; hence these clusters sit between small molecules and (black) CuO nanostructures.
Metal–oxo clusters are fascinating molecular analogues of metal oxide materials, with recent discoveries described as ‘molecular nanoparticles’.1 They have well-defined and tuneable structures making them excellent options as photocatalysts,2,3 molecules for energy conversion or storage,4 molecular magnets,5 or soluble precursors to semiconducting oxide materials.6,7 Metal–oxo clusters may absorb light in a similar manner to their bulk oxide counterparts, however, as most are smaller than the exciton Bohr radius of the parent material, quantum confinement theory predicts a size-dependent blue-shift of their absorption onset relative to the bulk oxide.8 This allows tuneability of optical properties which may be further optimised by adjusting surface interactions for efficient photochemical applications.
Cupric oxide (CuO) is an important semiconducting oxide material with applications in electrocatalysis (e.g. water splitting9,10 or biomass volarisation11), heterogeneous catalysis,12 and in photovoltaic cells.13 Cupric oxide is a black p-type (strongly correlated) semiconductor with a small band gap (reported values range from ∼1 – 2.1 eV)13–16 attributed to {O + Cu(3dx2−y2)} → Cu(3dx2−y2) transitions with significant oxygen to metal charge transfer character.15,16 CuO has a moderately large exciton Bohr radius (∼6 nm).17 Therefore it is logical that quantum size effects should bring the absorption onset of small Cu(II)-oxo clusters into the mid-visible region (e.g. 2–2.8 eV), potentially providing appropriate photovoltage for challenging photocatalytic processes driven by sunlight, such as water oxidation.
Despite potential uses in photocatalysis or as precursors for the deposit of ultrasmall copper or copper-oxide-based particles, Cu(II)-oxo clusters with a condensed Cu(II) + O/OH core remain remarkably rare,5,18–26 contrasting with the rich chemistry known for other metal–oxo clusters. Some interesting Cu–oxo structures have been reported with internal templates,20,21,25 multidentate ligands,5,19,21,26 or with mixed-metal systems;27 however, in these structures the incorporation of halides, heteroatom donors, or heterometals, will inherently change the electronic properties and alter the relationship with CuO. Pioneering work by Christou et al. reported the structure of Cu16-oxo clusters [Cu16O4(OH)4(OR)8(O2CtBu)12(ROH)], 1, R = nPr and 2, R = nBu, as rare examples of Cu–oxo clusters with exclusively O-donor ligands.18 The use of multidentate carboxylate ligands has also led to the discovery of the Cu-hydroxo nanodisc [Cu15(dhs)6(OH)6(H2O)10](H2O)20 (H4dhs = 2,3-dihydroxysuccinic acid),22 and the 1D metal-organic chain [(CO3)@Cu20(suc)6(OH)26(H2O)12·18H2O]n (suc = succinate) comprising of Cu cages with the (8-capped) Keggin structure encapsulating a carbonate anion.28
In previous reports,18,26 Cu–oxo clusters are synthesised from Cu(NO3)2 and ligands in alcohol solutions, using a base to activate the Cu salt into more reactive species. We hypothesised that the base initiates Cu(II)–OR formation which is then susceptible to hydrolysis and condensation reactions to build Cu–O–Cu connectivity, leading to clusters, but which may retain some surface alkoxides (Scheme 1). Therefore, the alcohol solvent plays an important role in the assembly of clusters, affecting solubility and also providing a steric influence on the cluster surface during growth. Furthermore, as water is necessary for hydrolysis and cluster growth, the amount of water in solution is also a critical factor, with uncontrolled hydrolysis likely leading to extended CuO materials.
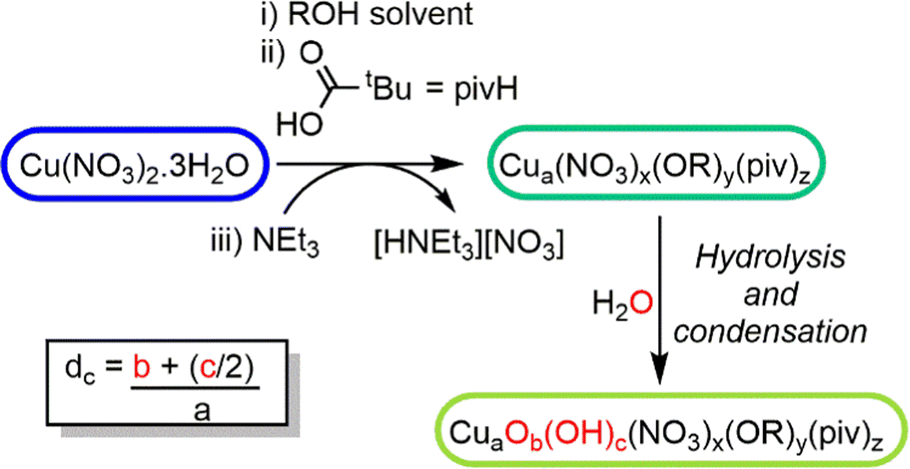 | ||
| Scheme 1 Proposed reaction scheme for the growth of Cu–oxo clusters via hydrolysis and condensation of a ‘pre-hydrolysis intermediate’. | ||
Using this approach with pivalic acid (tBuCO2H) as a supporting ligand and sterically unencumbered primary alcohols (R = Et, nPr, nBu) leads to previously reported green Cu16 clusters 1 and 2 (Fig. S1–S3, ESI†),18 or similarly structured ethoxide derivative 3, [Cu16O4(OH)4(OEt)8(O2CtBu)12(H2O)] which contains a symmetrically bound water molecule in its central pocket (Fig. 1 and Fig. S4, S5, ESI†). These structures all crystallise directly from alcohol solutions (or more rapidly from pentane, see ESI†), and have a degree of condensation (dc), defined as ({n(O2−) + 1/2n(OH−)}/n(Cu2+)), of 0.375, some way from that of CuO (dc = 1), indicating that their structures are mostly ‘surface’.
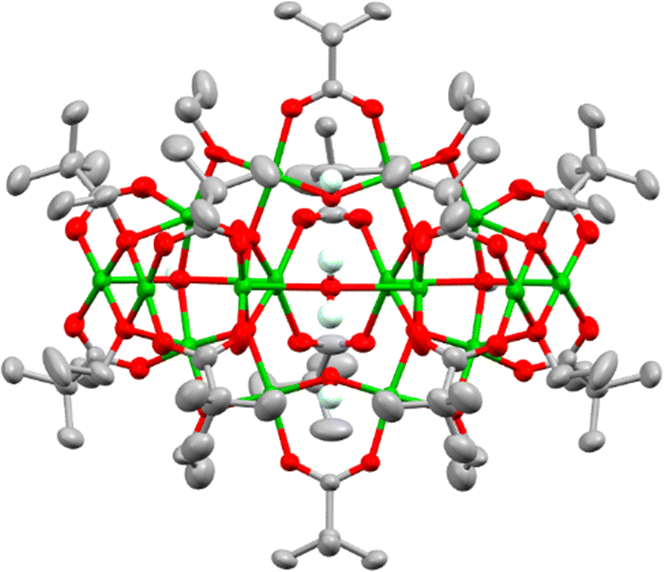 | ||
| Fig. 1 Solid-state structure of ethoxide capped Cu16-oxo cluster 3. Ellipsoids displayed at 50% probability, hydrogen atoms (except OH or H2O) omitted for clarity. Cu = green, O = red, C = grey, H = very pale blue. | ||
By switching to bulkier secondary alcohols (R = iPr or Cyp (C5H11)) grass-green crystals of larger clusters form after two weeks (in powdered form, or when dissolved, these compounds appear yellow/green). Isostructural forms were identified by X-ray crystallography, supported by bond-valence sum calculations, as [Cu31O12(OH)18(O2CtBu)18(NO3)2(ROH)6·n(ROH)] (R = iPr, n = 6, 4; R = Cyp, n = 12, 5) (Fig. 2 and Fig. S6–S8, ESI†). This structure has a pseudo-spherical Cu–O core (with 12 μ4-O2, 12 μ3-OH− and 6 μ2-OH−) with a maximum (Cu–O) core diameter of 1.5 nm and can thus be described as a ‘molecular nanoparticle’. However, the Cu–O core is not a clear representation of the repeating structure in CuO. In CuO the copper centres coordinate to four oxygens in a close to square planar geometry (Cu–O bond lengths = 1.91–2.02 Å),29 with longer distances (2.78 Å) to axially positioned oxygens. Similar coordination modes are found in 4 and 5 with comparable Cu–O bond lengths to the four nearest oxygens (range 1.88–2.28 Å) and most sites having a fifth oxygen occupying an apical site (range 2.29–2.89 Å, Supporting note 1, Fig S9, S10 and Table S1, ESI†). In contrast, the central Cu atom in 4 or 5 has a tilted octahedral geometry (Cu–O bond lengths: 2.05 to 2.22 Å). The cluster lies on a pseudo-C3 symmetry axis passing through the centrosymmetric octahedral Cu and the N atoms of the two nitrates (Fig. 2) with overall pseudo-D3d symmetry. The dc of 4 and 5 is 0.68, showing a much more condensed core and thus greater resemblance to CuO@carboxylate nanoparticles than the smaller clusters 1–3. The surface of 4 and 5, which can be considered a model for CuO@carboxylate nanoparticles,30 is capped by 18 anionic (μ2 or μ3) pivalate ligands, 18 hydroxides, six neutral ROH ligands and two loosely coordinated μ2-nitrate anions (Cu–ONO2 bond lengths 2.6–2.8 Å), which have disordered coordination geometries in the crystal structure and are each surrounded by the three coordinated and a further three non-coordinated ROH ligands (which likely engage in H-bonding interactions to the cluster surface, Fig. 2, Fig. S6 and S8, ESI†). Crucially these clusters have no remaining alkoxide groups (confirmed by bond valence sum calculations, Table S3, ESI†) which may provide enhanced stability to further hydrolysis and condensation reactions. The greater steric bulk of secondary alcohols appears to support condensation and enlargement of the core compared to less bulky primary alcohols. The synthesis of 4 and 5 is very condition-dependent, with brown precipitates containing CuO@pivalate nanoparticles (∼8–15 nm by Scherrer analysis) and Cu2(OH)3(NO3) (Fig S11 and S12, ESI†) alongside solutions containing the paddlewheel [Cu2(O2CtBu)4(NEt3)2] able to form as by-products if the reaction is conducted at higher concentrations; left open to ambient atmosphere; or allowed to sit for longer times. The by-products likely form due to uncontrolled hydrolysis. It appears essential to maintain good solubility of intermediates to allow 4/5 to self-assemble without the formation of insoluble by-products.
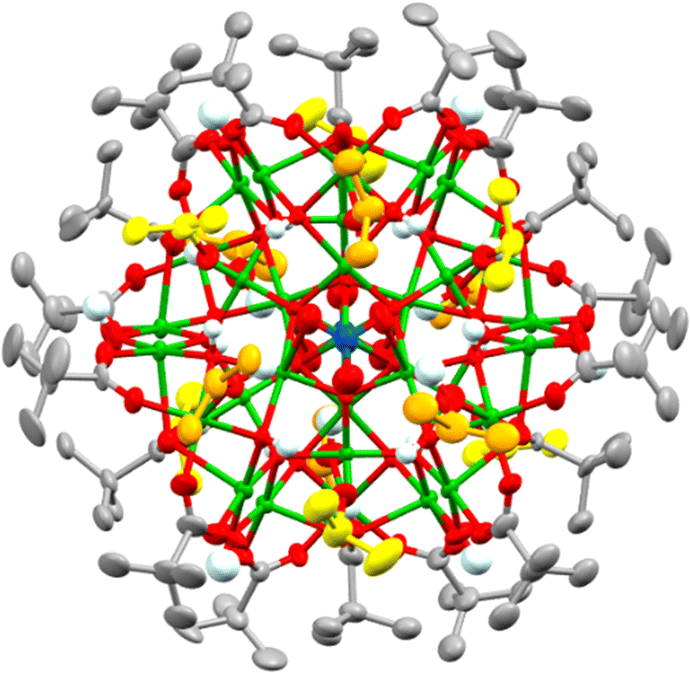 | ||
| Fig. 2 Solid-state structure of Cu31-oxo cluster 4. Cu = green, O = red, C = grey, N = blue, H = very pale blue (non-coordinated/H-bonded iPrOH Cs = yellow, coordinated iPrOH Cs = orange). Ellipsoids displayed at 50% and hydrogen atoms (except OH) omitted. | ||
Based on our observations and hypothesised reaction mechanism (Scheme 1), moisture plays a crucial role in cluster synthesis. However, the copper source Cu(NO3)2·3H2O contains three equivalents of water per copper atom, thus always provides an excess of water for hydrolysis and condensation reactions. Therefore, selective cluster formation is likely defined by crystallisation of the growing clusters from the reaction medium (for example when 3 is dissolved in dichloromethane or isolated as a powder it is susceptible to further hydrolysis when exposed to moisture). To test these concepts, the synthesis of clusters using anhydrous precursors was explored. Cu(OEt)2 was reacted with 1 equiv. of pivalic acid in dry toluene at 70 °C to produce previously reported loop-shaped [Cu6(OEt)6(O2CtBu)6], 6,18,31 (Fig. S13, ESI†). This cluster is a ‘pre-hydrolysis’ structure as it contains no oxo or hydroxide groups (dc = 0), and could potentially form at the beginning of the reaction between Cu(NO3)2·3H2O, pivalic acid and NEt3 in ethanol, i.e. before any hydrolysis (Scheme 1).32 The reaction of 6 with 2.7 equivalents of water in EtOH induces a slow colour change from blue-green to green, and a precipitate of 3 forms after two weeks (confirmed by diffraction after recrystallisation), demonstrating that water promotes cluster growth.32
Considering that water plays a crucial role in cluster formation, the reaction conditions to produce 4 were repeated, but, after 30 minutes of reaction (under air), the solution was degassed and placed under N2 over activated 3 Å molecular sieves. The flask was kept under air-free conditions and after one month, crystals of a new structure, 7, [Cu17O5(OH)6(OiPr)6(O2CtBu)10(NO3)2]·([NEt3H][NO3])2, reproducibly form. 7 has a dc of 0.47 (> 1–3 but < 4–5) indicating that this structure has formed under partial hydrolysis conditions, i.e. before 4 forms (Fig. 3 and Fig. S14–S16, ESI†). This also indicates that hydrolysis and cluster growth is a relatively slow process as it can be arrested by removing moisture from the reaction solution after 30 minutes of reaction. The structure of 7 maintains some Cu–OiPr functionality, suggesting that it may be susceptible to further reaction with water. 7 is a C-shaped cluster with an intriguing pocket which could act as a sterically encumbered reactive site. 7 contains 17 5-coordinate Cu centres which are supported by eight μ2-briging pivalate anions and two unusually coordinated pivalates that each coordinate to seven Cu centres (Fig. S17, ESI†). Two chelating nitrates (Cu–O = 2.02–2.65 Å) and four bridging isopropoxide groups coordinate around the edges of the cluster, whilst a combination of two isopropoxides and two hydroxides occupy the internal cleft of the C-shape in a disordered manner (Fig. S18, ESI†). Two further nitrate anions are connected via H-bonding to four Cu–OH sites which sit in a square on the top of the cluster.
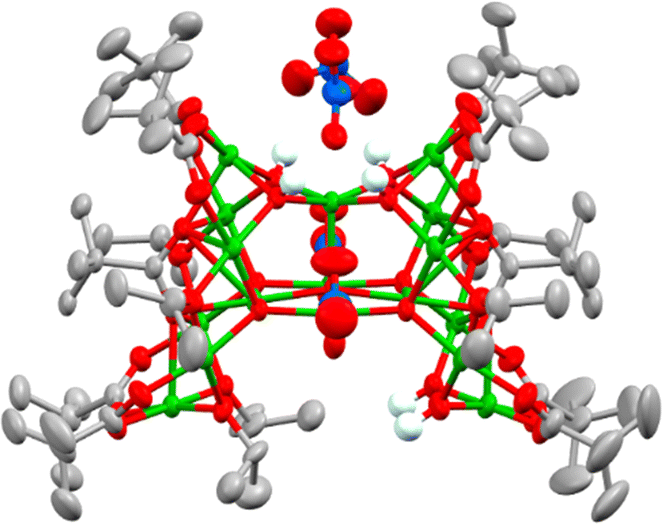 | ||
| Fig. 3 Solid-state structure of 7. Ellipsoids displayed at 50% probability, hydrogen atoms (except OH groups) and [NEt3H]+ cations omitted for clarity. Cu = green, O = red, C = grey, N = blue, H = very pale blue. The base of the structure (within the pocket) has disordered OiPr and OH groups. | ||
The relatively asymmetric shapes and range of different Cu carboxylate coordination geometries in these clusters highlights the flexibility of the Cu(II) centre and the unpredictability of the structures that it can form. This implies that many further possible cluster structures can be obtained by variation of the supporting ligands and degree of condensation.
The colours of these Cu–oxo clusters suggests a relationship with size (Fig. 4 and Fig. S19, S20, ESI†), as the compounds change from blue through green to yellow/green with increasing size, eventually reaching dark brown/black CuO nanoparticles (Fig. S21, ESI†). The UV/visible absorption spectra of 1–4, 6, the small Cu2 ‘paddlewheel’ [Cu2(O2CtBu)4(HOR)2] (R = iPr or H), 8 (Fig. S22, ESI†), and Cu(NO3)2 were recorded (Fig. 4 and Fig. S23–S31, ESI†). All display Beer–Lambert behaviour. The well-known paddlewheel structure (with two carboxylates per Cu) is blue in solution (Fig. 4) and has a high energy carboxylate (O) to metal (dx2–y2) charge transfer (LMCT) band in the UV region33,34 (8: λmax = 262 nm; ε ∼ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 M−1 cm−1) with a shoulder (“dimer band”) arising from the bridging carboxylate coordination mode at slightly lower energy, and weaker d–d transitions (to dx2–y2) in the visible region (λmax = 615 nm; ε ∼ 300 M−1 cm−1) which cause the blue colour. Compound 6 is blue/green in solution and shows a similar LMCT peak λmax = 261 nm, and a shoulder which now extends into the visible region, possibly from added contributions of alkoxide to Cu LMCT,8 and a weaker d–d absorption per Cu (Fig. 4). Upon growth of the cluster to (green) Cu16 (2) and (yellow/green) Cu31 (4), the shoulder region becomes increasingly significant, extending into the visible region (and hence absorbing blue photons), and beginning to resemble the spectrum of ∼4.4 nm brown CuO@carboxylate particles (Fig. S31, ESI†).30 The carboxylate based LMCT peak remains in 2 and 4 with similar maxima (253 & 256 nm respectively) in the UV region, but with diminishing maximum extinction coefficient, when defined per copper, as the clusters grow (and the Cu:ligand ratio decreases, Fig. S32, ESI†). In 4 the shoulder region merges with the d–d excitations causing absorption across the visible region. The exact nature of excitations in the 350–550 nm region remain unknown and will be the focus of future study, but it is interesting to speculate that 4 contains the same surface ligands (pivalate and iPrOH) as 8 (+ two weakly bound nitrates in 4), yet absorbs broadly across the visible region (N.B. Cu(NO3)2 dissolved in iPrOH only absorbs UV light, Fig. S30, ESI†), hence the extra absorption may be due to charge transfer excitations within the cluster core (e.g. with O to Cu character) rather than surface effects. Such excitations would be akin to the band-gap transitions that are responsible for the black colour in bulk CuO. Therefore, it is intriguing to consider these well-defined ∼1.5 nm clusters, with condensed Cu + O cores, as a bridge between small Cu-carboxylate molecules and larger CuO@carboxylate nanostructures.
300 M−1 cm−1) with a shoulder (“dimer band”) arising from the bridging carboxylate coordination mode at slightly lower energy, and weaker d–d transitions (to dx2–y2) in the visible region (λmax = 615 nm; ε ∼ 300 M−1 cm−1) which cause the blue colour. Compound 6 is blue/green in solution and shows a similar LMCT peak λmax = 261 nm, and a shoulder which now extends into the visible region, possibly from added contributions of alkoxide to Cu LMCT,8 and a weaker d–d absorption per Cu (Fig. 4). Upon growth of the cluster to (green) Cu16 (2) and (yellow/green) Cu31 (4), the shoulder region becomes increasingly significant, extending into the visible region (and hence absorbing blue photons), and beginning to resemble the spectrum of ∼4.4 nm brown CuO@carboxylate particles (Fig. S31, ESI†).30 The carboxylate based LMCT peak remains in 2 and 4 with similar maxima (253 & 256 nm respectively) in the UV region, but with diminishing maximum extinction coefficient, when defined per copper, as the clusters grow (and the Cu:ligand ratio decreases, Fig. S32, ESI†). In 4 the shoulder region merges with the d–d excitations causing absorption across the visible region. The exact nature of excitations in the 350–550 nm region remain unknown and will be the focus of future study, but it is interesting to speculate that 4 contains the same surface ligands (pivalate and iPrOH) as 8 (+ two weakly bound nitrates in 4), yet absorbs broadly across the visible region (N.B. Cu(NO3)2 dissolved in iPrOH only absorbs UV light, Fig. S30, ESI†), hence the extra absorption may be due to charge transfer excitations within the cluster core (e.g. with O to Cu character) rather than surface effects. Such excitations would be akin to the band-gap transitions that are responsible for the black colour in bulk CuO. Therefore, it is intriguing to consider these well-defined ∼1.5 nm clusters, with condensed Cu + O cores, as a bridge between small Cu-carboxylate molecules and larger CuO@carboxylate nanostructures.
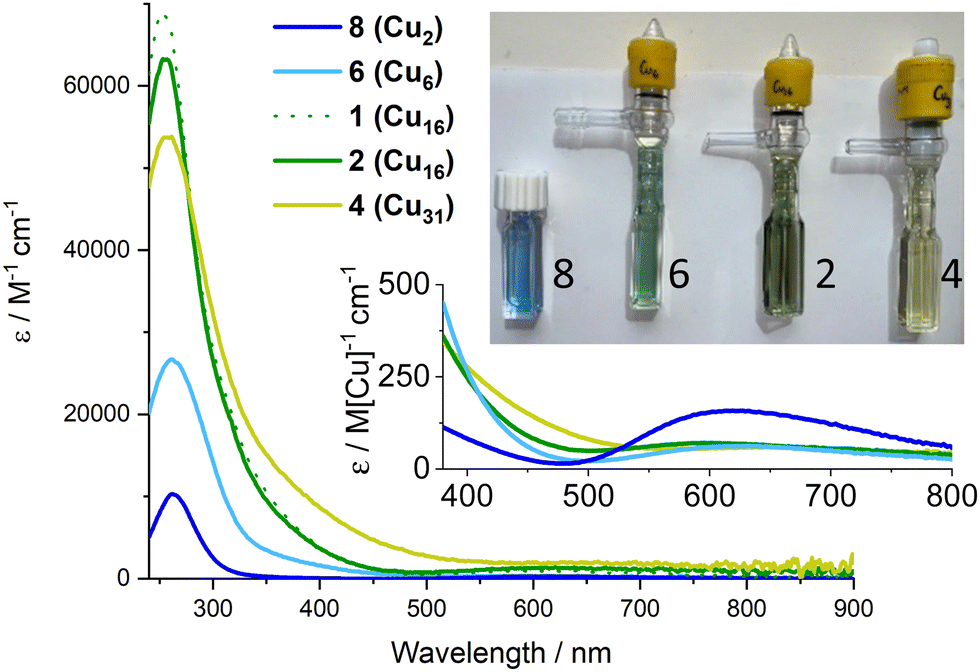 | ||
| Fig. 4 UV/vis spectra of 2, 4, 6 & 8 reported as molar extinction coefficients. Spectra recorded at 0.1–0.4 mM[Cu] in CH2Cl2 (or pentane for 2), inset shows magnified visible region as ‘per copper’ normalised extinction coefficients, spectra recorded at 1.7–3.3 mM[Cu]). Photos of solutions (with [Cu] = ∼0.24 mM). | ||
The size-colour correlation is consistent with the concept of quantum confinement, or considered from a molecular perspective, that as an increasing number of atomic orbitals closely interact the resulting HOMO–LUMO energy gap of the system becomes smaller and can absorb lower energies of light. This excitingly demonstrates the tuneability of the charge transfer absorption onset of Cu–oxo clusters across the visible region and the possibilities for manipulating frontier orbital energies and redox capability for useful photoredox reactivity.
In summary, new Cu–oxo clusters are reported, including Cu31 structures 4 and 5, which, to the best of our knowledge, are the largest and most condensed molecular Cu–oxo clusters discovered with only O-donor ligands. The high degree of condensation (0.68) and their size (1.5 nm Cu–O core) relates these well-defined clusters to CuO nanoparticles. Synthesis of these clusters operates via hydrolysis and condensation reactions, therefore, the amount of water influences products, and uncontrolled hydrolysis leads to CuO. By creating conditions which allow for a greater amount of hydrolysis to occur, i.e. maintaining cluster solubility with controlled water content, larger Cu–oxo clusters can be synthesised, and this will be explored in future studies. The colour of these clusters is influenced by their size, with the charge transfer absorption band becoming red-shifted on increasing size, consistent with concepts of quantum confinement and suggesting that Cu–oxo clusters and ultrasmall CuO nanoparticles have potential as highly tuneable systems for visible light driven photoreactivity.
The authors thank the Royal Society (URF\R1\191458), the Herchel Smith Fund and University of Warwick CDT in Analytical Science for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Russell-Webster, J. Lopez-Nieto, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2021, 60, 12591–12596 CrossRef CAS.
- S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
- M. Anjass, G. A. Lowe and C. Streb, Angew. Chem., Int. Ed., 2021, 60, 7522–7532 CrossRef CAS PubMed.
- D. S. Nesterov, J. Jezierska, O. V. Nesterova, A. J. L. Pombeiro and A. Ozarowski, Chem. Commun., 2014, 50, 3431–3434 RSC.
- Y. H. Lai, D. W. Palm and E. Reisner, Adv. Energy Mater., 2015, 5, 1501668 CrossRef.
- H. Lu, V. Andrei, K. J. Jenkinson, A. Regoutz, N. Li, C. E. Creissen, A. E. H. Wheatley, H. Hao, E. Reisner, D. S. Wright and S. D. Pike, Adv. Mater., 2018, 30, 1804033 CrossRef.
- T. Krämer, F. Tuna and S. D. Pike, Chem. Sci., 2019, 10, 6886–6898 RSC.
- K. S. Joya and H. J. M. de Groot, ACS Catal., 2016, 6, 1768–1771 CrossRef CAS.
- R. Siavash Moakhar, S. M. Hosseini-Hosseinabad, S. Masudy-Panah, A. Seza, M. Jalali, H. Fallah-Arani, F. Dabir, S. Gholipour, Y. Abdi, M. Bagheri-Hariri, N. Riahi-Noori, Y.-F. Lim, A. Hagfeldt and M. Saliba, Adv. Mater., 2021, 33, 2007285 CrossRef CAS PubMed.
- P.-C. Chuang and Y.-H. Lai, Catal. Sci. Technol., 2022, 12, 6375–6383 RSC.
- R. Poreddy, C. Engelbrekt and A. Riisager, Catal. Sci. Technol., 2015, 5, 2467–2477 RSC.
- P. Sawicka-Chudy, M. Sibiński, E. Rybak-Wilusz, M. Cholewa, G. Wisz and R. Yavorskyi, AIP Adv., 2020, 10, 010701 CrossRef CAS.
- B. K. Meyer, A. Polity, D. Reppin, M. Becker, P. Hering, P. J. Klar, T. Sander, C. Reindl, J. Benz, M. Eickhoff, C. Heiliger, M. Heinemann, J. Bläsing, A. Krost, S. Shokovets, C. Müller and C. Ronning, Phys. Status Solidi B, 2012, 249, 1487–1509 CrossRef CAS.
- Y. Wang, S. Lany, J. Ghanbaja, Y. Fagot-Revurat, Y. P. Chen, F. Soldera, D. Horwat, F. Mücklich and J. F. Pierson, Phys. Rev. B, 2016, 94, 245418 CrossRef.
- C. E. Ekuma, V. I. Anisimov, J. Moreno and M. Jarrell, Eur. Phys. J. B, 2014, 87, 23 CrossRef.
- B. Born, J. D. A. Krupa, S. Geoffroy-Gagnon, I. R. Hristovski, C. M. Collier and J. F. Holzman, ACS Photonics, 2016, 3, 2475–2481 CrossRef CAS.
- T.-F. Liu, T. C. Stamatatos, K. A. Abboud and G. Christou, Dalton Trans., 2010, 39, 3554–3556 RSC.
- C.-H. Chiang, Y.-W. Tzeng, C.-I. Yang, M. Nakano, W.-L. Wan, L.-L. Lai and G.-H. Lee, Dalton Trans., 2017, 46, 1237–1248 RSC.
- J. Chen, H. Zhou and F. Xu, Inorg. Chem., 2016, 55, 4695–4697 CrossRef CAS.
- M. Murugesu, R. Clérac, C. E. Anson and A. K. Powell, Inorg. Chem., 2004, 43, 7269–7271 CrossRef CAS PubMed.
- S.-M. Fang, Q. Zhang, M. Hu, E. C. Sañudo, M. Du and C.-S. Liu, Inorg. Chem., 2010, 49, 9617–9626 CrossRef CAS PubMed.
- V. Chandrasekhar, D. Sahoo, R. S. Narayanan, R. J. Butcher, F. Lloret and E. Pardo, Dalton Trans., 2013, 42, 8192–8196 RSC.
- Z. Chen, Y. Wang, L. Liu, Z. Zhang and F. Liang, Chem. Commun., 2012, 48, 11689–11691 RSC.
- M. Murugesu, R. Clérac, C. E. Anson and A. K. Powell, Chem. Commun., 2004, 1598–1599 RSC.
- I. A. Kühne, G. E. Kostakis, C. E. Anson and A. K. Powell, Chem. Commun., 2015, 51, 2702–2705 RSC.
- A. Kondinski and K. Y. Monakhov, Chem. – Eur. J., 2017, 23, 7841–7852 CrossRef CAS PubMed.
- F.-L. Liu, B. Kozlevčar, P. Strauch, G.-L. Zhuang, L.-Y. Guo, Z. Wang and D. Sun, Chem. – Eur. J., 2015, 21, 18847–18854 CrossRef CAS PubMed.
- S. Asbrink and A. Waskowska, J. Phys.: Condens. Matter, 1991, 3, 8173–8180 CrossRef.
- M. Estruga, A. Roig, C. Domingo and J. A. Ayllón, J. Nanopart. Res., 2012, 14, 1053 CrossRef.
- M. Masahiro, A. Hiroshi, N. Ryoji and H. Makoto, Chem. Lett., 1999, 57–58 Search PubMed.
- A previous report finds 6 as the product of the reaction of Cu(NO3)2·3H2O + pivalic acid + NEt3 in ethanol, whilst in our hands this reaction reproducibly forms 3 (see Dalton Trans., 2010, 39(15), 3554–3556).
- F. Tuczek and E. I. Solomon, Coord. Chem. Rev., 2001, 219–221, 1075–1112 CrossRef CAS.
- M. Alter, L. Binet, N. Touati, N. Lubin-Germain, A.-S. Le Hô, F. Mirambet and D. Gourier, Inorg. Chem., 2019, 58, 13115–13128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and synthesis procedures, diffraction and spectroscopy data. CCDC 2211399–2211403 and 2220157. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05478g |
| This journal is © The Royal Society of Chemistry 2023 |