
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn air-stable radical with a redox-chameleonic amide†
Jesse L.
Peltier
 a,
Melinda R.
Serrato
a,
Valentin
Thery
b,
Jacques
Pecaut
c,
Eder
Tomás-Mendivil
b,
Guy
Bertrand
a,
Rodolphe
Jazzar
a and
David
Martin
*b
a,
Melinda R.
Serrato
a,
Valentin
Thery
b,
Jacques
Pecaut
c,
Eder
Tomás-Mendivil
b,
Guy
Bertrand
a,
Rodolphe
Jazzar
a and
David
Martin
*b
aUCSD-CNRS Joint Research Chemistry Laboratory (IRL 3555), Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California 92093-0358, USA
bUniversity Grenoble Alpes, CNRS, DCM, Grenoble 38000, France. E-mail: david.martin@univ-grenoble-alpes.fr
cUniversity Grenoble Alpes, CEA, CNRS, INAC-SyMMES, UMR 5819, Grenoble 38000, France
First published on 13th December 2022
Abstract
An air-stable (amino)(amido)radical was synthesized by reacting a cyclic (alkyl)(amino)carbene with carbazoyl chloride, followed by one-electron reduction. We show that an adjacent radical center weakens the amide bond. It enables the amino group to act as a strong acceptor under steric contraint, thus enhancing the stabilizing capto-dative effect.
Glycyl radical enzymes are important biocatalysts that enable a variety of transformations; from the reduction of nucleotides to the breakdown of inactivated hydrocarbons.1 Their active resting state is generated by H atom abstraction at a glycine residue (Fig. 1a). The resulting C-radical A is highly sensitive to oxygen and the enzymatic processes work only under anaerobic conditions. Note that other reactive peptidyl radicals and related (amino)(amido) C-radicals B are rare in nature,1c,d but are commonly involved in synthetic radical peptidic chemistry.2
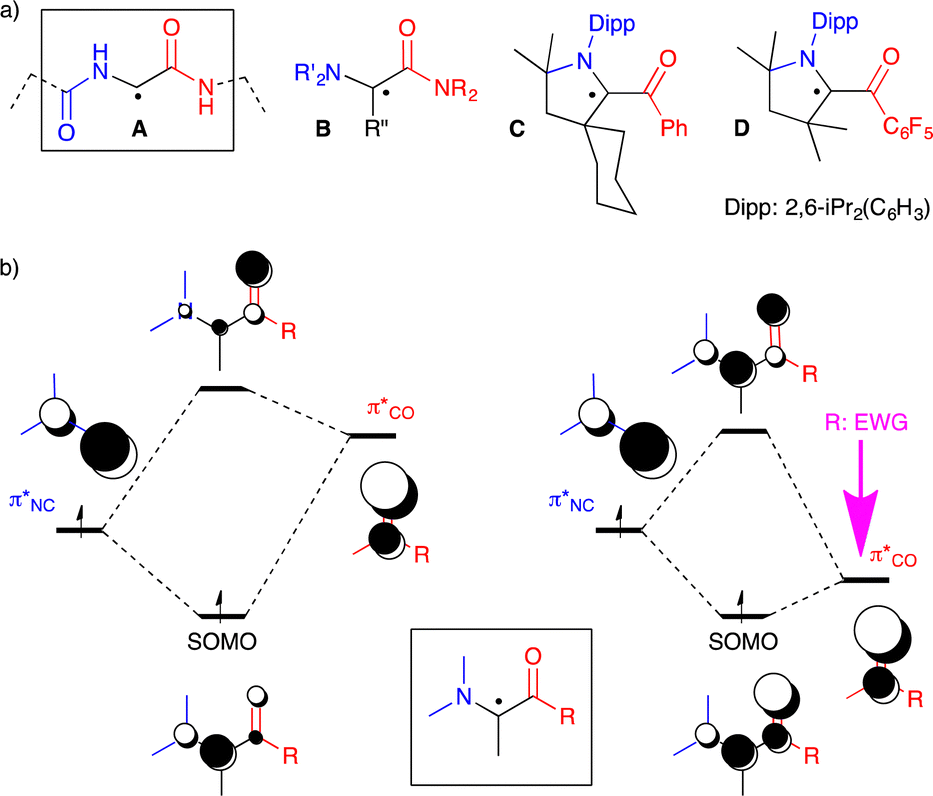 | ||
Fig. 1 (a) Glycyl radical pattern A in Enzymes, (amino)(amido) C-radical B, bottle-able push-pull C-radical C (air sensitive) and D (highly air-persistent); (b) schematic representations of SOMO of an (amino)(carbonyl) C-radical built from  and and 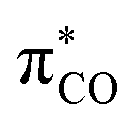 , left: “classical” case, right: R is an extreme electron-withdrawing group. , left: “classical” case, right: R is an extreme electron-withdrawing group. | ||
The persistence of the glycyl C-radical pattern in enzymes is usually attributed to the synergic combination of an electron-donating nitrogen (blue on Fig. 1) and an electron-withdrawing carbonyl group (red), a push-pull or captodative effect.3 The protein environment also precludes the formation of C–C dimers, which are usually obtained with simpler molecular models.3e–i In 2013, we took advantage of the bulky pattern of cyclic (alkyl)(amino)carbene (CAAC)4–6 to synthesize and isolate monomeric (amino)(carboxy) C-radical C under inert atmosphere.5a In addition, we showed that increasing the electron-withdrawing properties of the carbonyl substituent, such as in compound D, resulted in radicals with remarkable air-persistency.5d,7 A schematic molecular orbital analysis enables the rationalization of this effect. Indeed, the singly occupied molecular orbital (SOMO) is a bonding combination of 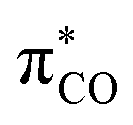 and
and  (Fig. 1b). An electron-withdrawing substituent on the carbonyl lowers the energy of the
(Fig. 1b). An electron-withdrawing substituent on the carbonyl lowers the energy of the  , thus increasing the weight of the CO fragment, which has major coefficient on oxygen. Therefore, the formal C-radical shifts to more of an O-centred radical, which is less reactive towards dioxygen.5d,8
, thus increasing the weight of the CO fragment, which has major coefficient on oxygen. Therefore, the formal C-radical shifts to more of an O-centred radical, which is less reactive towards dioxygen.5d,8
In this context, as illustrated by the high air-sensitivity of glycyl radical enzymes, amide patterns seem especially unfit for the design of bench-stable radicals; they are among both the poorest available N-donors and the weakest electron-withdrawing carbonyl groups. Herein, we challenge this paradigm and report an air-stable version of an amide-substituted captodative radical. We show that the adjacent radical centre weakens the amide bond and enables the N-group to act as a strong acceptor.
We initially considered a simple N,N′-dimethylamido group. The chloride salt of acylium 2a+ was synthesized by the addition of CAAC 1 to dimethylcarbamoyl chloride (Scheme 1). Cyclic voltammetry indicated two reversible reductions at −1.34 and −2.00 V (versus Fc/Fc+), corresponding to the formation of 2a˙ and the enolate 2a−, respectively (Fig. 2a). Radical 2a˙ was generated in situ by bulk electrolysis at −1.43 V. This highly air-sensitive radical was also synthesized by chemical reduction of acylium 2a+ with 0.5 equivalent of Zn(0) and isolated as a yellow solid in 88% yield. A single crystal X-ray diffraction study (Fig. 2b) revealed a dimethyl amino group with pronounced pyramidalization (sum of angles around N2: 331.6°). The lone pair of the amide nitrogen is not conjugated, but perpendicular to the carbonyl. As a result, the long C2–N2 distance (143.7 pm) is typical for a single bond and sharply contrasts with the usual bond length in planar acyclic amides (132–134 pm).9
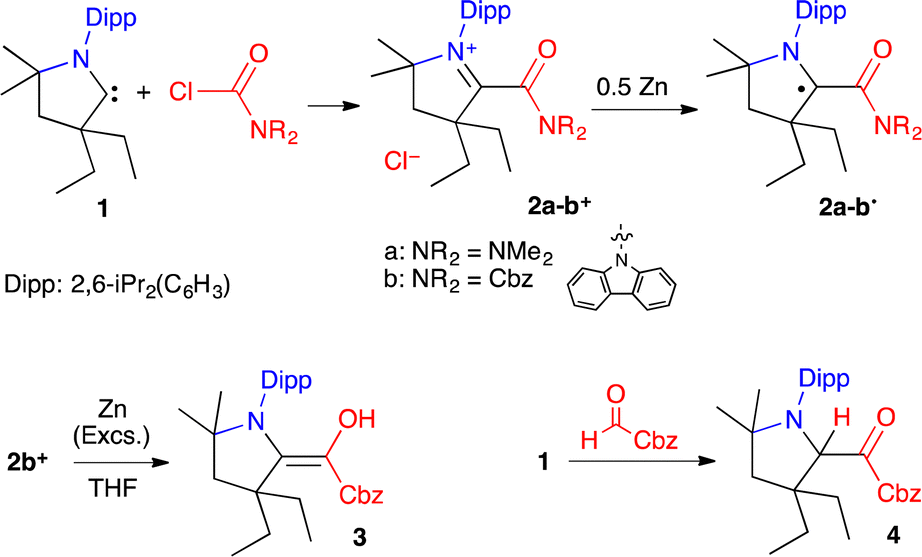 | ||
| Scheme 1 Synthesis of radicals 2a-b˙ and their derivatives. | ||
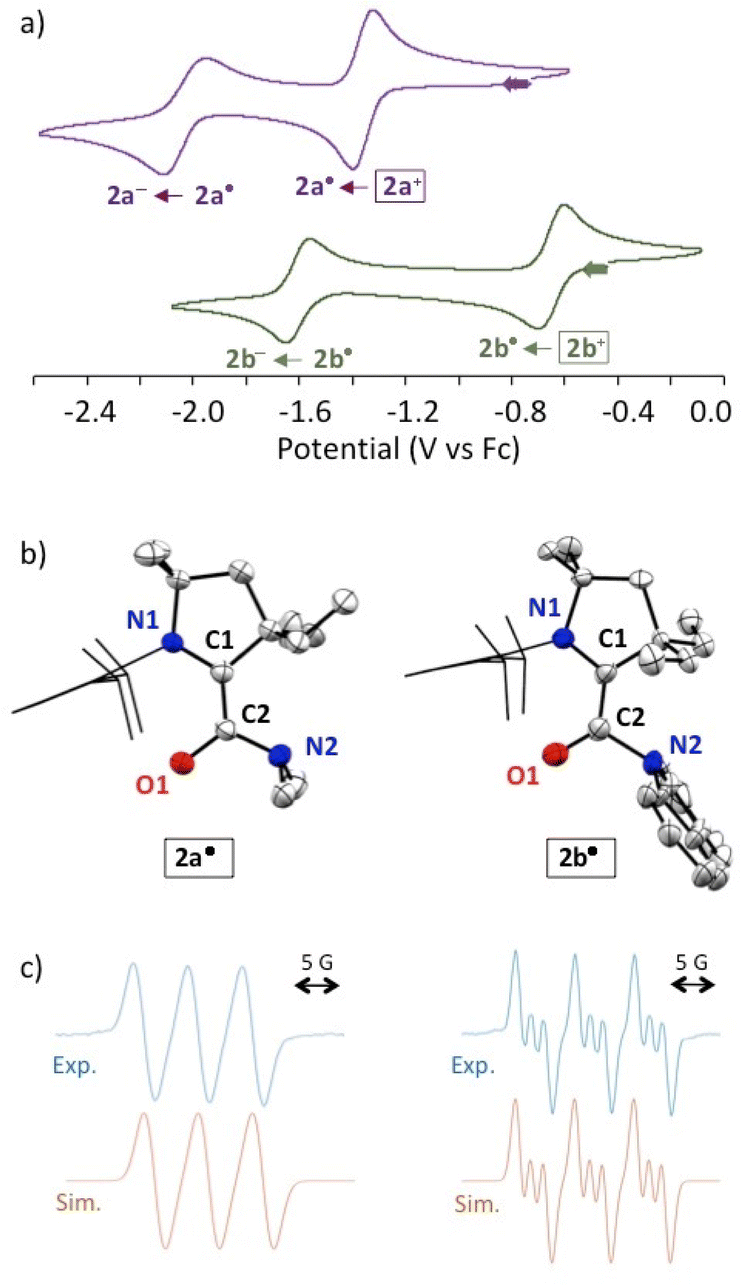 | ||
| Fig. 2 (a) Cyclic voltammograms of a 1 mM solution for both the chloride salt of 2a+ (top) and 2b+ (below) in 0.1 M nBu4NPF6 acetonitrile solution at 100 mV s−1 rates. (b) Solid state structures of radicals 2a˙ and 2b˙. Thermal ellipsoids are set to 50% probability. Molecules of solvent, hydrogen atoms and ellipsoids on 2,6-diisopropylphenyl groups are omitted for clarity. (c) top: X-band EPR spectra of 2a˙ (left) and 2b˙ (right) in acetonitrile at room temperature; below: corresponding simulated spectra with the following set of parameters: 2a˙, Lorentzian line-broadening parameter Lw = 0.264 and hyperfine coupling constant a(14N) = 15.8 MHz (1 nucleus); 2b˙, Lw = 0.143, a(14N) = 18.3 MHz (1 nucleus) and 4.0 MHz (1 nucleus). | ||
Acyclic twisted amide patterns usually require the deactivation of the nitrogen with an ancillary electron-withdrawing substituent or the incorporation into an aromatic ring.10,11 The local environment of N2 is more reminiscent of “anti-Bredt” amides or ureas, which feature a polycyclic saturated backbone with a bridgehead nitrogen.12,13 These compounds are not stable when there is a significant twisting around the (OC)–N bond, as they feature both an activated electrophilic carbonyl and a nucleophilic nitrogen centre. In radical 2a˙, the twist of the N,N′-di(methyl)amino group is maximal; however the amine acts as a strong electron-withdrawing group, which is a favourable electronic situation for a push-pull radical.5
We turned to a carbazole substituent to increase the electron-withdrawing capability of the carbonyl moiety. We synthesized acylium 2b+ (Scheme 1). Cyclic voltammetry featured two reversible processes at −0.63 and −1.59 V, which are significantly more positive values than in the case of 2a+ (Fig. 2). Radical 2b˙ was generated in situ by bulk electrolysis at −0.78 V. The radical was also synthesized by chemical reduction of acylium 2b+ with 0.5 equivalent of Zn(0) and isolated as a colourless solid in 84% yield. Of note, attempts to further reduce the radical with one equivalent of Zn(0) lead after work-up to the isolation of few crystals of the corresponding enaminol 3 (Scheme 1), which was characterized by X-ray diffraction (see ESI†). As in 2b˙, the carbazole is orthogonal to the carbonyl. This is in line with a previous study by Berkessel et al., which shows that strong electron-withdrawing groups stabilize Breslow-type enols.14 Interestingly, we were also able to isolate the corresponding keto tautomer 4 from the reaction of CAAC with N-formyl carbazole.15
As for 2a˙, a single crystal X-ray diffraction study of 2b˙ revealed a pyramidalized N2 centre (sum of angles around N2: 330.7°), a formal lone pair perpendicular to the carbonyl and a long C2–N2 distance (143.3 pm).16 Importantly, in marked contrast with sensitive radical 2a˙, 2b˙ is remarkably robust towards air in the solid state and in toluene. The observation of a fast decay by EPR monitoring required heating an aerated solution in ethanol at 60 °C.
DFT17 optimized structures of 2a-b˙ at the b3lyp/6-311g(d,p) level of theory matched the experimental solid-state geometries, as well as the EPR isotropic hyperfine coupling constants,18 (Fig. 2c; 2a˙, computed a(14N): 14 MHz, experimental: 15.8 MHz; 2b˙, computed a(14N): 16 and 3 MHz, experimental: 18.3 and 4.0 MHz). The distribution of the Mulliken spin density (see also the representation of SOMO in Fig. 3a) is similar for both radicals (2a˙: N1: 25%, C1: 41%, C2: 7%, O1: 26%; 2b˙: N1: 25%, C1: 37%, C2: 7%, O1: 30%). These values are reminiscent of the spin distribution of highly air persistent radical D, featuring a perfluorophenyl in place of the twisted amino groups. This suggests that the O-centred character of 2a-b˙ was sufficient to disfavour triplet oxygen addition at the C1 atom.5d,8 Accordingly, this reaction is predicted to be endergonic for 2a-b˙ by ΔG = +10.2 and +21.2 kcal mol−1, respectively. Thus, we considered that a single electron transfer to dioxygen was a more plausible initiation step for the pathway of decay of 2a˙ in the presence of air. Indeed, radical 2a˙ stands out with a very low oxidation potential (−1.34 V) when compared to previously reported CAAC-based (amino)(carboxy)radicals (from −0.2 V to −0.9 V).5 Note that the computed ionization potential fits well with values for parented radicals (2a˙: 5.1, 2b˙: 5.4, C: 5.1 and D: 5.5 eV). However, the conformational relaxation of 2a+, which follows the vertical ionization of 2a˙, is especially exothermic (2a: −28, 2b: −19, C: −19 and D: −15 kcal mol−1). Therefore, we concluded that the low oxidation potential of 2a˙ was also due to the singular stability of 2a+ compared to other acyliums of the series. Indeed, the di(methyl)amino group has a chameleonic behaviour: it is twisted and acts as a −I attractor in radical 2a˙, but it is a fully conjugated strong +M donor (stronger than the aromatic carbazole of 2b+) in acylium 2a+.
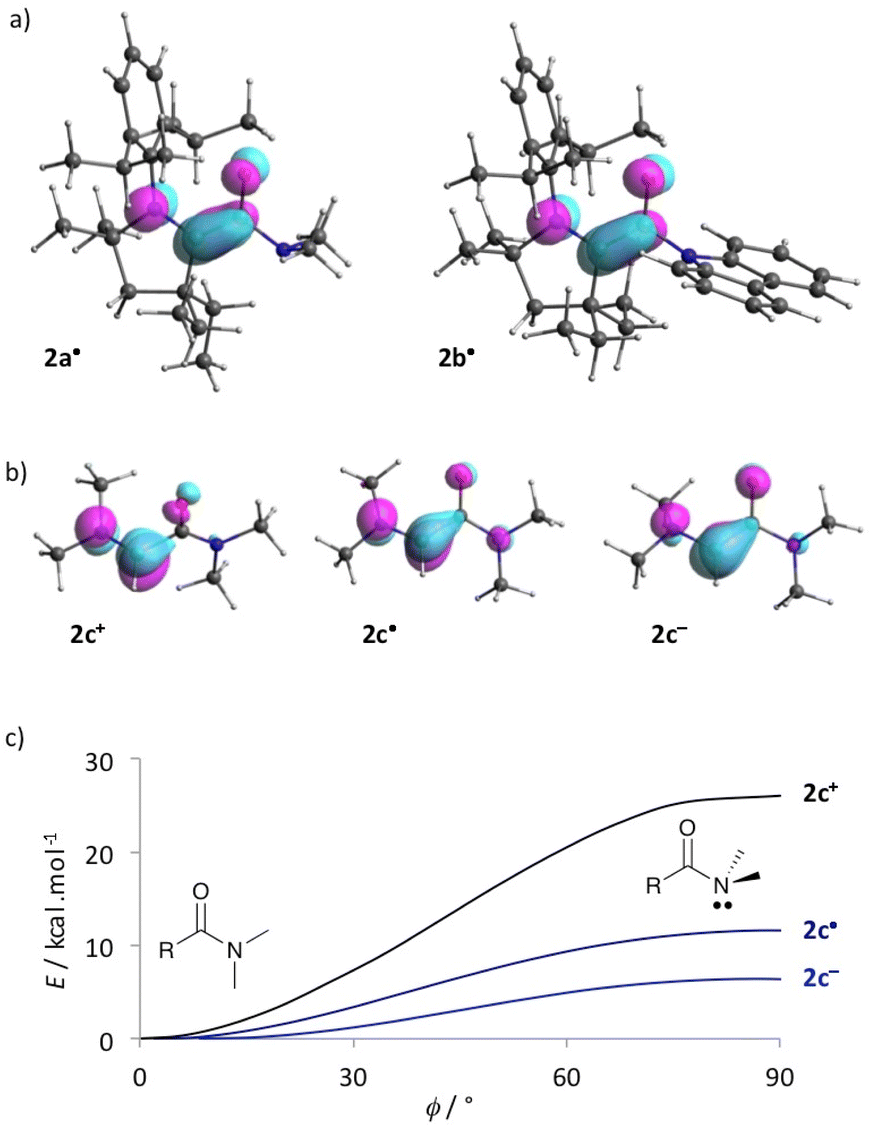 | ||
| Fig. 3 (a) Optimized DFT geometry of 2a-b˙ with representations of corresponding SOMO. (b) Optimized DFT geometry of model 2c+, 2c˙ and 2c− with representation of corresponding LUMO, SOMO and HOMO, respectively. (c) Energy in relaxed scan optimization of 2c+, 2c˙ and 2c− as a function of ϕ, the torsion angle between the formal N lone pair and the πCO molecular orbital. | ||
To get further insights, we considered simplified acylium, radical and enolate, 2c+, 2c˙ and 2c− respectively, which feature a dimethylaminocarbene in place of the bulky CAAC pattern. Note that in acyliums 2a–c+ the iminium moieties are perpendicular to the carbonyl, whereas the N–C–CO pattern is fully conjugated in radicals 2a–c˙ and enolates 2a–c−. Interestingly, the small model compound 2c˙ differs from CAAC-based radicals 2a–b˙ with a fully conjugated amide moiety and only a slight pyramidalization at the nitrogen is found in 2c−; the conformations of 2c+, 2c˙ and 2c− with formal N2 nitrogen lone pair perpendicular to the carbonyl are transition states (Fig. 3b). However, introducing a radical or an anion in α position of the carbonyl significantly weakens the amide bond. Indeed, the formal one electron reduction to afford 2c˙ (respectively 2c−) consists in populating the LUMO of 2c+ (SOMO of 2c˙, respectively) with anti-bonding character between C2 and N2. Accordingly, the energy barrier for full twisting dramatically decreases from 2c+ (ΔG≠ = +26.2 kcal mol−1) to 2c˙ (+7.1 kcal mol−1) and 2c− (+6.7 kcal mol−1).
Amido groups have been classified as latent rotational stereoelectronic chameleons by Alabugin et al.19 Misalignment of the nitrogen lone pair with the carbonyl usually requires polycyclic structures or high steric strain; however, the enhanced flexibility of an amide bond that results from an adjacent radical centre has gone unnoticed to date. Beyond implications for the design of bench-stable organic radicals, it is likely that natural evolution has already taken advantage of such redox-chameleonic behaviour.20 This effect should not be overlooked in future studies on glycyl enzymes or peptidyl radical chemistry.
This work was supported by the French-American cultural ex-change foundation (FACE), the NSF (CHE-1954380) and the French National Agency for Research (ANR-14-CE06-0013-01 and CBH-EUR-GS, ANR-17-EURE-0003). Thanks are due to the CECCIC and the ICMG platforms of Grenoble.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705–762 CrossRef CAS PubMed; (b) L. R. F. Backman, M. A. Funk, C. D. Dawson and C. L. Drennan, Crit. Rev. Biochem. Mol. Biol., 2017, 52, 674–695 CrossRef CAS PubMed; (c) J. Hioe, G. Savasci, H. Brand and H. Zipse, Chem. – Eur. J., 2011, 17, 3781–3789 CrossRef CAS PubMed; (d) J. Hioe and H. Zipse, Faraday Discuss., 2010, 145, 301–313 RSC; (e) W. Buckel and B. T. Golding, Annu. Rev. Microbiol., 2006, 60, 27–49 CrossRef CAS PubMed.
- (a) C. J. Easton, Chem. Rev., 1997, 97, 53–82 CrossRef CAS PubMed; (b) R. Andrukiewicz, R. Loska, V. Prisyahnyuk and K. Stalinski, J. Org. Chem., 2003, 68, 1552–1554 CrossRef CAS PubMed; (c) P. Renaud and L. Giraud, Synthesis, 1996, 913–926 CrossRef CAS; (d) C. Wyss, R. Batra, C. Lehmann, S. Sauer and B. Giese, Angew. Chem., Int. Ed. Engl., 1996, 35, 2529–2531 CrossRef CAS; (e) S. Sauer, C. Staehelin, C. Wyss and B. Giese, Chimia, 1997, 51, 23–24 CrossRef CAS; (f) A. Schneiker, S. Góbi, P. R. Joshi, G. Bazsó, Y.-P. Lee and G. Tarczay, J. Phys. Chem. Lett., 2021, 12, 6744–6751 CrossRef CAS PubMed; (g) F. J. A. Troyano, K. Merkens, K. Anwar and D. A. Gomez-Suarez, Angew. Chem., Int. Ed., 2021, 60, 1098–1115 CrossRef PubMed; (h) N. Venugopal, J. Moser, M. Vojtíčková, I. Císařová, B. König and U. Jahn, Adv. Synth. Catal., 2022, 364, 405–412 CrossRef CAS.
- (a) R. W. Baldock, P. Hudson and A. R. Katritzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1422–1427 RSC; (b) A. T. Balaban, M. T. Caproiu, N. Negoita and R. Baican, Tetrahedron, 1977, 33, 2249–2253 CrossRef CAS; (c) H. G. Viehe, E. Mértnyi, L. Stella and Z. Janousek, Angew. Chem., Int. Ed. Engl., 1979, 18, 917–932 CrossRef; (d) H. G. Viehe, Z. Janousek, R. Mértnyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 CrossRef CAS; (e) T. H. Koch, J. A. Olesen and J. DeNiro, J. Am. Chem. Soc., 1975, 97, 7285–7288 CrossRef CAS; (f) R. J. Himmelsbach, A. D. Barone, D. L. Kleyer and T. H. Koch, J. Org. Chem., 1983, 48, 2989–2994 CrossRef CAS; (g) G. Gaudiano, K. Sweeney, R. C. Haltiwanger and T. H. Koch, J. Am. Chem. Soc., 1984, 106, 7628–7629 CrossRef CAS; (h) O. Benson Jr., S. H. Demirdji, R. C. Haltiwanger and T. H. Koch, J. Am. Chem. Soc., 1991, 113, 8879–8886 CrossRef; (i) O. Benson Jr., G. Gaudiano, R. C. Haltiwanger and T. H. Koch, J. Org. Chem., 1988, 53, 3036–3045 CrossRef.
- (a) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (d) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (e) M. Melaimi, R. Jazzar, M. Soleihavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (f) S. K. Kushvaha, A. Mishra, H. W. Roesky and K. C. Mondal, Chem. – Asian J., 2022, 17, e202101301 Search PubMed; (g) H. Kimm and E. Lee, Bull. Korean Chem. Soc., 2022 DOI:10.1002/bkcs.12620.
- CAAC-based organic radicals: (a) J. K. Mahoney, D. Martin, C. Moore, A. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed; (b) L. Jin, M. Melaimi, L. L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC; (c) Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 CrossRef CAS PubMed; (d) J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (e) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (f) D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed; (g) J. K. Mahoney, R. Jazzar, G. Royal, D. Martin and G. Bertrand, Chem. – Eur. J., 2017, 23, 6206–6212 CrossRef CAS PubMed; (h) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (i) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed; (j) P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef PubMed; (k) P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 CrossRef CAS PubMed; (l) V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109–1117 CrossRef CAS PubMed; (m) J. Messelberger, A. Grünwald, S. J. Goodner, F. Zeilinger, P. Pinter, M. E. Miehlich, F. W. Heinemann, M. M. Hansmann and D. Munz, Chem. Sci., 2020, 11, 4138–4149 RSC.
- For stable organic radicals based on other types of N-heterocyclic carbenes, see also: (a) J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS PubMed; (b) C. L. Deardorff, R. E. Sikma, C. P. Rhodes and T. W. Hudnall, Chem. Commun., 2016, 52, 9024–9027 RSC; (c) L. Y. M. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Zikhovic, H. M. Ronnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS PubMed; (d) D. Rottschafer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef PubMed; (e) N. M. Gallagher, H.-Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS PubMed; (f) Y. Kim, J. E. Byeon, G. Y. Jeong, S. S. Kim, H. Song and E. Lee, J. Am. Chem. Soc., 2021, 143(23), 8527–8532 CrossRef CAS PubMed; (g) J. Zhao, X. Li and Y.-F. Han, J. Am. Chem. Soc., 2021, 143, 14428–14432 CrossRef CAS PubMed; (h) L. Delfau, S. Nichilo, F. Molton, J. Broggi, E. Tomás-Mendivil and D. Martin, Angew. Chem., Int. Ed., 2021, 60, 26783–26789 CrossRef CAS PubMed; (i) Y. Kim and E. Lee, Chem. – Eur. J., 2018, 24, 19110–19121 CrossRef CAS PubMed.
- Stabilized oxyallyl radical cations are also parented: (a) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014–7017 CrossRef CAS PubMed; (b) T. Schulz, C. Farber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC; (c) V. Regnier and D. Martin, Org. Chem. Front., 2015, 2, 1536–1545 RSC; (d) V. Regnier, F. Molton, C. Philouze and D. Martin, Chem. Commun., 2016, 52, 11422–11425 RSC; (e) M. Devillard, V. Regnier, M. Tripathi and D. Martin, J. Mol. Struct., 2018, 1172, 3–7 CrossRef CAS; (f) M. Tripathi, V. Regnier, Z. Ziani, M. Devillard, C. Philouze and D. Martin, RSC Adv., 2018, 8, 38346–38350 RSC; (g) E. Tomás-Mendivil, M. Devillard, V. Regnier, J. Pecaut and D. Martin, Angew. Chem., Int. Ed., 2020, 59, 11516–11520 CrossRef PubMed.
- The low reactivity of O-centered radicals with dioxygen is usually paralleled with the extreme weakness of O–O bonds. For a discussion, see: (a) R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC; (b) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- G. Meng, J. Zhang and M. Szostak, Chem. Rev., 2021, 121, 12746–12783 CrossRef CAS PubMed.
- For a unique case of twisted N-alkyl amide, in the solid-state structure of dimeric assemblies, see: S. E. Snyder, B.-S. Huang, Y. W. Chu, H.-S. Lin and J. R. Carey, Chem. – Eur. J., 2012, 18, 12663–12671 CrossRef CAS PubMed.
- H. K. Hall and A. El-Shekeil, Chem. Rev., 1983, 83, 549–555 CrossRef CAS.
- (a) K. Tani and B. M. Stoltz, Nature, 2006, 441, 731–734 CrossRef CAS PubMed; (b) J. Clayden and W. J. Moran, Angew. Chem., Int. Ed., 2006, 45, 7118–7120 CrossRef CAS PubMed; (c) T. Ly, M. Krout, D. K. Pham, K. Tani, B. M. Stoltz and R. R. Julian, J. Am. Chem. Soc., 2007, 129, 1864–1865 CrossRef CAS PubMed.
- M. Paul, J.-M. Neudörfl and A. Berkessel, Angew. Chem., Int. Ed., 2019, 58, 10596–10600 CrossRef CAS PubMed.
- (a) D. Martin, V. Lavallo, Y. Canac and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 5023–5030 CrossRef CAS PubMed; (b) M. Paul, M. Breugst, J.-M. Neudorfl, R. B. Sunoj and A. Berkessel, J. Am. Chem. Soc., 2016, 138, 5044–5051 CrossRef CAS PubMed.
- CCDC 2191398 and 2191542–2191545 contain the supplementary crystallographic data for this paper†.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Calculations were performed with the Gaussian suite of programs: Gaussian09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009. See ESI for complete citation Search PubMed.
- Experimental values for isotropic hyperfine constants were extracted from EPR spectra by fitting with the EasySpin simulation package: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- S. Z. Vatsadze, Y. D. Loginova, G. dos Passos Gomes and I. V. Alabugin, Chem. – Eur. J., 2017, 23, 3225–3245 CrossRef CAS PubMed.
- O. A. Levitskiy, A. V. Bogdanov, I. A. Klimchuk and T. V. Magdesieva, Chem. – Eur. J., 2020, 26, 6793–6804 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical and computational data. CCDC 2191398 and 2191542–2191545. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc05404c |
| This journal is © The Royal Society of Chemistry 2023 |