
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure-sensitive epoxidation of dicyclopentadiene over TiO2 catalysts†
Sang-Ho
Chung‡
 a,
G. Hwan
Park‡
bc,
Niels
Schukkink
a,
Hyoyoung
Lee
*bc and
N. Raveendran
Shiju
*a
a,
G. Hwan
Park‡
bc,
Niels
Schukkink
a,
Hyoyoung
Lee
*bc and
N. Raveendran
Shiju
*a
aVan’t Hoff Institute for Molecular Sciences, University of Amsterdam, P.O. Box 94157, 1090 GD Amsterdam, The Netherlands. E-mail: n.r.shiju@uva.nl
bCenter for Integrated Nanostructure Physics, Institute for Basic Science, Sungkyunkwan University, Suwon 440-746, South Korea. E-mail: hyoyoung@skku.edu
cDepartment of Chemistry, Sungkyunkwan University, Suwon 440-746, South Korea
First published on 19th December 2022
Abstract
Epoxidation of dicyclopentadiene (DCPD) is studied on a series of TiO2 catalysts using hydrogen peroxide as an oxidant. DCPD derivatives have applications in several areas including polymer, pharmaceutical and pesticide products. The control of selectivity leading to the desired product is important for many of these applications. Using experimental and computational studies, we show that the surface crystalline phases of TiO2 play crucial roles not only in the formation of peroxo species but also in the selective epoxidation of two different C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in DCPD.
C double bonds in DCPD.
Given the versatility of the two different double bonds in its chemical structure, dicyclopentadiene (DCPD) is one of the most interesting cyclic olefin compounds. Numerous DCPD derivatives can be found in the pharmaceutical, pesticide, and polymer industries.1 Amongst the DCPD derivatives, DCPD epoxides find their main uses in adhesives and insulation materials.2
Some heterogeneous catalysts have been studied for DCPD epoxidation, such as H3PW12O40 on SBA-15 and on chloromethylated polystyrene resin,3,4 and the dispersion of phosphotungstic acid is responsible for the catalytic performance.4 Metal complexes intercalated in Zn/Al layered double hydroxide structures (Sulfonate-salen-MIII, M = Mn or Fe) showed higher activity than Fe.5 Besides the catalytic activity (conversion rate of DCPD), product selectivity is another crucial factor in the epoxidation of DCPD. The epoxidation of DCPD yields two different mono-epoxides (endo-4-oxatetracyclo-[6.2.1.0.2,603,5]undec-9-ene (P1) and endo-9-oxatetracyclo-[5.3.1.0.2,608,10]undec-3-ene (P2)), depending on the location of the epoxide group (in the cyclopentene ring or in the norbornene ring, respectively) (Fig. 1(a)). Due to the difficulty in the product separation unit, the development of a selective epoxidation of DCPD has been encouraged,6 but the product selectivity typically did not rely on the chemical properties of the metal center.3–5
 | ||
| Fig. 1 (a) Reaction scheme of DCPD epoxidation using H2O2 as an oxidant and reaction products P1 and P2. Catalytic performances of different TiO2 catalysts in the epoxidation of DCPD: (b) TiO2-anatase and TiO2-rutile; (c) TiO2-blue and TiO2-black. | ||
TiO2 has been widely used as a catalyst for various reactions such as photocatalysis,7 CO oxidation,8 and H2O2 decomposition.9 In particular, the crystallinity of the TiO2 catalysts (anatase and rutile) is responsible for their geometric and electronic properties10 and plays key roles in view of the product selectivities in the reactions of the decomposition of hydrogen sulphide11 and the photo-oxidation of water.12
Our preliminary results demonstrated that the molecular oxygen (as well as the dissolved oxygen in the liquid phase) could not activate the double bonds of DCPD in methanol at 333 K. For example, insignificant conversion of DCPD (<1%) was obtained at 9 bar of pure oxygen gas in an autoclave. Thus, we studied selective DCPD epoxidation on a series of TiO2 catalysts using hydrogen peroxide (H2O2) as an oxidant. Indeed, the epoxidation of cyclic olefin can proceed with H2O2 in two sequential steps: (i) the formation of peroxo-species on the metal sites and (ii) the oxygen transfer from the surface to an olefin to form an epoxide.13
Fig. 1(b) displays the DCPD epoxidation results of two different crystalline phases of TiO2. TiO2-anatase effectively converted DCPD into the related mono-epoxides, showing ca. 2 times higher activity than TiO2-rutile (CDCPD = 13% and 7% in 6 h, respectively). The selectivity of the epoxidation products (towards P1 and P2) is greatly influenced by the crystalline phase of the TiO2 catalysts. For example, on TiO2-anatase, both P1 and P2 were produced with higher selectivity toward P1 (Table 1). This indicates that the double bond in the cyclopentene ring is preferably reacted on the TiO2 anatase phase, which is in line with the previous results over the Ti-incorporated SBA-15 catalyst.14,15 The DCPD di-epoxide was not observed in this study, suggesting that the epoxidation sites were utilized for the reactant (DCPD) and not occupied by the mono-epoxides, similar to the results of Bhattacharjee et al.5 Interestingly, despite its lower catalytic performance, TiO2-rutile solely produced P2, i.e., the double bond in the norbornene moiety is selectively reacted to form mono-epoxide.
| Catalyst | BET surface area (m2 g−1) | Average pore size (nm) | Normalized activitya (molDCPD m−2 h−1) | S P1/P2 |
|---|---|---|---|---|
| a For TiO2-anatase and TiO2-rutile, the normalized activity was calculated in 6 h of reaction. For TiO2-blue and TiO2-black, the activity values were calculated in 8 h, which is ca. 6 h from the point of observed conversion. | ||||
| TiO2-anatase | 60.8 | 21.8 | 28 | 1.7 |
| TiO2-rutile | 2.4 | 16.8 | 138 | 0 |
| TiO2-blue | 58.6 | 29.2 | 37 | 1.1 |
| TiO2-black | 2.6 | 16.3 | 350 | 0 |
We further prepared two additional TiO2 catalysts (TiO2-blue and TiO2-black),16–18 and the colors of the catalysts are attributed to the oxygen vacancies, based on the crystalline phases (anatase and rutile) (Fig. S1, ESI†).19 At the initial reaction stage (until ca. 2 h), no conversion of DCPD was observed over TiO2-blue and TiO2-black. This suggests that (i) DCPD is not reactive with H2O2 in solution and (ii) a certain delay (or lag-phase) is necessary to initiate DCPD epoxidation over the catalysts with oxygen vacancies. Since the textural properties of TiO2-blue and TiO2-black are similar to those of TiO2-anatase and TiO2-rutile, respectively (Table 1 and Fig. S1, S2, ESI,† as also reported by previous works16–18,20), we expected that the observed lag-phase is related to the surface oxygen vacancies, which might need to be modified by the oxygen from H2O2. In terms of the surface area normalized activity (Table 1), TiO2-blue and TiO2-black showed superior catalytic performance compared to TiO2-anatase and TiO2-rutile. We expect that the few nanometer layers of the disordered TiO2 surface21 can be attributed to the enhanced epoxidation performance. Similarly, the formation of reactive oxygen species is preferred on the amorphous ZrO2 than the crystalline monoclinic-ZrO2.22 Amorphous Nb2O5 and Ta2O5 also showed higher performance in the catalytic oxidation of glycerol and cyclohexene than their crystalline forms.23,24 For TiO2-blue, both P1 and P2 were produced with higher selectivity to P1 than P2. We suggest that the newly formed oxygen functionalities are highly reactive to convert the double bond in the norbornene ring as well as the one in the cyclopentene ring, similar to the fact that the oxygen vacancies are known to be responsible for the altered activity in oxidation reactions.25 Meanwhile, only P2 was observed on TiO2-black, indicating that the selectivity towards P1 or P2 is largely dependent on the surface crystalline phase.
After the epoxidation of DCPD, the color of the spent TiO2-anatase catalyst was changed from white to yellow, indicating the formation of the additional surface oxygen functional groups on titania.26,27 To identify the oxygen functionalities responsible for the catalytic activity, we characterized TiO2 catalysts with Raman spectroscopy (Fig. 2). For TiO2-anatase and TiO2-blue, the Raman features at 148, 395, 515 and 630 cm−1 are attributed to the anatase phase (the modes of Eg, B1g, A1g or B1g, and Eg, respectively).28 Meanwhile, the five Raman bands are observed for TiO2-rutile and TiO2-black at 140, 235, 445, 610 and 825 cm−1, due to the modes of B1g, multi-phonon process, Eg, A1g and B2g, respectively. After the treatment of TiO2 with H2O2, the formation of oxo species on TiO2 (yellow coloration of TiO2) by H2O2 was observed with the possibility of three different forms on the Ti metal centers (oxo, peroxo, and superoxo species).29 The O–O stretching frequencies are typically observed between 800 and 930 cm−1, depending on the coordination with the environment.30 For example, the Raman band of H2O2 is typically positioned at 880 cm−1.31 On TiO2-anatase, the Raman bands of peroxo species were observed at 871 cm−1, indicating that the peroxo species are coordinated to the Ti sites (Fig. 3(a)).32 On the contrary, for TiO2-blue, the Raman band of peroxo species was observed at a higher wavenumber (910 cm−1) (Fig. 2(b)), related to the perturbation of the chemical structure of the adsorbed molecule on the catalyst surface.33 For the rutile phase TiO2 catalysts (e.g., TiO2-rutile and TiO2-black), however, no additional Raman bands are observed after treatment with H2O2 (Fig. 2(b) and (d)), possibly due to (i) the low surface areas of the rutile phase TiO2 catalysts and (ii) the short lifetime of peroxo intermediates on the rutile phase catalysts.34
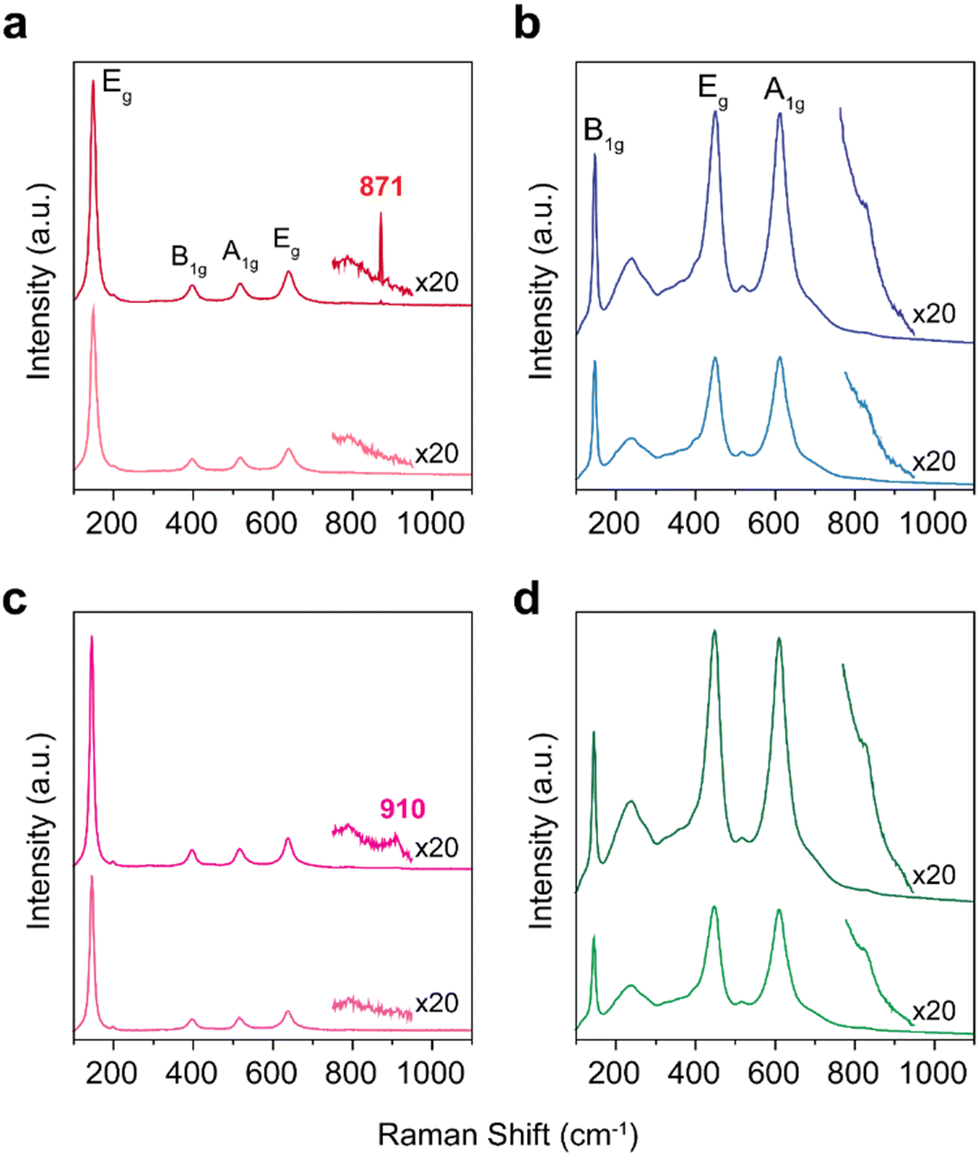 | ||
| Fig. 2 Raman spectra of TiO2 catalysts. (a) TiO2-anatase, (b) TiO2-rutile, (c) TiO2-blue and (d) TiO2-black. In each figure, the Raman spectra of H2O2 treated samples are displayed on top of the spectra of the untreated samples. | ||
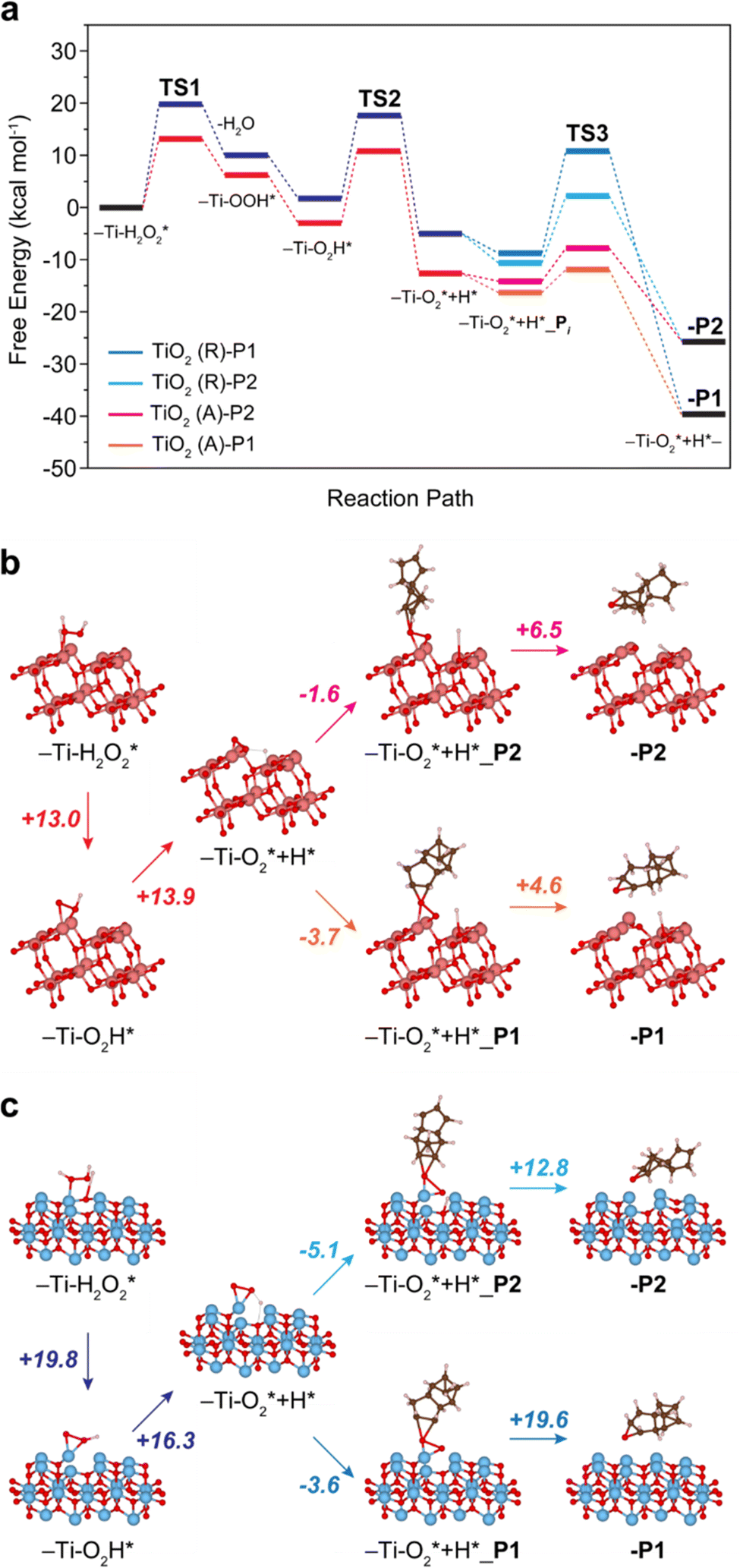 | ||
| Fig. 3 DFT-calculated free-energy profile (kcal mol−1) for the DCPD epoxidation with H2O2 on TiO2-anatase (b, orange, magenta) and TiO2-rutile (c, navy, blue). The free energies are calculated based on the models in Fig. S5 and S6 (ESI†). The numbers in Fig. 4b and c denote the energy barriers. Coordinates of -P2 and -P1 in (b) and (c) are provided in Tables S2–S5, ESI† as representative structures. | ||
We have explored the reaction pathway of DCPD epoxidation and the selectivity differences among the crystalline TiO2 phases using density functional theory (DFT) calculation (Fig. S3 and S4, ESI†). Fig. 3(a) shows the calculated free energy profiles for DCPD epoxidation with H2O2 catalyzed by the TiO2-anatase and TiO2-rutile. Raman spectroscopy indicates that peroxo species are formed on the surface of TiO2, and the reaction starts from the TiO2 surface with adsorbed H2O2 (Ti–H2O2*). Formation of the surface peroxo species (Ti–O2*) proceeds via the generation of hydroperoxo compounds. Once the surface of TiO2 adsorbed H2O2, it firstly generates η1-coordinated Ti-hydroperoxo compounds, Ti-η1(OOH), following the release of a water molecule for TiO2-anatase and TiO2-rutile, which requires +13.0 (Fig. 3(b)) and +19.8 kcal mol−1 (Fig. 3(c)) at Transition State 1 (TS1), respectively. Subsequently, the thermodynamically more stable η2-coordinated Ti-hydroperoxo compound (Ti-η2(OOH)) is formed. The protonated Ti-peroxo species (Ti–O2*) can be formed from Ti-η2(OOH) via hydrogen transfer to the adjacent Ti atom overcoming moderate energy barriers of +13.9 (Fig. 3(b)) and +16.3 kcal mol−1 (Fig. 3(c)) at TS2 (Fig. 3(a)), respectively. Along the overall reaction steps, TiO2-anatase (Fig. 3(a), red color) has lower free energies than TiO2-rutile (Fig. 3(a), blue color), indicating a higher DCPD conversion rate of TiO2-anatase, which is in good agreement with our experimental results (Fig. 1(b)).
The two different CC bonds in DCPD (cyclopentene or norbornene moiety) have different reactivity to TiO2 surface structures. Thus, the selectivity towards P1 or P2 strongly depends on the direction of oxygen transfer from Ti-peroxo (Ti–O2*). Adsorption of the DCPD molecule on the surface of TiO2-rutile is exothermic for both cyclopentene and norbornene with a very small energy difference (−3.6 and −5.1 kcal mol−1 (Fig. 3(c)), respectively). However, O–O bond cleavage with cyclopentene requires a significantly higher energy barrier compared with norbornene (+19.6 and +12.8 kcal mol−1 (Fig. 3(c)) at TS3 (Fig. 3(a)), respectively). This leads to the selective, one-sided oxygen transfer to the CC double bond in norbornene, which yields nearly 100% P2 selectivity. On the other hand, in the case of TiO2-anatase, P1 is preferably formed since the adsorption towards the norbornene moiety requires a higher energy barrier for O–O cleavage than cyclopentene (+6.5 and +4.6 kcal mol−1, respectively) (see Fig. 3(b)).
In summary, the surface crystalline phase of TiO2 catalysts plays a crucial role in selective DCPD epoxidation, not only in the conversion rate of DCPD but also in the product selectivity towards mono-epoxides in cyclopentene and the norbornene moiety. Moreover, the surface oxygen vacancies of TiO2-blue and TiO2-black are responsible for the lag-phases at the initial reaction stages, indicating that the formation of the surface peroxo functionalities (Ti–O2*) is the rate-determining step.
We acknowledge the Dutch research council (NWO) for the funding (LIFT project 731.017.412), the Institute for Basic Science (IBS-R011-D1), and the Korea Medical Device Development Fund (KMDF_PR_20200901_0004). We thank V. Lachman, N. J. Geels, P. F. Collignon and M.C. Mittelmeijer–Hazeleger (UvA) for the technical support.
Conflicts of interest
There are no conflicts to declare.References
- T. T. P. Cheung, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2001, vol. 2 Search PubMed.
- M. Worzakowska, Polym. Bull., 2012, 68, 775–787 CrossRef CAS.
- R. Gao, Q. Zhu, W. L. Dai and K. Fan, RSC Adv., 2012, 2, 6087–6093 RSC.
- Y. Shen, X. H. Lu, C. C. Wei, X. T. Ma, C. Peng, J. He, D. Zhou and Q. H. Xia, Mol. Catal., 2017, 433, 185–192 CrossRef CAS.
- S. Bhattacharjee and J. A. Anderson, J. Mol. Catal. A: Chem., 2006, 249, 103–110 CrossRef CAS.
- M. Sheng and R. Rosenthal, US Pat., 3 631 072, 1971 Search PubMed.
- J. Yan, G. Wu, N. Guan, L. Li, Z. Li and X. Cao, Phys. Chem. Chem. Phys., 2013, 15, 10978–10988 RSC.
- J. Li, G. Lu, G. Wu, D. Mao, Y. Guo, Y. Wang and Y. Guo, Catal. Sci. Technol., 2014, 4, 1268–1275 RSC.
- W. F. Huang, P. Raghunath and M. C. Lin, J. Comput. Chem., 2011, 32, 1065–1081 CrossRef CAS PubMed.
- V. Paunović, M. Rellán-Piñeiro, N. López and J. Pérez-Ramírez, Catal. Today, 2021, 369, 221–226 CrossRef.
- R. W. Siegel, J. Mater. Res., 1992, 7, 2840–2845 CrossRef.
- T. Ohno, D. Haga, K. Fujihara, K. Kaizaki and M. Matsumura, J. Phys. Chem. B, 1997, 101, 6415–6419 CrossRef CAS.
- N. S. Antonova, J. J. Carbó, U. Kortz, O. A. Kholdeeva and J. M. Poblet, J. Am. Chem. Soc., 2010, 132, 7488–7497 CrossRef CAS PubMed.
- Y. C. Lin, C. C. Chang, K. H. Sung, J. F. Lee and S. Cheng, Microporous Mesoporous Mater., 2018, 272, 276–285 CrossRef CAS.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103–8112 CrossRef CAS.
- Y. Kim, H. M. Hwang, L. Wang, I. Kim, Y. Yoon and H. Lee, Sci. Rep., 2016, 6, 25212–25221 CrossRef CAS PubMed.
- C. T. K. Nguyen, N. Quang Tran, S. Seo, H. Hwang, S. Oh, J. Yu, J. Lee, T. Anh Le, J. Hwang, M. Kim and H. Lee, Mater. Today, 2020, 35, 25–33 CrossRef CAS.
- S. Oh, J. S. J.-H. J. Kim, H. M. Hwang, D. Kim, J. S. J.-H. J. Kim, G. H. Park, J. S. J.-H. J. Kim, Y. H. Lee and H. Lee, J. Mater. Chem. A, 2021, 9, 4822–4830 RSC.
- A. Naldoni, M. Altomare, G. Zoppellaro, N. Liu, Š. Kment, R. Zbořil and P. Schmuki, ACS Catal., 2019, 9, 345–364 CrossRef CAS PubMed.
- J. Lee, X. Liu, A. Kumar, Y. Hwang, E. Lee, J. Yu, Y. D. Kim and H. Lee, Chem. Sci., 2021, 12, 9619–9629 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- K. Sobańska, P. Pietrzyk and Z. Sojka, ACS Catal., 2017, 7, 2935–2947 CrossRef.
- M. Ziolek, I. Sobczak, P. Decyk, K. Sobańska, P. Pietrzyk and Z. Sojka, Appl. Catal., B, 2015, 164, 288–296 CrossRef CAS.
- M. Ziolek, I. Sobczak, P. Decyk and L. Wolski, Catal. Commun., 2013, 37, 85–91 CrossRef CAS.
- A. Y. Zhang, T. Lin, Y. Y. He and Y. X. Mou, J. Hazard. Mater., 2016, 311, 81–90 CrossRef CAS PubMed.
- A. H. Boonstra and C. A. H. A. H. A. Mutsaers, J. Phys. Chem., 1975, 79, 1940–1943 CrossRef CAS.
- J. Zou, J. Gao and Y. Wang, J. Photochem. Photobiol., A, 2009, 202, 128–135 CrossRef CAS.
- W. F. Zhang, Y. L. He, M. S. Zhang, Z. Yin and Q. Chen, J. Phys. D: Appl. Phys., 2000, 33, 912–916 CrossRef CAS.
- G. L. Gutsev, B. K. Rao and P. Jena, J. Phys. Chem. A, 2000, 104, 11961–11971 CrossRef CAS.
- J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568–572 CrossRef CAS PubMed.
- A. A. Mikhaylov, A. G. Medvedev, A. V. Churakov, D. A. Grishanov, P. V. Prikhodchenko and O. Lev, Chem. – Eur. J., 2016, 22, 2980–2986 CrossRef CAS PubMed.
- L. Bonato, M. Virot, T. Dumas, A. Mesbah, P. Lecante, D. Prieur, X. Le Goff, C. Hennig, N. Dacheux, P. Moisy and S. I. Nikitenko, Chem. – Eur. J., 2019, 9580–9585 CrossRef CAS PubMed.
- K. Tomobe, E. Yamamoto, D. Kojić, Y. Sato, M. Yasui and K. Yasuoka, Sci. Adv., 2017, 3, e1701400 CrossRef PubMed.
- Á. Ganyecz, P. D. Mezei and M. Kállay, Comput. Theor. Chem., 2019, 1168, 112607 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05305e |
| ‡ These authors contributed to the manuscript equally. |
| This journal is © The Royal Society of Chemistry 2023 |