
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chain-extension in hyperbranched polymers alters tissue distribution and cytotoxicity profiles in orthotopic models of triple negative breast cancers†
Cara
Moloney‡
 *a,
Fatemeh
Mehradnia‡
b,
Robert J.
Cavanagh
a,
Asmaa
Ibrahim
a,
Amanda K.
Pearce
b,
Alison A.
Ritchie
a,
Philip
Clarke
a,
Ruman
Rahman
a,
Anna M.
Grabowska
a and
Cameron
Alexander
*b
*a,
Fatemeh
Mehradnia‡
b,
Robert J.
Cavanagh
a,
Asmaa
Ibrahim
a,
Amanda K.
Pearce
b,
Alison A.
Ritchie
a,
Philip
Clarke
a,
Ruman
Rahman
a,
Anna M.
Grabowska
a and
Cameron
Alexander
*b
aSchool of Medicine, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: cara.moloney@nottingham.ac.uk
bSchool of Pharmacy, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
First published on 4th August 2023
Abstract
The therapeutic efficacy of nanomedicines is highly dependent on their access to target sites in the body, and this in turn is markedly affected by their size, shape and transport properties in tissue. Although there have been many studies in this area, the ability to design nanomaterials with optimal physicochemical properties for in vivo efficacy remains a significant challenge. In particular, it is often difficult to quantify the detailed effects of cancer drug delivery systems in vivo as tumour volume reduction, a commonly reported marker of efficacy, does not always correlate with cytotoxicity in tumour tissue. Here, we studied the behaviour in vivo of two specific poly(2-hydroxypropyl methacrylamide) (pHPMA) pro-drugs, with hyperbranched and chain-extended branched architectures, redox-responsive backbone components, and pH-sensitive linkers to the anti-cancer drug doxorubicin. Evaluation of the biodistribution of these polymers following systemic injection indicated differences in the circulation time and organ distribution of the two polymers, despite their very similar hydrodynamic radii (∼10 and 15 nm) and architectures. In addition, both polymers showed improved tumour accumulation in orthotopic triple-negative breast cancers in mice, and decreased accumulation in healthy tissue, as compared to free doxorubicin, even though neither polymer–doxorubicin pro-drug decreased overall tumour volume as much as the free drug under the dosing regimens selected. However, the results of histopathological examinations by haematoxylin and eosin, and TUNEL staining indicated a higher population of apoptotic cells in the tumours for both polymer pro-drug treatments, and in turn a lower population of apoptotic cells in the heart, liver and spleen, as compared to free doxorubicin treatment. These data suggest that the penetration of these polymer pro-drugs was enhanced in tumour tissue relative to free doxorubicin, and that the combination of size, architecture, bioresponsive backbone and drug linker degradation yielded greater efficacy for the polymers as measured by biomarkers than that of tumour volume. We suggest therefore that the effects of nanomedicines may be different at various length scales relative to small molecule free drugs, and that penetration into tumour tissue for some nanomedicines may not be as problematic as prior reports have suggested. Furthermore, the data indicate that dual-responsive crosslinked polymer-prodrugs in this study may be effective nanomedicines for breast cancer chemotherapy, and that endpoints beyond tumour volume reduction can be valuable in selecting candidates for pre-clinical trials.
Introduction
Triple negative breast cancer (TNBC), classified via the lack of expression of estrogen and progesterone receptors (ER and PR) and low levels of human epidermal growth factor receptor 2 (HER2) expression, is one of the most aggressive forms of breast cancer and accounts for ca. 10–15% of all diagnosed cases.1 Patients presenting with TNBC tend to have a poor prognosis due to the lack of sensitivity towards conventional therapies used to treat other forms of breast cancer, such as endocrine and molecular targeted therapy, and typically exhibit high levels of metastasis and recurrence.2,3 Although there is currently no standard-of-care treatment for TNBC, patients typically receive surgery, radiation and conventional chemotherapies.4 Doxorubicin (Dox) is a commonly used anthracycline chemotherapy in the treatment of breast cancer,5 with several Food and Drug Administration (FDA) approved treatments on the market. However, various severe side-effects are frequently observed following Dox treatment, particularly renal and cardio-toxicity.6 Furthermore, the effectiveness of Dox is limited by a series of resistance mechanisms which reduce its pharmacological potential.7Some of the issues associated with administration of chemotherapeutics have been addressed through altering the pharmacokinetic profiles of these drugs via encapsulation in, or conjugation to, a carrier. A successful clinical example is Doxil®, a liposomal formulation containing Dox, which was one of the first approved nanomedicines.8,9 However, there is still a demand for more potent TNBC therapies, and it has been suggested that polymer pro-drugs might have additional advantages over liposomal formulations to overcome barriers to cancer therapies.10 Conjugation with polymers can increase solubility and stability of chemotherapeutics in aqueous solutions, leading to increased efficacy against tumours, both in vivo and in vitro.11 In addition to this, the linkages between polymers and drugs can be tailored to be stimuli-responsive in the tumour microenvironment or to incorporate tumour specific targeting moieties, leading to enhanced levels of active drug at the target site.12,13 Dox is a good candidate to be applied in a polymer pro-drug system, as it possesses numerous functional chemistries amenable to conjugation, which has led to a range of reported polymer–Dox linkages, including carbamate,14 urea15 and hydrazone,16 to a range of different polymer types.
In particular, much development has been undertaken into poly(N-(2-hydroxypropyl)methacrylamide), or pHPMA, conjugated to Dox, i.e. pHPMA–Dox pro-drug systems.17,18 Following initial reports in the 1970s on the potential use of HPMA in biological applications,19,20 a variety of pHPMA conjugates have been prepared to treat multiple types of cancer.21,22 Furthermore, several HPMA–Dox formulations have reached clinical trials, including the candidate polymer therapeutics PK1 and PK2. PK1 consists of Dox covalently bound to HPMA via a peptidyl linker, cleavage of which by lysosomal enzymes allows for release within tumour cells. The distribution half-life of PK1 was found to be increased as compared to free Dox (1.8 h and 5 min, respectively), indicating better circulation times for the polymer-conjugate. In phase II clinical trials, PK1 showed tolerable toxicities and partial responses in 6/62 patients.23 PK2 has a similar structure to PK1, with the addition of a galactosamine linker which can target liver cells, and phase I clinical studies showed moderate side-effects and partial responses in several patients with this pHPMA pro-drug too.24 However, while these studies highlighted the potential of polymer pro-drugs as clinical chemotherapeutic therapies, particularly in reducing side-effects, PK1 and PK2 were not advanced further as the improvements in efficacy were not deemed sufficient compared to existing Dox formulations, to justify full commercial development.25 Accordingly, there is a need to understand better how polymer–drug conjugate formulations might be improved in order to prepare pro-drugs which might perform better clinically.
One significant problem in development of drug delivery systems is that many pre-clinical models used to evaluate their efficacy are based on measuring reduction in the volumes of tumours, which may or may not be in physiologically relevant sites in the animal, as the primary therapeutic endpoint. Tumour volume is a convenient parameter to measure and is quicker and easier to evaluate compared to overall survival, or analysis of individual tissue samples. However, while undoubtedly useful, tumour volume does not give a detailed mechanistic understanding of cancer therapy, and thus for structure–function studies of drugs and drug delivery systems, other assays are required. Similarly, reliance on overall survival as a sole metric without knowledge of drug distribution through tissue parenchyma, risks an underestimation of potential drug efficacy (i.e., a given tumour may be sensitive, if exposed to a sufficient amount of drug). Of particular note are studies related to the transport and penetration of drug carriers into tumours, as it has been suggested that the lack of efficacy of some nanomedicines is due to their inability to distribute to cells away from the tumour vasculature.26 It is clear that size, shape and chemical functionality can all alter the ability of materials to transport in blood vessels and in tissue. However, the subtle interplay of these factors, combined with the dynamic and varied environments through which nanoparticles travel in the body, make it challenging for detailed predictions of particle distribution to be made.27,28 Nevertheless, important studies have shown that polymer or nanoparticle size, surface-displayed functionality and ability to break down into smaller fragments when in target sites, are all important factors enhancing penetration into tumours.29–35 Recent work has elegantly shown that pHPMA polymers with a variety of side-chain and backbone architectures can exhibit marked variations in their ability to transport into tumours and other tissues.36 Up to ∼4–5 fold differences in tumour accumulation in a murine EL4 T cell lymphoma model were observed for pHPMA polymers varying in dendron or dendritic architectures, even though the primary polymer chemistries and therapeutic payloads were very similar. These variations in tumour accumulation were also manifest in tumour reduction and overall survival. The authors report a dendron-based pHPMA as showing the highest tumour retention and best overall efficacy in vivo, despite the polymers not presenting as the most potent in 2D in vitro experiments.36
In a previous study, we injected mice bearing orthotopic TNBC with redox- and pH-responsive pHPMA–Dox polymer pro-drugs spanning a range of sizes and architectures and noted some intriguing differences between organ accumulation and tumour volume reduction for two polymers of very similar size and overall chemistry, but different molecular architectures. A hyperbranched pHPMA of nominal hydrodynamic diameter dH ∼ 10 nm was shown to exhibit greater accumulation in the kidneys compared to the liver, while a chain-extended hyperbranched polymer, ‘pseudo-star’ pHPMA of nominal dH ∼ 15 nm was retained to a greater extent in the liver.37 Both polymers were well-tolerated by mice in terms of body weight, showed similar efficacy in terms of tumour reduction when dosed at 2 mg kg−1 over a period of 20 days, and were more effective than free Dox over this time frame. However, when dosed at 4 mg kg−1 over 10 days, the hyperbranched pHPMA–Dox polymer was more potent than the related chain-extended hyperbranched pHPMA–Dox polymer, and only the hyperbranched pHPMA–Dox polymer out-performed the free drug.37
Here we describe some detailed further studies into the behaviour of these hyperbranched and chain-extended hyperbranched pHPMA polymers when dosed in mice carrying orthotopic TNBC xenografts. We show that with a reduced dosing (1.7 mg kg−1) twice weekly for 4 weeks, the differences in organ accumulation were retained for these polymers, but they also showed marked enhancements in tumour accumulation and penetration compared to free drug. Importantly, we observed significant increases in apoptosis in tumours but reduced toxicities in non-target organs compared to free Dox. These data suggest that molecular architectures of polymer pro-drugs can be altered to affect their biodistribution and organ accumulation, and that the chemistries can be controlled to allow deep penetration into tumour tissue and enhanced cell kill in diseased regions with low toxicity to healthy organs.
Experimental
Preparation of polymer formulations
In this study, four pHPMA polymer formulations were employed – these are coded as HB-SS-HH-Dox, HB-SS-HH-Cy5, CE-HB-SS-Dox and CE-HB-SS-Cy5 and the key attributes of these polymers are shown in Table 1. The preparation of these polymers has been previously reported by us37 and is described in detail in the ESI,† with 1H NMR spectra and GPC spectra reported in Fig. S1 and S2.†| Polymer | M n (kDa)/GPC | Đ |
d
H![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (DLS) (nm) (DLS) (nm) |
Zeta potential (mV) | Dye/drug content (wt%) |
|---|---|---|---|---|---|
| a Molar masses for Dox-conjugated polymers were calculated based on GPC data for the precursors and NMR integrals. | |||||
| HB-SS-HH-Cy5 | 52 | 1.5 | 10 (PDI = 0.14) | −24.1 | 0.63 |
| HB-SS-HH-Dox | 58a | 1.5 | 12 (PDI = 0.25) | −15.3 | 11 |
| CE-HB-SS-Cy5 | 122 | 1.6 | 15 (PDI = 0.15) | −15.3 | 0.27 |
| CE-HB-SS-Dox | 126a | 1.6 | 17 (PDI = 0.31) | −16.6 | 9 |
In vivo evaluation of polymers
:50.
Evaluation of effect of polymers on tissues
Statistical analysis
All experiments were performed at least three times and expressed as mean ± SD. Data were analysed for statistical significance using one-way ANOVA. In all cases, differences were considered significant when ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05.Results and discussion
In vivo biodistribution of Cy-5 labelled polymers
Two pHPMA polymers were studied in the initial biodistribution studies, coded as HB-SS-HH and CE-HB-SS to indicate that they were of hyperbranched or pseudo-star chain-extended hyperbranched polymer architectures, and with disulfide bonds in their backbones. The synthesis and full characterisation of these polymers have been reported by us previously,37 with information provided in the ESI.† HB-SS-HH is a hyperbranched HPMA structure with a central disulfide bridge, and with a hydrazide co-monomer for linkage to Dox via a pH-responsive hydrazone link when activated. CE-HB-SS is formed by chain extending HB-SS-HH polymer with HPMA, resulting in a non-degradable pHPMA corona being formed around the redox-responsive core, but, as with HB-SS-HH, contains a pH-responsive hydrazone link to Dox when activated. Representative structures of the Cy5 and Cy5 with Dox HPMA polymers are reported in Fig. 1a and b, respectively. TEM images and schematic representations of HB-SS-HH and CE-HB-SS are reported in Fig. 1c and d.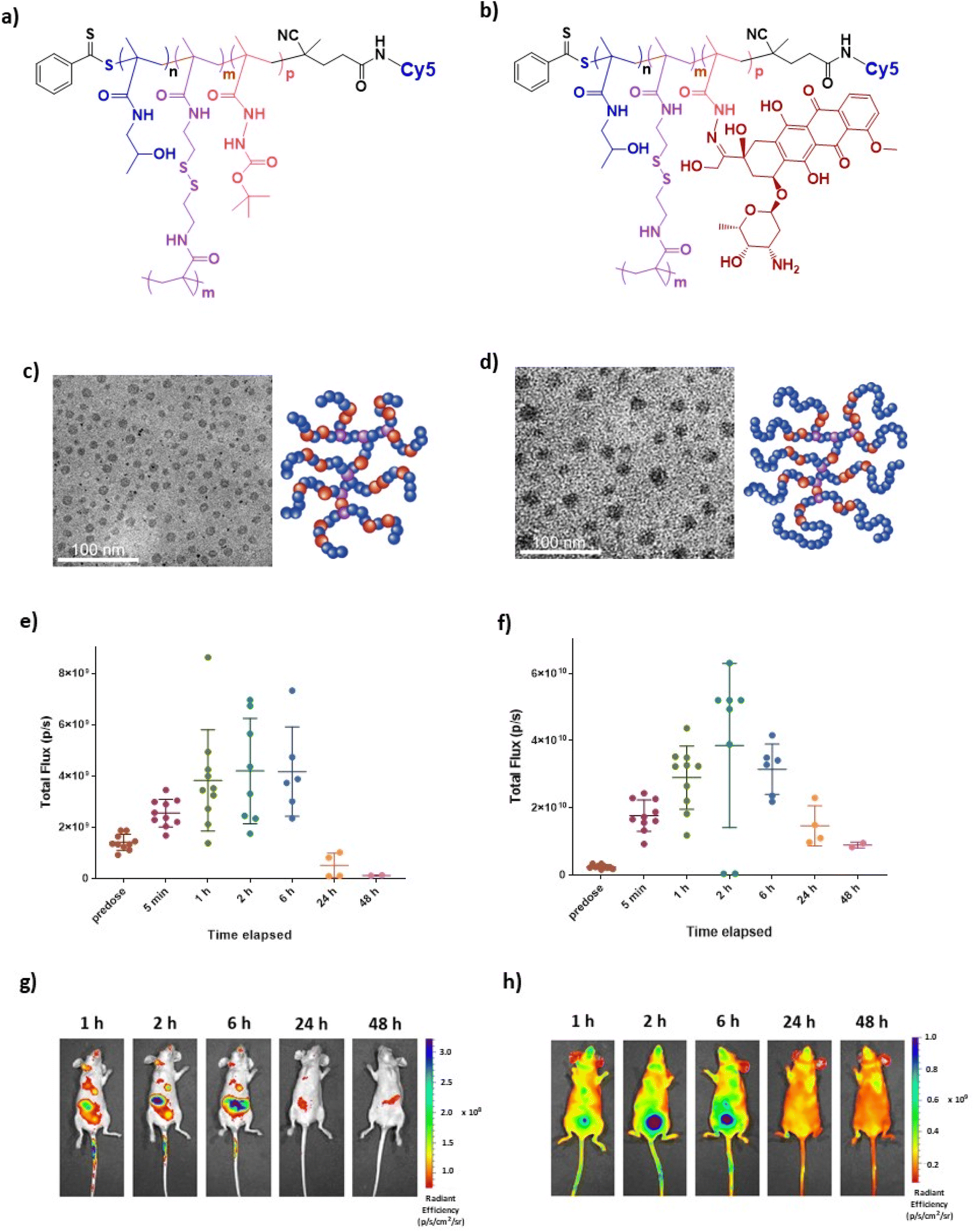 | ||
| Fig. 1 Representative structures of (a) Cy5 conjugated and (b) Cy5 and Dox conjugated HPMA polymers. (c and d) TEM images and schematic representations of polymer architectures for HB-SS-HH and CE-HB-SS, respectively. (e and f) Recorded fluorescent signal intensities measured using IVIS imaging up to 48 hours post-injection of 100 μL of 2 mg mL−1 HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively, in healthy mice. (g and h) Recorded IVIS images at 1, 2, 6, 24 and 48 hours post-injection of HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively. | ||
Cy5 labelled polymers were prepared to enable monitoring of the biodistribution of both polymers in healthy mice using IVIS imaging. Following intravenous administration of 100 μL of HB-SS-HH-Cy5 and CE-HB-SS-Cy5 NPs at doses of 2 mg mL−1 polymer, mice were imaged using an IVIS Spectrum at several timepoints. The recorded fluorescence, resulting from Cy5 signals, over 48 hours post-injection are reported in Fig. 1e and f for HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively, with representative IVIS images at 1, 2, 6, 24 and 48 hours post-injection reported in Fig. 1g and h. At the beginning of the experiment, n = 10 subjects were dosed with the fluorescently labelled polymers and imaged at designated timepoints. Following 1-hour post-injection, n = 2 subjects were sacrificed to enable organ specific studies (see below), and this was repeated for the remainder of the timepoints. For HB-SS-HH-Cy5, peak fluorescence due to circulating polymer was observed between 2 and 6 hours post-injection, while for CE-HB-SS-Cy5, the detected maximum concentration was observed 2 hours post-injection. However, CE-HB-SS-Cy5 was retained to a higher extent at 24 hours, and given that the weight fraction of Cy5 in CE-HB-SS was ∼50% that of Cy5 in HB-SS-HH (Table 1), the data suggest that the chain extended architecture yielded longer circulation times and reduced elimination compared to the hyperbranched polymer. In both cases, clearance was noted 48 hours post-injection, an observation which was employed to determine the dosing schedule for the subsequent efficacy studies.
In order to investigate further the effects of polymer structures on biodistribution, at selected timepoints (1, 2, 6, 24 and 48 hours post-injection) subjects were sacrificed, organs were excised and imaging was performed using an IVIS Spectrum. The levels of deposition within clearance pathways such as the liver, spleen, lung and kidneys were investigated, and the recorded fluorescence over 48 hours post-injection was determined as shown in Fig. 2a and b for HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively. It was noted that polymer HB-SS-HH-Cy5, with lower molecular weight and physical size, exhibited low accumulation in liver, kidney and lung and moderate accumulation in spleen (radiance values of 1.7 × 104, 8.2 × 104, 3.7 × 104 and 1.1 × 105 photons s−1 cm−2, respectively, 1 hour post injection). For the chain-extended hyperbranched polymer, CE-HB-SS-Cy5, higher fluorescence signals were recorded across all organs at all timepoints, which is in agreement with the signals reported for the whole animals as reported in Fig. 1e–h. In contrast to HB-SS-HH-Cy5, polymer CE-HB-SS-Cy5 exhibited highest accumulation in the kidney and lung, with lower accumulation levels noted for the liver and spleen (8.1 × 105, 6.3 × 105, 2.2 × 105, 3.0 × 105 photons s−1 cm−2, respectively, 1 hour post injection). The deposition levels within the heart were also of note due to the cardiotoxicity associated with Dox administration, with moderate accumulation noted for both polymers (1.3 × 105 and 3.5 × 105 photons s−1 cm−2 for HB-SS-HH-Dox and CE-HB-SS-Dox, respectively, 1 hour post injection). Other organs of interest (pancreas, lymph node, brain and bladder) were also investigated, and the recorded fluorescence is reported in Fig. S3a and b.† Representative IVIS images of all organs at selected timepoints are shown in Fig. S3c and d.† High accumulation was apparent in the pancreas and brain, with moderate accumulation in the bladder and lymph node for both polymers (for HB-SS-HH-Dox, 3.5 × 105, 3.7 × 105, 1.8 × 105 and 2.0 × 105 photons s−1 cm−2, respectively; for CE-HB-SS, 5.1 × 105, 5.7 × 105, 3.9 × 105 and 6.1 × 105 photons s−1 cm−2, respectively, 1 hour post injection).
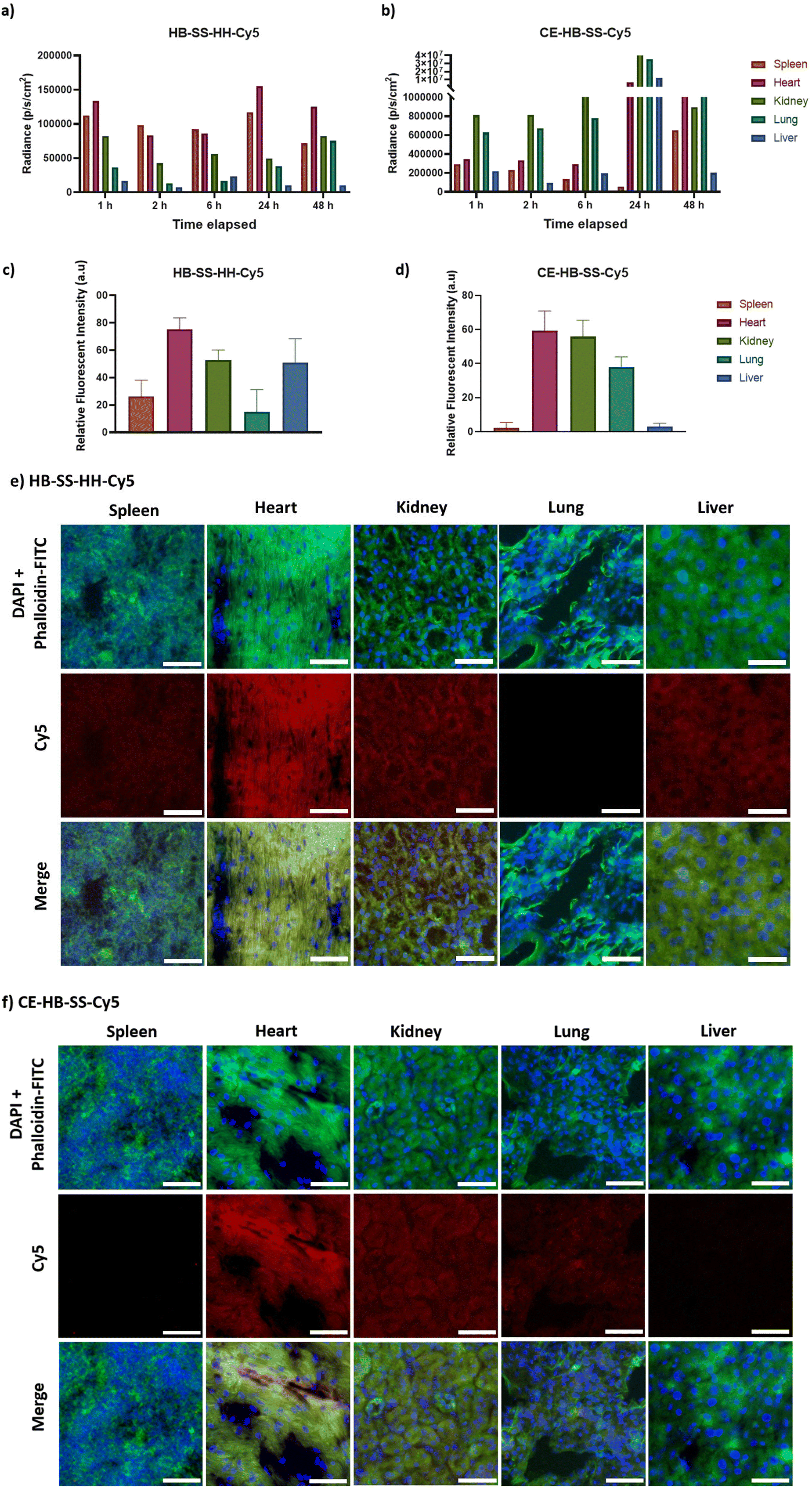 | ||
| Fig. 2 (a and b) Recorded fluorescence intensities measured for selected organs (spleen, heart, kidney, lung and liver) using IVIS imaging up to 48 hours post injection of 100 μL of 2 mg mL−1 HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively, in healthy mice. (c and d) Quantified Cy5 signal in excised organs of interest 1 hour post-injection for HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively. Following excision, organs were sectioned to 10 μm thickness and analysis was carried out in several distinct regions of the sections. (e and f) Representative fluorescence microscopy images of corresponding organ sections 1 hour post-injection indicating polymer deposition of HB-SS-HH-Cy5 and CE-HB-SS-Cy5, respectively (scale bar 50 μm; 20×; blue = DAPI, green = phalloidin-FITC, red = Cy5). | ||
To investigate organ-level distribution further, excised organs 1 hour post injection were sectioned and imaged using fluorescence microscopy. The Cy5 signal, which is indicative of polymer accumulation, was quantified and the results are in agreement with the IVIS analysis, depicting different biodistribution patterns of each pHPMA polymer (Fig. 2c and d for main organs, Fig. S3e and f† for additional organs). HB-SS-HH-Cy5 exhibited highest accumulation levels in the heart, kidney and liver 1 hour post-injection (mean relative fluorescent intensities of 75, 53 and 51 a.u., respectively), while CE-HB-SS-Cy5 was present at highest levels in the heart, kidneys and lungs (60, 55 and 38 a.u., respectively). Representative fluorescent images have been reported in Fig. 2e and f (Fig. S3g and h† for additional organs). The relative levels of kidney and liver accumulation were slightly at variance for these polymers in this study compared to our previous work at earlier time-points,37 which we attribute to the changes in dosing regimens, but overall these data indicated greater organ retention for CE-HB-SS-Cy5, in agreement with the whole animal data.
Effects of polymer–doxorubicin dosing an orthotopic MB-MDA-231 breast cancer model
The anti-tumour efficacy and toxicity of the pHPMA–Dox conjugates were investigated in an orthotopic MB-MDA-231 fLuc breast cancer model. Tumour-bearing mice CD-1 NuNu mice were treated with 100 μL I.V. injections of saline (control), free Dox, and NP suspensions of HB-SS-HH-Dox, CE-HB-SS-Dox, HB-SS-HH-Cy5 and CE-HB-SS-Cy5 (1.7 mg kg−1 Dox dose for all groups) twice weekly for up to 4 weeks. The dosing regime was chosen in order to probe the effects of differences in circulation time for the polymers compared to free drug and at the lowest dose with which we considered we might observe therapeutic effects while minimising overall toxicity. In addition, we were interested in the effects the polymer–drug conjugates might have on non-target sites, as therapeutic window is crucial in any development of these materials for real clinical application. Accordingly, the overall dose selected (1.7 mg kg−1 Dox) was less than that we used in our prior work and several-fold lower than those adopted in other studies (5–7.5 mg kg−1 Dox dose).36 The mice were observed daily for clinical symptoms, with no adverse effects noted, and the tumour volume and body mass were recorded over the course of the study. No significant variance in tumour volume or mouse weight was noted following HB-SS-HH-Cy5 and CE-HB-SS-Cy5 treatment as compared to the control group (Fig. S4a and b†). Additionally, no significant decrease in weight between Dox treated and control groups was noted, indicating no dose-related toxicity (Fig. 3a). Although there was an overall increase in mean tumour volume, as measured by a callipers and by the recorded bioluminescence of the tumours, across all treated groups and the control group, no significant differences were recorded for treatments with HB-SS-HH-Dox, CE-HB-SS-Dox and free drug (Fig. 3b and c). Photos of the tumours excised at the end of the study highlighted the variability of tumour size within each treatment group (Fig. 3d). Taken together, while there was no toxicity observed following treatment with Dox–polymers and free Dox, there was no apparent anti-tumour effect as determined from tumour volume as compared to the control group.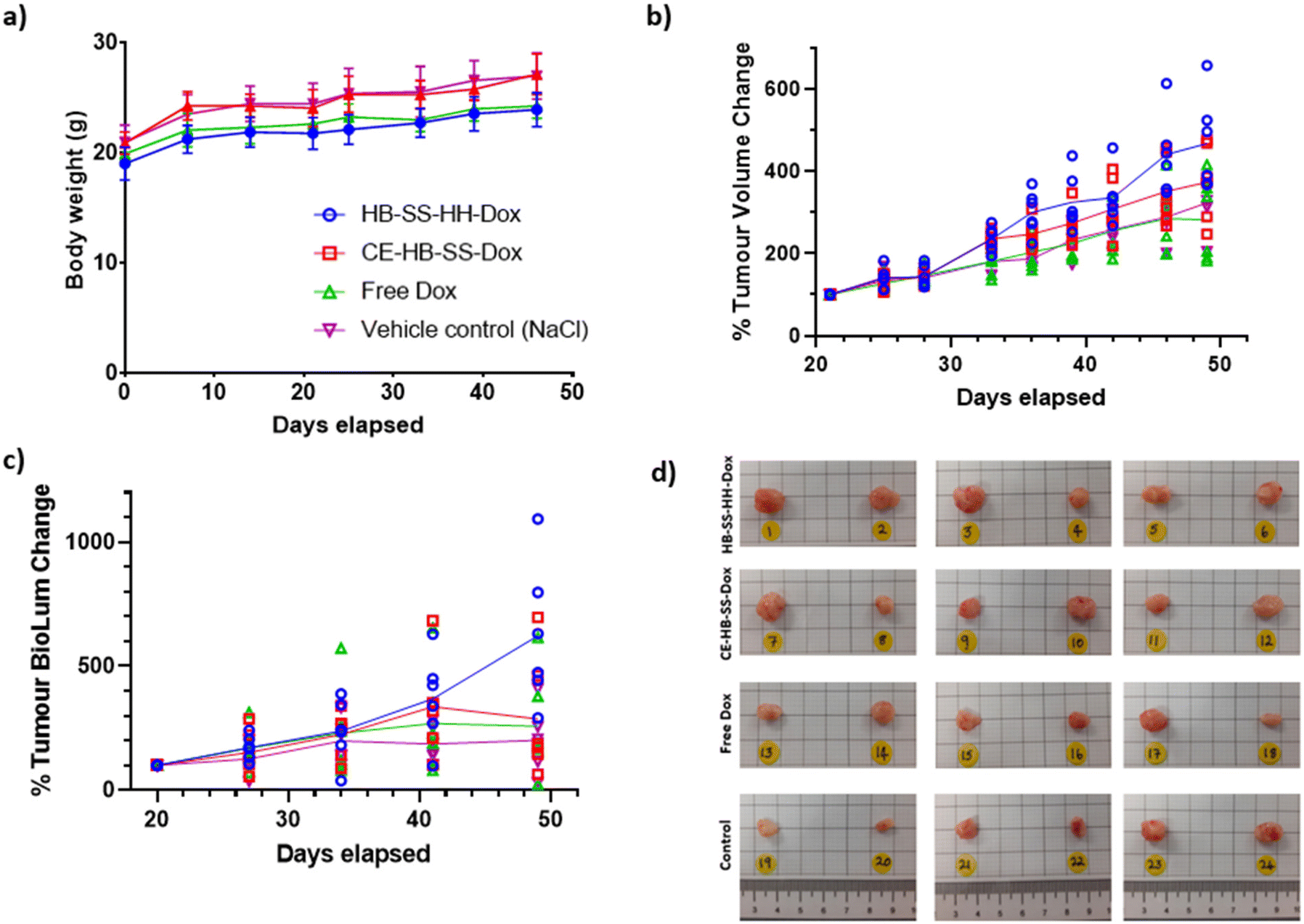 | ||
| Fig. 3 Recorded (a) body weight of subjects, (b) tumour volume change as a percentage of pre-treatment, and (c) recorded bioluminescence change for tumour as a percentage of pre-treatment for control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox (1.7 mg kg−1 Dox dose for each) treatment groups (n = 6) over the course of the study. (d) Images of tumours removed at the end of the study for HB-SS-HH-Dox, CE-HB-SS-Dox, free Dox and control groups. | ||
Although there was an apparent lack of efficacy of the polymer–Dox conjugates against MB-MDA-231 tumours by conventionally reported methods, a deeper analysis was undertaken into the tissue level effects. Tumour deposition levels of Dox were quantified using fluorescence microscopy (Fig. 4a), with a significant increase in Dox deposition observed within groups treated with the polymer–drug conjugates as compared to the free Dox treated group, as demonstrated by the increase in recorded radiance (57.7 ± 4.6 a.u. for HB-SS-HH-Dox, 49.8 ± 2.9 a.u. for CE-HB-SS-Dox and 43.5 ± 4.1 a.u. for free Dox). A significantly higher level of Dox in the tumours was observed for HB-SS-HH-Dox as compared to CE-HB-SS-Dox, indicating that the smaller polymer, both by molecular weight and hydrodynamic size, exhibits enhanced tumour penetration. Representative fluorescence microscopy images (Fig. 4b) highlight the increased accumulation of Dox when dosing the polymer–Dox conjugates as compared to free Dox.
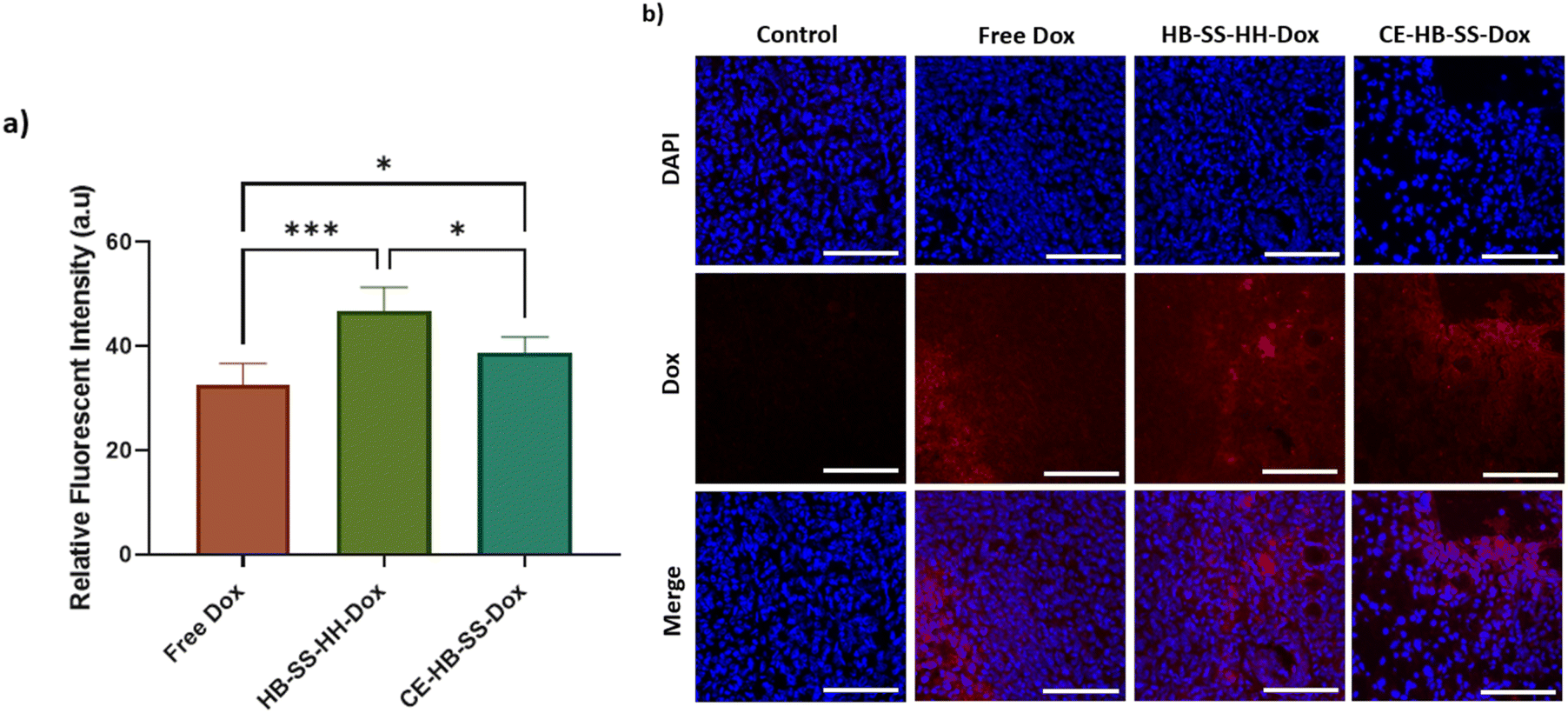 | ||
| Fig. 4 (a) Recorded relative fluorescent intensities of tumour sections at the end of the study for control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox treated groups (****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05 one-way ANOVA with Tukey's post-analysis). Following excision, organs were sectioned to 10 μm thickness and analysis was carried out in several distinct regions of the sections. (b) Representative fluorescence images of tumour sections indicating enhanced levels of Dox deposition for polymer–Dox conjugates as compared to free Dox treated groups (scale bar = 100 μm; 20×; blue = DAPI, red = Dox). | ||
In addition to increased tumour deposition of polymer–Dox as compared to free Dox, differences were noted during histopathological evaluation of H&E-stained tumour sections. For both HB-SS-HH-Dox and CE-HB-SS-Dox treated groups, areas of extensive necrosis and inflammatory cell infiltration (black arrows) are evident both at 10× and 40× magnification (Fig. 5). In the Dox treated group, scattered apoptotic cells are present (green circles), as evident by the dense eosinophilic cytoplasm, cellular shrinkage, and clear halo surrounding cells. Other organs which are typically associated with Dox toxicity, i.e. the heart, liver and spleen, were also evaluated (Fig. 6). The heart tissue in the control and Dox–polymer treated groups exhibited normal architecture, with some tissue degeneration noted for the free Dox treated group. In the free Dox treated groups, there is distortion of normal hepatic architecture, and the liver cells exhibit hydropic degeneration (yellow arrows), which is denoted by swelling of hepatocytes. In liver sections for HB-SS-HH-Dox and CE-HB-SS-Dox, the tissue largely exhibited normal architecture, although a small portion of hepatic cells in the HB-SS-HH-Dox treated group showed degeneration. Similarly, the splenic tissue of mice treated with free Dox has a distorted architecture compared with the control, HB-SS-HH-Dox, and CE-HB-SS-Dox treated mice, which have organised areas of white pulp and red pulp. These data suggest that Dox conjugation to HPMA polymers improved accumulation in tumour cells while simultaneously decreasing accumulation in healthy tissues. The enriched drug concentration in tumours consequently resulted in enhanced apoptosis, with fewer toxic side effects noted in other organs.
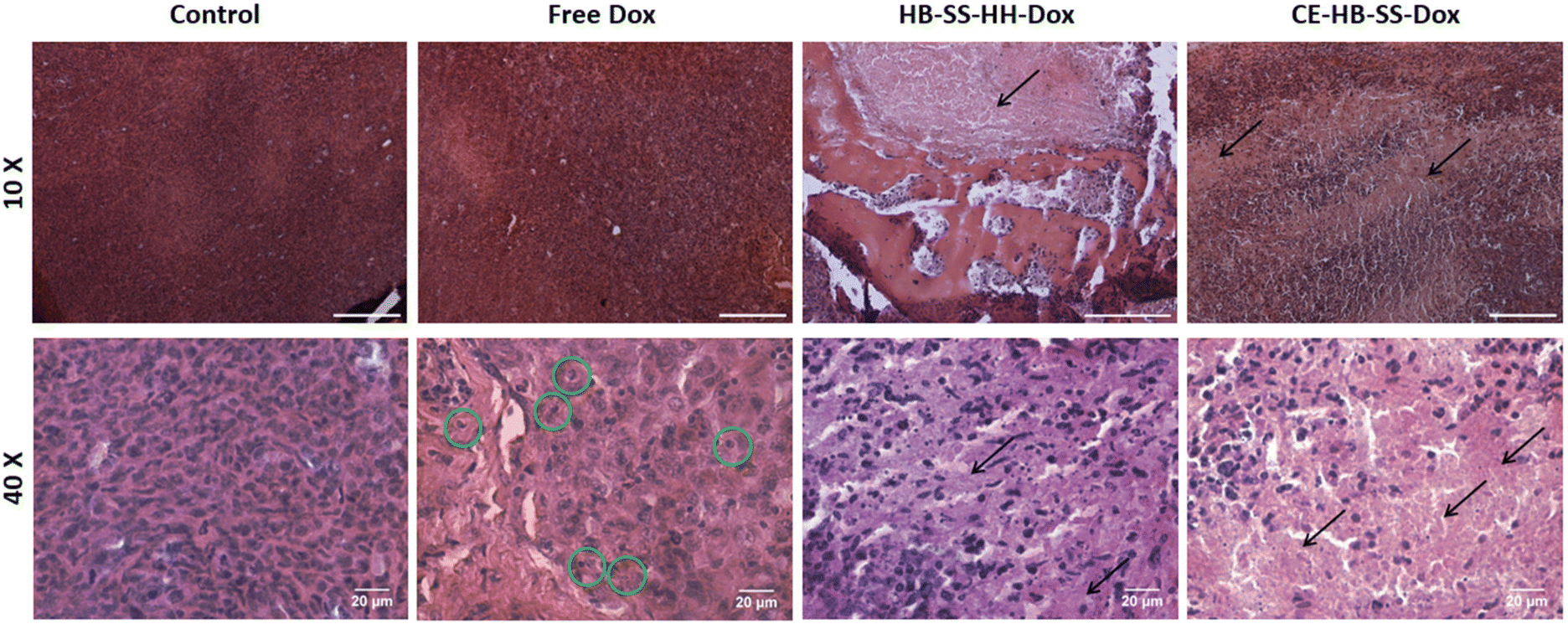 | ||
Fig. 5 Representative H&E images of tumours removed at the end of the study for control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox treated groups taken at 10× magnification (top row, scale bar = 200 μm) and 40× magnification (bottom row, scale bar = 20 μm). Areas of necrosis and inflammatory cell infiltration have been highlighted with black arrows (←) and scattered apoptotic cells have been highlighted with green circles ( ). ). | ||
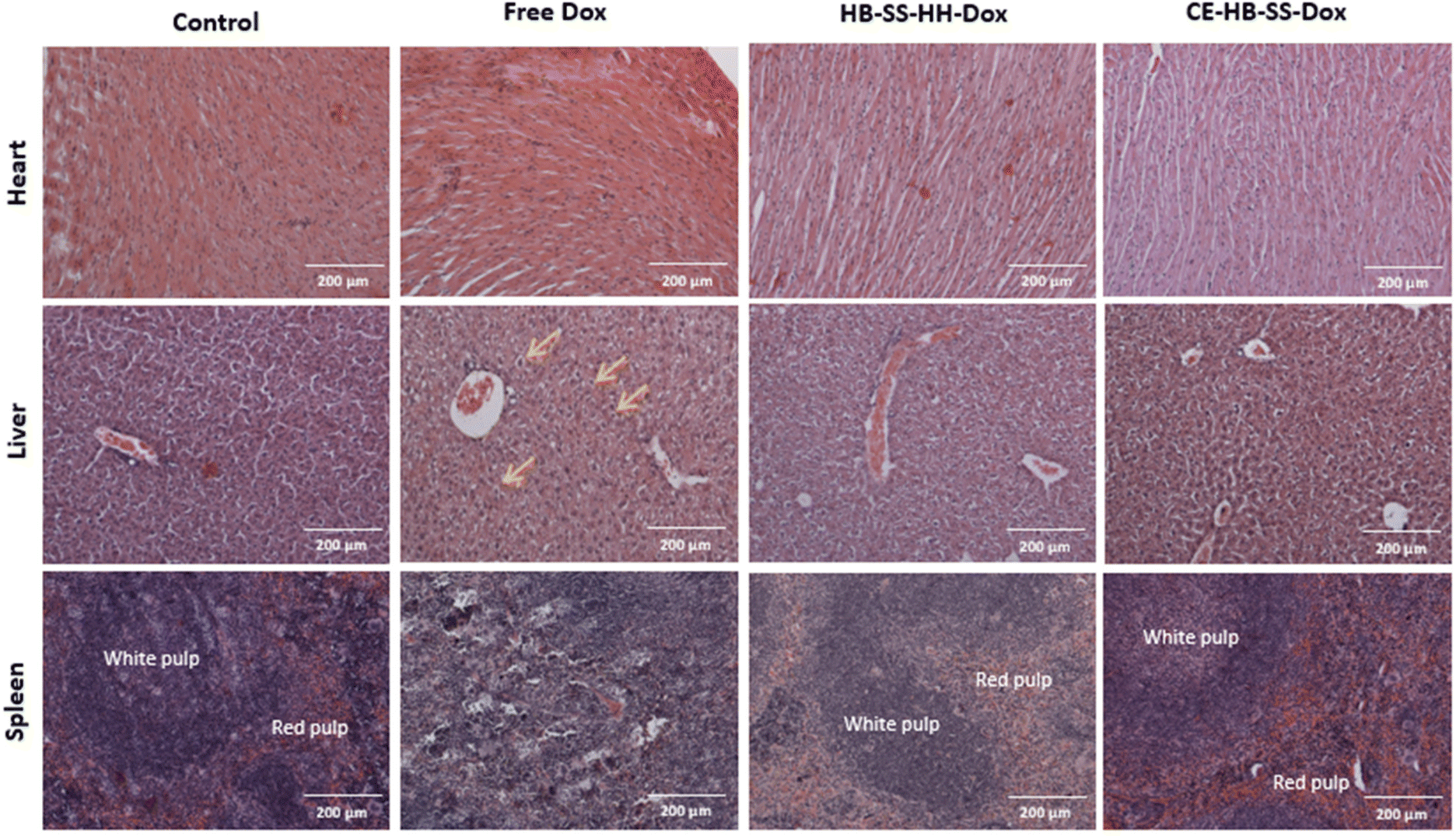 | ||
Fig. 6 Representative H&E images of heart, liver and spleen removed at the end of the study for control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox treated groups taken at 10× magnification (scale bar = 200 μm). In liver tissue, areas of hydropic degeneration have been indicated with yellow arrows ( ), and in splenic tissue, areas of organised red and white pulp have been labelled. ), and in splenic tissue, areas of organised red and white pulp have been labelled. | ||
Further experiments with the Cy5-labelled materials showed, as expected, that the polymers exhibited little toxicity to the tumours without drug molecules present (Fig. S5†). Additionally, the Cy5-labelled polymers exhibited little to no toxicity in the heart, liver, spleen, pancreas, kidney, brain, bladder, lung and lymph node, with normal architecture retained (Fig. S6 and S7†), indicating that the polymers were well-tolerated.
Levels of apoptosis in tumours and selected main organs following treatment with control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox were quantified using a terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay (Fig. 7a and b). This assay involves the labelling of double-stranded DNA breaks, which are associated with apoptosis, with a marker that can be visualized via microscopy.40 A co-stain was used to indicate regions of healthy cells, i.e. those with intact DNA. In this assay, healthy cells are stained green while apoptotic regions are stained brown, as shown in Fig. 7c for magnified images (10×) and Fig. S8† for whole section images. Images of the tumours showed increased levels of apoptosis, in comparison to the control, following treatments with free Dox and polymer–Dox conjugates (Fig. 7a). Significantly higher levels of apoptosis were observed following polymer–Dox treatment than with free Dox treatment (43.8 ± 3.4% for HB-SS-HH-Dox, 31.5 ± 6.3% for CE-HB-SS-Dox and 21.4 ± 3.3% for free Dox). A significantly higher level of apoptosis was observed for HB-SS-HH-Dox as compared to CE-HB-SS-Dox, which correlates with the enhanced tumour deposition as noted in Fig. 4a.
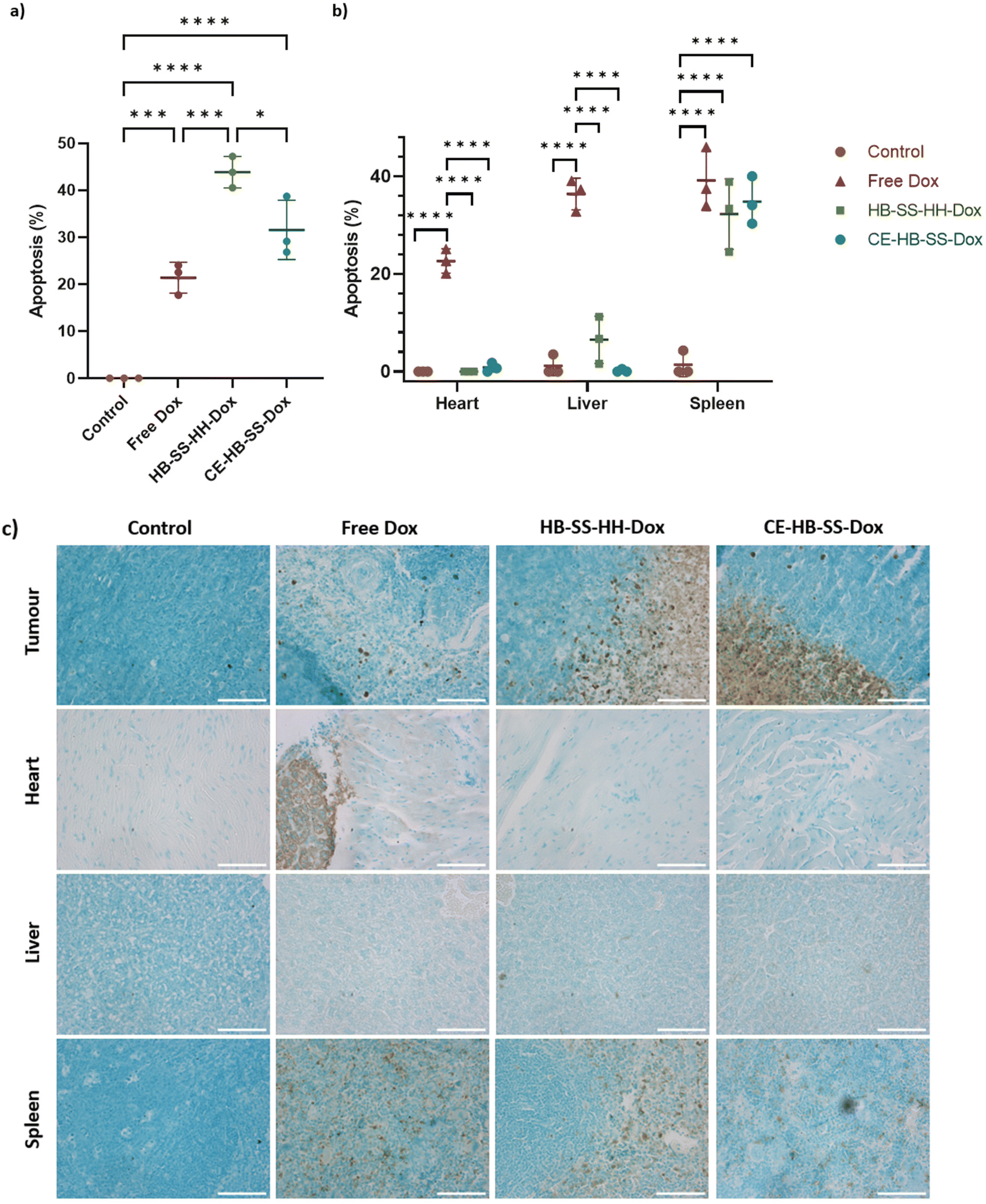 | ||
| Fig. 7 (a) Quantification of apoptosis levels using TUNEL assay for control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox treated groups for (a) tumours, and (b) heart, liver and spleen, removed at the end of the study (****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05 one-way ANOVA with Tukey's post-analysis). (c) Representative images following TUNEL assay staining for sections of tumour, heart, liver and spleen, across control, free Dox, HB-SS-HH-Dox and CE-HB-SS-Dox treated groups, taken at 10× magnification (green/blue = healthy cells, brown = apoptotic cells). | ||
Analysis of tissue from the heart indicated a significant increase in the levels of apoptosis for the free Dox treatment as compared to the control and the polymer–Dox treatments as highlighted in Fig. 7b (22.6 ± 2.5% for free Dox treatment, <1% for control and polymer–Dox treatments). Importantly, the levels of apoptosis in the heart following treatment with HB-SS-HH-Dox and CE-HB-SS-Dox were comparable to that of the control group, with no significant increase noted. This observation indicates low levels of cardio-toxicity of both polymer prodrug systems in this model which, in agreement with the histopathological evaluation of H&E-stained tissues, suggests an important advantage for the pHPMA–Dox conjugates over the free drug, given the high levels of apoptosis noted for the free Dox treatment and the well documented side-effects observed following Dox treatments.41 Similarly for the liver, free Dox treatment exhibited a significant increase in apoptosis levels as compared to the control and polymer–Dox treatments (36.4 ± 3.2% for free Dox, 6.5 ± 4.8% for HB-SS-HH-Dox, and ≤1% for CE-HB-SS-Dox and control). Despite the apparent slightly elevated apoptosis levels observed for HB-SS-HH-Dox, this observation was found to be not statistically significant, indicating low hepatotoxicity of both of the polymer prodrugs in contrast to the reported hepatotoxicity of Dox treatments.42 Finally, a significant increase in levels of apoptosis was noted in the spleen for all Dox treatment groups as compared to the control group (39.2 ± 6.2% for free Dox, 32.3 ± 7.3% for HB-SS-HH-Dox, 34.8 ± 4.9% for CE-HB-SS-Dox and 1.4 ± 2.5% for control group). The conjugation of Dox to the polymer structures decreased the observed splenic toxicity, an observation which has previously been reported in the literature.43
The results combined from biodistribution, tissue and organ accumulation/deposition and quantification of toxicity showed some significant potential advantages of the pHPMA polymer Dox conjugates over the free drug and indicated the importance of evaluating anti-cancer efficacy by methods beyond those measuring tumour volume. The data also suggested that transport of the polymer pro-drugs was different from free drug not only in circulation but also in tissue. In previous work we have shown that polymer nanoparticles with size ranges of 30–160 nm can traffic through 3D cancer cell spheroids more effectively than small molecule drugs,37,44,45 and others have described particle ‘hopping’ and transcytosis mechanisms which may enhance penetration of nanoparticles compared to conventional drug compounds.46–50 It is likely that the enhanced penetration of the pHPMA pro-drugs in the tumour regions was affected, at least in part, by transcytosis of the polymers from the cells adjacent to vasculature and that this was a more effective process than passive diffusion of Dox into these regions. However, there will have been other factors involved in the intra- and inter-cellular trafficking, and also in the anti-cancer activities of the polymer–doxorubicin conjugates. There have been many studies describing the effects of nanoparticle size and shape in terms of transport into tumours,34,48,51–54 and factors such as surface hydrophilicity, protein corona formation and variation have long been known to influence the biodistribution and end-fate of polymers and nanoparticles.55,56 However, for the pHPMA polymers in this study, there were two additional specific factors to consider, i.e. the reducible disulfide links in the pHPMA branching regions and the acid-cleavable hydrazone link between doxorubicin and the polymer main chains. The hydrodynamic diameters of the polymers as measured by DLS after their preparation were 10 nm (HB-SS-HH) and 15 nm (CE-HB-SS), and their sizes as measured by TEM were 9.0 ± 1.8 nm (n = 30) and 14.8 nm ± 3.6 nm (n = 21), respectively. In the absence of reducing conditions the particle sizes did not change over 24 h but in the presence of 10 mM concentrations of glutathione (GSH), both polymers displayed particle sizes below 10 nm as measured by DLS within 24 h.37 While it is known that GSH is present at much higher concentrations in the intracellular space than in the bloodstream, it is not possible to discount the enhanced penetration of the polymers in this study in tumours to partial fragmentation in hypoxic regions of the tumours. In addition, in some solid tumours, the extracellular space can be more acidic, and any acid-catalysed hydrolysis of the hydrazine linkage of Dox to the pHPMA backbones may have altered the conformation and thus apparent ‘size’ of the polymers in these regions. Previous reports have indicated changes in conformation of pHPMA polymer pro-drugs due to drug conjugation,57 and thus any release of Dox, even if incomplete from each polymer chain, may have altered the apparent size and shape of the pHPMA materials and affected their penetration through the tissue. It is further possible that gradual release of Dox in this way accounted for the greater extent of apoptosis and necrosis in the tumours as opposed to other organs with more ‘normal’ pH and redox environment. In turn, this may partly account for the apparent lack of correlation between apoptosis and overall tumour volume reduction by the polymer pro-drugs and free Dox in this study. As noted, we dosed at a lower level of Dox equivalent than in our prior work in order to probe effects of polymer size and shape rather than to maximise cytotoxicity, and therefore it is perhaps not too surprising that although we saw enhanced apoptosis in tumour tissue with the polymer pro-drugs compared to free doxorubicin, the overall levels were not sufficient to induce tumour shrinkage.
It is also important to frame some of the discussions with the significant caveats around the actual sizes and shapes of the pHPMA materials in vivo. Although we saw no evidence for size changes or aggregation of the polymers in vitro, in transit in the body the particles may have adsorbed proteins the bloodstream or bound to ECM components in tissue, altering their effective size and shape in specific biological locations. It has previously been shown that some pHPMA polymer nanoparticles are highly resistant to protein corona formation,58 and so we think it unlikely that the polymers in this study would have an adsorbed protein layer, however a size measurement made in buffer or in a dehydrated state in TEM cannot be directly related to the size of the polymers in plasma. Furthermore, under flow these polymers may not have retained their ‘hyperbranched’ and ‘pseudo-star’ morphologies, and adopted more anisotropic architectures in regions of high blood flow or interstitial pressures.59 The sizes of both HB-SS-HH and CE-HB-SS as measured by DLS and TEM are above the normal accepted threshold for renal clearance, but DLS sizes are based on an algorithm which assumes spherical and non-interacting particles, which may not be the case in vivo. Therefore, within certain tissues such as the kidney, it may have been possible for the smaller HB-SS-HH polymer to have deformed to non-spherical structures with at least one dimension below 10 nm, accounting for the more rapid elimination of this polymer compared to CE-HB-SS. Changes in polymer architecture between branched and star architectures have previously been shown to alter renal accumulation for polymers with molar masses above the renal exclusion limit59 and it is possible that a similar phenomenon may have occurred here, albeit for a hyperbranched compared to a chain-extended hyperbranched polymer. Finally, the changes in tissue penetration for the two polymers, with HB-SS-HH showing greater tumour accumulation compared to CE-HB-SS, may be considered a function of multiple parameters, including polymer size, deformability under flow, and degradation kinetics. Polymer CE-HB-SS was not only higher in molar mass than HB-SS-HH (126 kDa compared to 58 kDa) but the longer chains extending away from the disulfide functional cross-links may have rendered this polymer less susceptible to reductive degradation in vivo than HB-SS-HH, due to reduced access of glutathione into the ‘core’ of CE-HB-SS. If these hypotheses are correct, polymer CE-HB-SS would be expected to fragment less in a reductive tumour microenvironment than the smaller and possibly more ‘open’ polymer HB-SS-HH. However, a full analysis of polymer conformation and variations of degradation kinetics in individual tissues in vivo is not currently possible with existing imaging techniques. Therefore, in the absence of definitive data, it is possible only to infer that the behaviour of the materials, as reported by their penetration into tissue and the effects of drugs released by them in situ, are related to their original structures and architectures.
Nevertheless, the results obtained indicated that the polymer prodrugs were better at inducing apoptotic changes in cells as compared to free drugs, and thus might display improved therapeutic properties if developed further. It could also be suggested that the physical properties of the polymer prodrugs, such as shape, size, architecture and molar mass, had an impact on the therapeutic effect of the systems as highlighted by the increased tumour deposition and hence, significantly increased levels of apoptosis observed for the smaller hyperbranched HB-SS-HH-Dox as compared to the CE-HB-SS. We are currently preparing variants of these polymers with a range of release mechanisms and drug cargoes to probe these phenomena further.
Conclusions
This study has shown that polymer prodrug pHPMA nanoparticles exhibit some potential as therapeutics for triple negative breast cancer. No significant side effects were noted in mice following treatment with either HB-SS-HH-Cy5 or CE-HB-SS-Cy5, and very low organ damage was apparent, as determined by H&E analysis. In vivo efficacy studies revealed no significant change in tumour volume between the control group or the free Dox, HB-S-HH-Dox and CE-HB-SS-Dox treated groups, but tissue level analysis indicated variance between the efficacy of polymer–Dox conjugates and free Dox. Higher levels of Dox in tumours were noted by fluorescence microscopy for polymer–Dox treated groups as compared to the free Dox treated group, which in turn resulted in an increase in the levels of apoptosis, as determined qualitatively by H&E analysis and quantitatively by a TUNEL assay. Furthermore, a decrease in cardio-, hepato- and splenic toxicity was noted, and importantly levels of cardiotoxicity for the polymer–Dox conjugates were comparable to the control group. These data demonstrate that the 3D architecture, molar mass and size of HPMA-based nanoparticles are important factors governing the biological activity of polymer–Dox conjugates. In particular, polymer HB-SS-HH-Dox, the smaller of the polymer–Dox conjugates studied here in terms of hydrodynamic diameter and molar mass, exhibited higher levels of tumour deposition and increased apoptosis compared to its CE-HB-SS-Dox counterpart. Overall, the enhanced apoptosis induction in cancerous cells and reduced cardio-, hepato- and splenic toxicity of the pHPMA-based polymer pro-drugs suggest that these materials should be investigated further across different dosing regimes and pre-clinical models for enhanced anti-cancer efficacy.Data availability
All relevant data are available on request via the University of Nottingham or cameron.alexander@nottingham.ac.uk.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank UKRI/EPSRC for funding [grants EP/N006615/1, EP/H005625/1, EP/N03371X/1, EP/N006615/1 and EP/S009000/1], the Royal Society [Wolfson Research Merit Award WM150086 to CA]. We acknowledge Dr David Onion for expert assistance in FACS data analysis. We also thank Tom Hyde, Esme Ireson and Paul Cooling for expert technical support. The Nanoscale & Macroscale Research Centre (NMRC) is acknowledged for providing the facilities for TEM and related analysis. We thank the School of Life Sciences imaging facility (SLIM) and their staff for use of their facilities. The animal experiments were approved by the UK Home Office under the Licence number PPL P435A9CF8.References
- L. Cao and Y. Niu, Triple Negative Breast Cancer: Special Histological Types and Emerging Therapeutic Methods, Cancer Biol. Med., 2020, 17, 293–306 CAS.
- L. Yin, J.-J. Duan, X.-W. Bian and S.-C. Yu, Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress, Breast Cancer Res., 2020, 22, 61 CrossRef PubMed.
- G. Bianchini, J. M. Balko, I. A. Mayer, M. E. Sanders and L. Gianni, Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease, Nat. Rev. Clin. Oncol., 2016, 13, 674–690 CrossRef CAS PubMed.
- P. F. Monteiro, M. Gulfam, C. J. Monteiro, A. Travanut, T. Fedatto Abelha, A. K. Pearce, C. Jerôme, A. M. Grabowska, P. A. Clarke, H. M. Collins, D. M. Heery, P. Gershkovich and C. Alexander, Synthesis of Micellar-like Terpolymer Nanoparticles with Reductively Cleavable Cross-links and Evaluation of Efficacy in 2D and 3D Models of Triple Negative Breast Cancer, J. Controlled Release, 2020, 323, 549–564 CrossRef CAS PubMed.
- L. Smith, M. B. Watson, S. L. O'Kane, P. J. Drew, M. J. Lind and L. Cawkwell, The Analysis of Doxorubicin Resistance in Human Breast Cancer Cells Using Antibody Microarrays, Mol. Cancer Ther., 2006, 5, 2115–2120 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems, J. Pharm. Pharmacol., 2013, 65, 157–170 CrossRef CAS PubMed.
- C. J. Lovitt, T. B. Shelper and V. M. Avery, Doxorubicin Resistance in Breast Cancer Cells is Mediated by Extracellular Matrix Proteins, BMC Cancer, 2018, 18, 41 CrossRef PubMed.
- A. Gabizon, R. Shiota and D. Papahadjopolous, Pharmacokinetics and Tissue Distribution of Doxorubicin Encapsulated in Stable Liposomes With Long Circulation Times, J. Natl. Cancer Inst., 1989, 81, 1484–1488 CrossRef CAS PubMed.
- F. M. Muggia, J. D. Hainsworth, S. Jeffers, P. Miller, S. Groshen, M. Tan, L. Roman, B. Uziely, L. Muderspach, A. Garcia, A. Burnett, F. A. Greco, C. P. Morrow, L. J. Paradiso and L. J. Liang, Phase II Study of Liposomal Doxorubicin in Refractory Ovarian Cancer: Antitumor Activity and Toxicity Modification by Liposomal Encapsulation, J. Clin. Oncol., 1997, 15, 987–993 CrossRef CAS PubMed.
- I. Ekladious, Y. L. Colson and M. W. Grinstaff, Polymer-drug Conjugate Therapeutics: Advances, Insights and Prospects, Nat. Rev. Drug Discovery, 2019, 18, 273–294 CrossRef CAS PubMed.
- Y. Li, C. Ye, C. Cai, M. Zhao, N. Han, Z. Liu, J. Zhai and J. Yin, Design and Synthesis of Polymer Prodrugs for Improving Water-Solubility, Pharmacokinetic Behavior and Antitumor Efficacy of TXA9, Pharm. Res., 2020, 37, 66 CrossRef CAS PubMed.
- Z. Gu, Y. Dong, S. Xu, L. Wang and Z. Liu, Molecularly Imprinted Polymer-Based Smart Prodrug Delivery System for Specific Targeting, Prolonged Retention, and Tumor Microenvironment-Triggered Release, Agnew. Chem., Int. Ed., 2021, 60, 2663–2667 CrossRef CAS PubMed.
- M. Zhou, L. Wen, C. Wang, Q. Lei, Y. L. Li and X. Yi, Recent Advances in Stimuli-Sensitive Amphiphillic Polymer-Paclitaxel Prodrugs, Front. Bioeng. Biotechnol., 2022, 10, 875034 CrossRef PubMed.
- C. E. Vasey, R. J. Cavanagh, V. Taresco, C. Moloney, S. Smith, R. Rahman and C. Alexander, Polymer Pro-Drug Nanoparticles for Sustained Release of Cytotoxic Drugs Evaluated in Patient-Derived Glioblastoma Cell Lines and In Situ Gelling Formulations, Pharmaceutics, 2021, 13, 208 CrossRef CAS PubMed.
- F. Sodano, R. J. Cavanagh, A. K. Pearce, L. Lazzarato, B. Rolando, A. Fraix, T. F. Abelha, C. E. Vasey, C. Alexander, V. Taresco and S. Sortino, Enhancing Doxorubicin Anticancer Activity with a Novel Polymeric Platform Photoreleasing Nitric Oxide, Biomater. Sci., 2020, 8, 1329 RSC.
- A. K. Pearce, J. D. Simpson, N. L. Fletcher, Z. H. Houston, A. V. Fuchs, P. J. Russell, A. K. Whittaker and K. J. Thurecht, Localised delivery of doxorubicin to prostate cancer cells through a PSMA-targeted hyperbranched polymer theranostic, Biomaterials, 2017, 141, 330–339 CrossRef CAS PubMed.
- A.-K. Noack, H. Lucas, P. Chytil, T. Etrych, K. Mader and T. Mueller, Intratumoral Distribution and pH-Dependent Drug Release of High Molecular Weight HPMA Copolymer Drug Conjugates Strongly Depend on Specific Tumor Substructure and Microenvironment, Int. J. Mol. Sci., 2020, 21, 6029 CrossRef CAS PubMed.
- P. Chytil, L. Kostka and T. Etrych, HPMA Copolymer-Based Nanomedicines in Controlled Drug Delivery, J. Pers. Med., 2021, 11, 115 CrossRef PubMed.
- J. Kopecek and H. Bazilova, Poly[N-(2-hydroxypropyl)methyacrylamide] – I. Radical Polymerization and Copolymerization, Eur. Polym. J., 1973, 9, 7–14 CrossRef CAS.
- M. Bohdanecky, H. Bazilova and J. Kopecek, Poly[N-(2-hydroxypropyl)methyacrylamide] – II: Hydrodynamic Properties of Dilute Solutions, Eur. Polym. J., 1974, 10, 405–410 CrossRef CAS.
- J. Yang, R. Zhang, H. Pan, Y. Li, Y. Fang, L. Zhang and J. Kopecek, Backbone Degradable N-(2-Hydroxypropy)methacrylamide Copolymer Conjugates with Gemcitabine and Paclitaxel: Impact of Molecular Weight on Activity toward Human Ovarian Carcinoma Xenografts, Mol. Pharm., 2017, 14, 1384–1394 CrossRef CAS PubMed.
- H. Pan, M. Sima, S. C. Miller, P. Kopeckova, J. Yang and J. Kopecek, Efficiency of High Polymer Weight Backbone Degradable HPMA Copolymer-Prostaglandin E1 Conjugate in Promotion of Bon Formation in Ovariectomized Rats, Biomaterials, 2013, 34, 6528–6538 CrossRef CAS PubMed.
- L. W. Seymour, D. R. Ferry, D. J. Kerr, D. Rea, M. Whitlock, R. Poyner, C. Boivin, S. Hesslewood, C. Twelves, R. Blackie, A. Schatzlein, D. Jodrell, D. Bissett, H. Calvert, M. Lind, A. Robbins, S. Burtles, R. Duncan and J. Cassidy, Phase II Studies of Polymer-Doxorubicin (PK1, FCE28068) in the Treatment of Breast, Lung and Colorectal Cancer, Int. J. Oncol., 2009, 34, 1629–1636 CrossRef CAS PubMed.
- L. W. Seymour, D. R. Ferry, D. Anderson, S. Hesslewood, P. J. Julyan, R. Poyner, J. Doran, A. M. Young, S. Burtles and D. J. Kerr, Hepatic Drug Targeting: Phase 1 Evaluation of Polymer-Bound Doxorubicin, J. Clin. Oncol., 2002, 20, 1668–1676 CrossRef CAS PubMed.
- R. Williams, Discontinued Drugs in 2008: Oncology Drugs, Expert Opin. Invest. Drugs, 2009, 18, 1581–1594 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat. Nanotechnol., 2011, 6(12), 815–823 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, The entry of nanoparticles into solid tumours, Nat. Mater., 2020, 566–575 CrossRef CAS PubMed.
- W. S. Saw, T. Anasamy, Y. Y. Foo, Y. C. Kwa, C. S. Kue, C. H. Yeong, L. V. Kiew, H. B. Lee and L. Y. Chung, Delivery of Nanoconstructs in Cancer Therapy: Challenges and Therapeutic Opportunities, Adv. Ther., 2021, 4(3), 2000206 CrossRef CAS.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, The Role of Micelle Size in Tumor Accumulation, Penetration, and Treatment, ACS Nano, 2015, 9(7), 7195–7206 CrossRef CAS PubMed.
- Q. Xu, L. M. Ensign, N. J. Boylan, A. Schon, X. Gong, J. C. Yang, N. W. Lamb, S. Cai, T. Yu, E. Freire and J. Hanes, Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo, ACS Nano, 2015, 9(9), 9217–9227 CrossRef CAS PubMed.
- J. Zhen, S. Tian, Q. Liu, C. Zheng, Z. Zhang, Y. Ding, Y. An, Y. Liu and L. Shi, Nanocarriers responsive to a hypoxia gradient facilitate enhanced tumor penetration and improved anti-tumor efficacy, Biomater. Sci., 2019, 7, 2986 RSC.
- Q. Zhou, S. Shao, J. Wang, C. Xu, J. Xiang, Y. Piao, Z. Zhou, Q. Yu, J. Tang, X. Liu, Z. Gan, R. Mo, Z. Gu and Y. Shen, Enzyme-activatable polymer–drug conjugate augments tumour penetration and treatment efficacy, Nat. Nanotechnol., 2019, 799–809 CrossRef CAS PubMed.
- J. Liu, H. J. Li, Y. L. Luo, C. F. Xu, X. J. Du, J. Z. Du and J. Wang, Enhanced Primary Tumor Penetration Facilitates Nanoparticle Draining into Lymph Nodes after Systemic Injection for Tumor Metastasis Inhibition, ACS Nano, 2019, 13(8), 8648–8658 CrossRef CAS PubMed.
- L. D. Deng, Z. J. Feng, H. Z. Deng, Y. J. Jiang, K. Song, Y. L. Shi, S. Q. Liu, J. H. Zhang, S. P. Bai, Z. H. Qin and A. J. Dong, Rational Design of Nanoparticles to Overcome Poor Tumor Penetration and Hypoxia-Induced Chemotherapy Resistance: Combination of Optimizing Size and Self-Inducing High Level of Reactive Oxygen Species, ACS Appl. Mater. Interfaces, 2019, 11(35), 31743–31754 CrossRef CAS PubMed.
- Z. Xiang, X. Yang, G. Jiang, D. Fan, L. Geng, H. Wang, Z. Hu and Q. Fang, Design of a Simple and Practical Nanosystem Coordinates Tumor Targeting and Penetration for Improved Theranostics, Adv. Ther., 2019, 2(2), 1800107 CrossRef.
- L. Kostka, L. Kotrchová, V. Šubr, A. Libánská, C. A. Ferreira, I. Malátová, H. J. Lee, T. E. Barnhart, J. W. Engle, W. Cai, M. Šírová and T. Etrych, HPMA-based star polymer biomaterials with tuneable structure and biodegradability tailored for advanced drug delivery to solid tumours, Biomaterials, 2020, 235, 119728 CrossRef CAS PubMed.
- A. K. Pearce, A. B. Anane-Adjei, R. J. Cavanagh, P. F. Monteiro, T. M. Bennett, V. Taresco, P. A. Clarke, A. A. Ritchie, M. R. Alexander, A. M. Grabowska and C. Alexander, Effects of Polymer 3D Architecture, Size and Chemistry on Biological Transport and Drug Delivery In Vitro and in Orthotopic Triple Negative Breast Cancer Models, Adv. Healthcare Mater., 2020, 9, 2000892 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, NIH Image to ImageJ: 25 years of image analysis, Nat. Methods, 2012, 9(7), 671–675 CrossRef CAS PubMed.
- K. Kyrylkova, S. Kyryachenko, M. Leid and C. Kioussi, Detection of Apoptosis by TUNEL Assay, in Odontogenesis. Methods in Molecular Biology, ed. C. Kioussi, Humana Press, 2012, pp. 41–47 Search PubMed.
- M. Volkova and R. Russell, Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment, Curr. Cardiol. Rev., 2011, 7, 214–220 CrossRef CAS PubMed.
- Y. Kalender, M. Yel and S. Kalender, Doxorubicin Hepatotoxicity and Hepatic Free Radical Metabolism in Rats: The Effects of Vitamin E and Catechin, Toxicology, 2005, 209, 39–45 CrossRef CAS PubMed.
- J. Liang, R. Guo, M. Xuan, Q. Sun and W. Wu, An Acid-Sensitive Nanofiber Conjugate Based on a Short Aromatic Peptide for Targeted Delivery of Doxorubicin in Liver Cancer, Int. J. Nanomed., 2022, 17, 2961–2973 CrossRef PubMed.
- V. Taresco, T. F. Abelha, R. J. Cavanagh, C. E. Vasey, A. B. Anane-Adjei, A. K. Pearce, P. F. Monteiro, K. A. Spriggs, P. Clarke, A. Ritchie, S. Martin, R. Rahman, A. M. Grabowska, M. B. Ashford and C. Alexander, Functionalized Block Co-Polymer Pro-Drug Nanoparticles with Anti-Cancer Efficacy in 3D Spheroids and in an Orthotopic Triple Negative Breast Cancer Model, Adv. Ther., 2020, 4(1), 2000103 CrossRef.
- W.-H. Hsu, P. Sanchez-Gomez, E. Gomez-Ibarlucea, D. Ivanov, R. Rahman, A. M. Grabowska, N. Csaba, C. Alexander and M. Garcia-Fuentes, Structure-optimized interpolymer polyphosphazene complexes for effective gene delivery against glioblastoma, Adv. Ther., 2019, 2(3), 1800126 CrossRef.
- H. Lu, J. Su, R. Mamdooh, Y. Li and M. H. Stenzel, Cellular Uptake of Gold Nanoparticles and Their Movement in 3D Multicellular Tumor Spheroids: Effect of Molecular Weight and Grafting Density of Poly(2-hydroxyl ethyl acrylate), Macromol. Biosci., 2020, 20(1), 1–11 CrossRef PubMed.
- H. Lu, R. H. Utama, U. Kitiyotsawat, K. Babiuch, Y. Jiang and M. H. Stenzel, Enhanced transcellular penetration and drug delivery by crosslinked polymeric micelles into pancreatic multicellular tumor spheroids, Biomater. Sci., 2015, 3(7), 1085–1095 RSC.
- W. Fan, Q. Wei, J. Xiang, Y. Tang, Q. Zhou, Y. Geng, Y. Liu, R. Sun, L. Xu, G. Wang, Y. Piao, S. Shao, Z. Zhou, J. Tang, T. Xie, Z. Li and Y. Shen, Mucus Penetrating and Cell-Binding Polyzwitterionic Micelles as Potent Oral Nanomedicine for Cancer Drug Delivery, Adv. Mater., 2022, 34(16), 2109189 CrossRef CAS PubMed.
- Q. Zhou, C. Dong, W. Fan, H. Jiang, J. Xiang, N. Qiu, Y. Piao, T. Xie, Y. Luo, Z. Li, F. Liu and Y. Shen, Tumor extravasation and infiltration as barriers of nanomedicine for high efficacy: The current status and transcytosis strategy, Biomaterials, 2020, 240, 119902 CrossRef CAS PubMed.
- D. P. McIntosh, X. Y. Tan, P. Oh and J. E. Schnitzer, Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: a pathway to overcome cell barriers to drug and gene delivery, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(4), 1996–2001 CrossRef CAS PubMed.
- J. L. Y. Wu, B. P. Stordy, L. N. M. Nguyen, C. P. Deutschman and W. C. W. Chan, A proposed mathematical description of in vivo nanoparticle delivery, Adv. Drug Delivery Rev., 2022, 189, 114520 CrossRef CAS PubMed.
- J. W. Myerson, A. C. Anselmo, Y. Liu, S. Mitragotri, D. M. Eckmann and V. R. Muzykantov, Non-affinity factors modulating vascular targeting of nano- and microcarriers, Adv. Drug Delivery Rev., 2016, 99, 97–112 CrossRef CAS PubMed.
- Q. Hu, C. J. F. Rijcken, E. van Gaal, P. Brundel, H. Kostkova, T. Etrych, B. Weber, M. Barz, F. Kiessling, J. Prakash, G. Storm, W. E. Hennink and T. Lammers, Tailoring the physicochemical properties of core-crosslinked polymeric micelles for pharmaceutical applications, J. Controlled Release, 2016, 244, 314–325 CrossRef CAS PubMed.
- T. Lammers, F. Kiessling, W. E. Hennink and G. Storm, Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress, J. Controlled Release, 2012, 161(2), 175–187 CrossRef CAS PubMed.
- D. Y. Chen, N. Parayath, S. Ganesh, W. M. Wang and M. Amiji, The role of apolipoprotein- and vitronectin-enriched protein corona on lipid nanoparticles for in vivo targeted delivery and transfection of oligonucleotides in murine tumor models, Nanoscale, 2019, 11(40), 18806–18824 RSC.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Delivery Rev., 2016, 99(Pt A), 28–51 CrossRef CAS PubMed.
- A. Paul, M. J. Vicent and R. Duncan, Using small-angle neutron scattering to study the solution conformation of N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin conjugates, Biomacromolecules, 2007, 8(5), 1573–1579 CrossRef CAS PubMed.
- I. Alberg, S. Kramer, M. Schinnerer, Q. Z. Hu, C. Seidl, C. Leps, N. Drude, D. Moeckel, C. Rijcken, T. Lammers, M. Diken, M. Maskos, S. Morsbach, K. Landfester, S. Tenzer, M. Barz and R. Zentel, Polymeric Nanoparticles with Neglectable Protein Corona, Small, 2020, 16(18), 1907574 CrossRef CAS PubMed.
- A. Duro-Castano, R. M. England, D. Razola, E. Romero, M. Oteo-Vives, M. Angel Morcillo and M. J. Vicent, Well-Defined Star-Shaped Polyglutamates with Improved Pharmacokinetic Profiles As Excellent Candidates for Biomedical Applications, Mol. Pharm., 2015, 12(10), 3639–3649 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm00609c |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |