
Inhibition and eradication of bacterial biofilm using polymeric materials
Arnab
Banerjee
a,
Pampa
Chowdhury
a,
Kamal
Bauri
b,
Biswajit
Saha
*c and
Priyadarsi
De
 *a
*a
aPolymer Research Centre and Centre for Advanced Functional Materials, Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur-741246, Nadia, West Bengal, India. E-mail: p_de@iiserkol.ac.in
bDepartment of Chemistry, Raghunathpur College, Raghunathpur-723133, Purulia, West Bengal, India
cDepartment of Chemical and Biomedical Engineering, FAMU-FSU College of Engineering, Tallahassee, Florida 32310, USA. E-mail: bsaha@eng.famu.fsu.edu
First published on 17th October 2022
Abstract
Biofilms, ubiquitous in nature, are three-dimensional complex microbial communities sheathed in a self-secreted extracellular polymeric matrix. Infections caused by these communities have sprouted as serious threats to global healthcare systems due to their intrinsic tolerance toward conventional antibiotics. There is a huge demand for alternative “cutting-edge” materials featuring strong antibiofilm abilities to mitigate and/or exterminate pre-matured biofilms. Natural or synthetic macromolecule-based compounds have evolved as one of the most sought-after materials because of their unique stimulus-directed selective targeting efficiency to the bacterial cell, antibiotic-encapsulation ability endowing them with a synergistic effect, and highly dense embedded cationic functionalities that promote accumulation within the biofilm. In this comprehensive review, we aim to highlight the progress made in inhibiting or eradicating bacterial biofilms using various forms of polymeric material including cationic and charge-switchable macromolecules, conjugated polymers, polymeric metal nanocomposites, hydrogels, and supramolecular polymers. We particularly emphasize understanding the underlying antibiofilm mechanisms of each presented example ushered in by state-of-the-art synthetic strategies. Lastly, focusing on bench-to-bedside, the review is concluded by providing some forthcoming aspects and possible future development directions to expand polymer-based antibiofilm research, keeping their clinical translations in mind.
1. Introduction
Since the inception of penicillin in 1941, the infections caused by bacteria have presented a major challenge and burden to healthcare systems owing to their high tolerance to antibiotics, as classified by the World Health Organization (WHO). They have accounted for approximately 7 million deaths per year that could rise to 10 million in 2050 if effective therapies are not discovered.1 The key contributor (around 80%)2 to human-related antibiotic resistance and infections is associated with bacteria/fungi-formed biofilms, spatially organized three-dimensional (3D) microbial communities surrounded by self-made extracellular polymeric substances (EPS), composed of fibrin, extracellular DNA, nucleic acid, and exopolysaccharides.3,4 Interestingly, 90% of the biofilm volume is mainly occupied by the EPS matrix; the remaining 10% is the dry weight of the microbes. The EPS matrix plays a critical role in the survival of bacteria: they are often vascularized by an extensive network of water channels that are used to transport oxygen and nutrients to the bacteria and reject metabolic waste.5 Moreover, the matrix empowers biofilms with great mechanical plasticity and allows them to grow even under hostile conditions. For instance, biofilms alter their metabolism by switching from respiration to fermentation if the required oxygen supply is limited. Whilst biofilms have found their way to providing a handful of biotechnological solutions such as biodiesel production,6 water remediation,7 and biofertilizers,8 they simultaneously cause serious threats to public health,9,10 especially to immunocompromised patients who require medical implants, organ transplants, artificial limbs, or external life support.A vast majority of the research in this field essentially emphasizes the inhibition of biofilm development or its extermination at an early stage. Pre-matured stubborn biofilms, however, are difficult to eliminate because of their intrinsic resistance to antibiotics. Hence, it is imperative to decipher the opportunities for therapeutic interventions. Based on the biofilm life cycle, the inhibition and/or abolition of these communities can be achieved by reducing the generation of EPS, targeting the pathogenic environments (low pH, hypoxia), degrading the EPS matrix, and physical removal. Unlike planktonic bacteria, the implementation of these excision methods for aged biofilms is allied with a few shortcomings, viz. painful ultrasound surgery, high rates of recurrence,11 antibiotic-mediated inefficient biochemical clearance,12,13etc. This fact raises the question of how the antibiotic tolerance level of biofilm and planktonic bacteria differs. Answering this requires deeper insights into the 3D biofilms that involve the following facets: (a) the impermeability of antibiotics restricts drug diffusion, which is referred to as the “barrier effect”,14,15 (b) positively charged antibiotics often being absorbed by negatively charged EPS,16 and (c) the existence of antibiotic-resistant bacterial species. This shortfall of antibiotics has driven an urgent search for “cutting-edge” alternative antibiofilm materials, thus becoming a need of the hour.
In this spotlight, diverse materials including small molecules,17,18 metal oxides, carbon nanomaterials,19 inorganic nanoparticles,20,21 and natural or synthetic macromolecular species22 have received prodigious attention from researchers as antibacterial/antibiofilm agents. Amongst these, polymer-based materials have seized special attention, bestowed by their stimulus-directed strong targeting efficacy to bacterial cells, antibiotic encapsulation ability to endow a synergistic effect, and most importantly embedded cationic functionalities which promote their internalization within the biofilm, thereby exerting antibiofilm activity. Of note, they can also act as surface ligands to improve the stability and antibiofilm efficacy of oxidation-susceptible antibacterial silver nanoparticles (AgNPs). Because of these fascinating features, tremendous growth towards the fabrication of potent polymeric antibiofilm agents has been witnessed over the past few years and eventually emerged as state-of-the-art materials. Such efforts have been reviewed extensively very recently,23,24 highlighting the use of polymeric nanomaterials or coatings,25,26 peptides, and natural proteins for the prevention and disruption of biofilms. In this paper, the prime intention is to review the antibiofilm performances of other dimensions of polymeric materials ranging from cationic macromolecules, biopolymers, and polymer nanocomposites to hydrogels (Fig. 1). To comprehensively portray the advancements made so far, we divided the whole review into two sections – biopolymer and synthetic macromolecule-based antibiofilm agents – and then subdivided them in terms of various polymeric forms. We then enrich the discussion by carrying out a detailed interrogation of each presented example to illustrate the antibiofilm mechanism ushered in by bespoke synthetic strategies. The review is finally concluded by providing some forthcoming prospects and plausible scope for the expansion of antibiofilm research using polymeric materials.
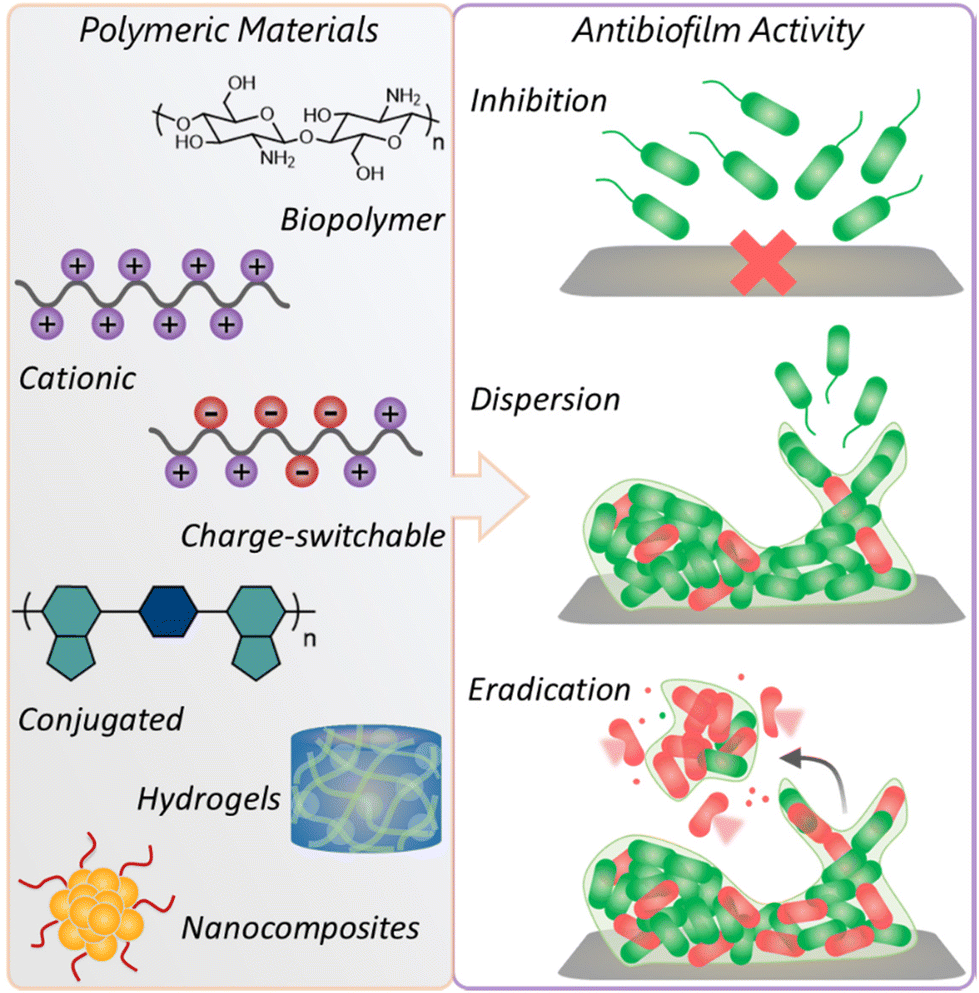 | ||
| Fig. 1 Schematic illustration demonstrating diverse classes of polymeric materials and their antibiofilm activity. | ||
2. Overview of biofilms
2.1 Biofilm characteristics
Biofilms are described as “aggregates” of microorganisms where bacterial cells are immersed in self-secure 3D matrixes of EPS that are adhering to one other and/or a surface. Liquid–solid and liquid–air interfaces,27 as well as the intersection of biotic and abiotic surfaces, are all potential sites for the development of biofilms, which can sometimes span different biological kingdoms and consist of either a single species (monomicrobial biofilms) or a variety of diverse species (polymicrobial biofilms). From inside to outside, a mature biofilm architecture respectively comprises a matrix layer, conditioned layer, connecting layer and biofilm layer. The formation and proliferation of such a 3D-structure is a dynamic process that involves four complicated steps: reversible surface colonization, microcolony formation, maturation, and senescence (dispersion and ageing).Cell–cell signalling pathways collectively referred to as quorum sensing (QS)34,35 play an integral part in the creation, maturity, and dispersion of biofilms. QS is a mechanism through which the bacterial cells communicate with each other and coordinate their behaviour. The main functions of QS are horizontal gene transfer, cell division and maintenance, host–pathogen interactions, and biofilm movement/formation. More importantly, it controls the gene expression in response to variations in nearby microbial population density, driven by the recognition and excretion of signalling molecules called QS molecules or autoinducers.36,37 Aryl homoserine lactones in Gram-negative bacteria, oligopeptides in Gram-positive bacteria, and universal boron-containing autoinducer (autoinducer-2) are the major QS molecules in bacterial pathways. Recognition of such QS molecules allows individual microorganisms to monitor the population density of other species in their local environment and react if this is below or above a certain autoinducer threshold by regulating gene expression. Thereby it promotes phenotypic diversity to prompt several biological processes, such as controlling sporulation, swarming motility, and virulence.37
From a detailed structural perspective, it is established that the bulk of the biomass in a biofilm is made up of a hydrated crosslinked network EPS. In fact, it is thought to be the key factor influencing the biofilm's physicochemical characteristics. These components help to anchor the biofilm to the surface and trap nutrients,38 offer structural support, allow the absorption of organic small molecules and inorganic ions, display enzymatic activity, enable cell-to-cell communication amongst the biofilm microbes and horizontal gene transfer and so forth. Along with performing these tasks, it further protects the microbes from radiation, desiccation, and unforeseen environmental situations. Interestingly, the biofilm-residing bacteria are capable of resisting up to 1000-fold concentrations of small antibiotics, contrary to their planktonic form, because of the EPS matrix.39
2.2 Challenges and strategies for biofilm clearance
Bacterial biofilms use a variety of strategies to avoid antibiotic treatment and mechanical stresses. Individual resistance and tolerance mechanisms alter, based on the antimicrobial agent and the biofilm's development circumstances. Biofilm-associated resistance mechanisms are often unique from the genetic mutations or gene transfer processes identified in traditional antimicrobial resistance pathways in terms of the structural and environmental aspects of biofilm. The key difficulties and problems associated with the biofilm clearance are the following: (i) The EPS matrix serves as a barrier, a place to store and shelter antibiotic-degrading enzymes, and harmful organisms, respectively, and thus retards the penetration of bactericides into the biofilm.33 (ii) eDNA present in the EPS matrix is one of the major contributors to drug resistance as it easily absorbs microbes to induce rapid DNA exchange.40 (iii) Different chemical environments such as pH fluctuation and anaerobic zones affect the stability and efficiency of the employed antibiotics. (iv) Apart from the physical and chemical resistance mechanisms, biofilms also contain a specific subpopulation of phenotypically diverse persister cells,41,42 a particular kind of bacterium. The activity of traditional antibiotics gets weakened while reaching their intended target owing to the prevention by the persister cells. Notably, such cells displayed a non-metabolic behaviour that often entirely diminished the potency of biocides. These aforementioned factors demonstrate the inefficiency of traditional strategies for biofilm removal, and thus new alternative approaches to combat bacterial biofilm-caused infections are emerging, vide infra.2.3 Existing methods for biofilm removal
In the following section, a brief overview of currently existing antibiofilm methods is discussed. Their mode of action and advantages associated with biofilm eradication are summarized in Table 1.| Methods | Examples/specifications | Mode of activity | Advantages |
|---|---|---|---|
| Electrochemical methods | Wireless electroceutical dressings (Ag/Zn redox fabric), nonthermal plasma | Production of H2O2, ROS, superoxide radicals and other species | • Cheap and environment friendly |
| • Destruction of both protein and DNA in recalcitrant biofilm | |||
| • No cytotoxicity and hemocompatibility | |||
| Antimicrobial compounds | Silver oxynitrate, nitroxoline, tert-butyl benzoquinone, etc. | • Binding of Ag+ ion with reduced thiols | • Selective permeability to bacterial cells and damage persister cells |
| • Positive charge-promoted membrane perturbation | • Various stimulus-triggered treatments are feasible | ||
| • Destruction of biofilm anchoring proteins | |||
| Drug delivery methods | Amikacin encapsulated in liposomes (Arikace), dextran-based hydrogels, etc. | Release of encapsulated model drugs via stimulus-responsive degradation of self-assembled nanocarriers | • Minimal risk of passive targeting |
| • Avoid unwanted interactions with EPS |
Even though these methods offer bright future perspectives in combating biofilm-caused infections, their implementation is not very advantageous because the compound/drug that directly kills microorganisms risks triggering the emergence of resistance, rendering them ineffective in the long run, as seen with antibiotics. Hence, it is difficult to predict and compare the potentiality of the aforesaid biofilm removal methods by considering their long-term consequences, including the impacts of beneficial organisms in the microbiome.
3. Biopolymer-based antibiofilm agents
The usage of naturally occurring biopolymers with rational structural modifications as biocidal materials has garnered significant attention from researchers. Typically, their excellent biocompatibility, biodegradability, non-toxicity, and capability of rupture and penetration of the bacterial cell membrane engender biocidal properties.60,61 Amongst all, chitosan (CS), dextran and glucose-derived polymers have mainly emerged as promising antibacterial materials. The bactericidal effect of these biopolymers was examined in terms of their diverse structural forms like linear polymers, hydrogels, nanocarriers, nanocomposites, etc. that followed different antibiofilm mechanisms (Table 2).| Antibiofilm agents | Examples | Characteristics | Mode of activity | Ref. |
|---|---|---|---|---|
| Biopolymers | Chitosan-derivatives | Excellent biocompatibility, biodegradability, water-solubility, non-toxicity | • Dispersion of the biofilm matrix | 62, 64, 65, 68 and 72–77 |
| Dextran-derived hydrogels | • Reduction in EPS production | |||
| Glucose-derived polymers | • Damage of bacterial membranes and leakage of cell constituents | |||
| Synthetic macromolecules | Cationic and charge-switchable polymers | Synthetic mimics of AMPs, high proteolytic stability, stimulus-responsiveness, cost-effective preparation | • Predominantly rupturing of bacterial membranes | 84, 85, 88, 91, 92, 95, 96, 99, 101–104, 107, 108 and 112–115 |
| • Release of antibiotics | ||||
| Conjugated polymers | Toxic ROS production upon illumination | • ROS-promoted cell deactivation | 118, 120, 122 and 123 | |
| • Photothermal ablation | ||||
| Polymeric nanocomposites | Ultrahigh surface area, small volume, multifunctional | • Release of Ag+ ions | 129, 131 and 133–138 | |
| • AMF-triggered local hyperthermia | ||||
| Polymeric hydrogels | Excellent oxygen permeability, stimulus-responsiveness, reservoir for Ag+ ions and antibiotics | • Release of Ag+ ions | 144, 147–150, 153, 156 and 157 | |
| • NIR/CuS-mediated local hyperthermia | ||||
| Other polymers | Porphyrin-derived polymer | Photodynamic therapy | Production of toxic ROS | 158 |
| Magnetic mesoporous silica nanoparticles (MSNs) | Multistimulus-responsive nanocarrier | AMF-triggered release of encapsulated antibiotics | 161 | |
| Core-substituted naphthalene diimide-based 2D nanosheets | Well-defined internal order, controllable morphology | Rupturing of bacterial membranes | 162 |
Sahariah and co-workers synthetically modified CSs with different cationic and hydrophobic moieties to establish their structure–activity relationship against Staphylococcus aureus (S. aureus) biofilm (Fig. 2).62 A total of 6 series of CS-derivatives were prepared using tertiary butyldimethylsilyl (TBDMS) protection63 that allowed controlled N-selective modification of the amino group with various cationic and lipophilic functionalities. Unlike unmodified CS, these CS-derivatives showed excellent water-solubility, enabling bioactivity at a neutral pH, and moderate to strong bactericidal activity against S. aureus. In terms of the determined minimum inhibitory concentrations (MIC) and minimum bactericidal concentration (MBC) values, N-ethyl-N,N-dimethyl CS (MIC = 4 μg mL−1; MBC = 16 μg mL−1) showed much higher antibacterial activity against planktonic bacteria than the other synthesized CS compounds. On the other hand, the CS derivative having higher hydrophobicity, N-acetyl-N-stearoyl-N′,N′′,N′′-trimethyl CS, exhibited maximum S. aureus biofilm obliteration ability with a very low minimum biofilm eradication concentration (MBEC) of 128 μg mL−1. Interestingly, during biofilm elimination, the CS derivatives were not only accumulated at the surface but also dispersed throughout the biofilm matrix, corroborated by deeper level confocal laser scanning microscopy (CLSM) imaging of biofilms presenting dead cells stained in red. The enhanced effect of N-acetyl-N-stearoyl-N′,N′′,N′′-trimethyl CS arose from its suitable decoration with acetyl and stearoyl moieties along with the quaternary ammonium groups. These facts suggest that both hydrophobic (acetyl, stearoyl) and cationic groups are necessary to achieve effective antibiofilm activity.
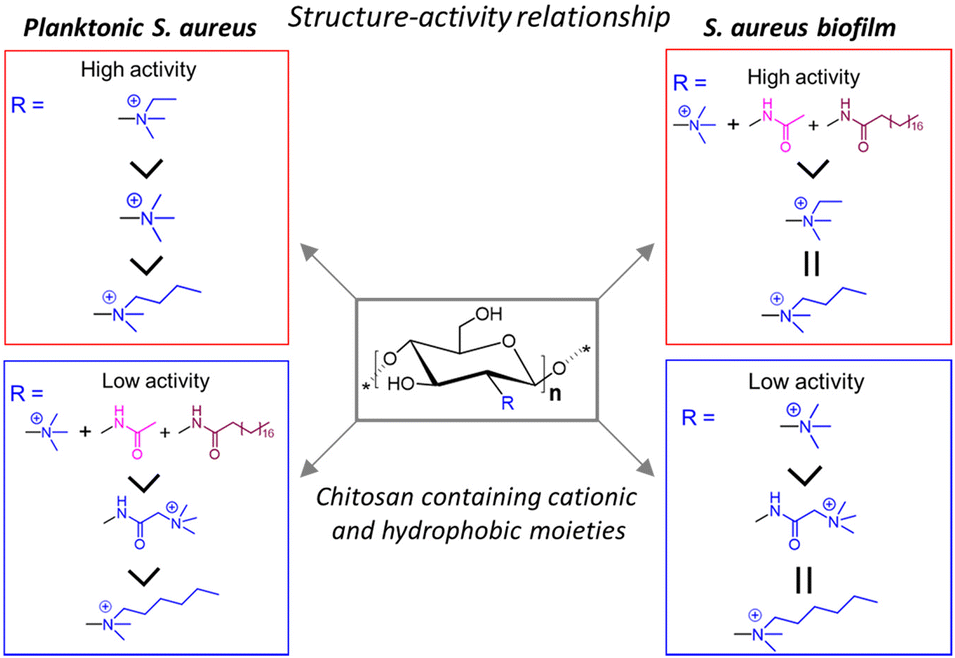 | ||
| Fig. 2 Structure–activity relationship of quaternary ammoniumyl CS-derivatives against planktonic S. aureus and S. aureus biofilms. “Reprinted from ref. 62. with permission from [American Chemical Society], Copyright [2018]”. | ||
A similar strategy was adopted by Liu et al.64 where two oppositely charged modified CS derivatives (cationic water-soluble chitosan (WCS) and anionic sulfonated chitosan (SCS)) were used to examine their ability to suppress Pseudomonas aeruginosa (P. aeruginosa) adhesion and biofilm development. Both WCS and SCS exhibited good bactericidal effects with an MIC value of 1 mg mL−1; however, the MBC value for SCS (8 mg mL−1) was significantly lower than that of WCS (16 mg mL−1). Concurrently, >90% of biofilm inhibition occurred following treatment with 8 mg mL−1 SCS, slightly higher compared with WCS with the same concentration. The CLSM and scanning electron microscopy (SEM) images demonstrated the appearance of dead cells within the biofilm and the formation of irregular morphologies with bacterial membrane damage, respectively, implying the inhibition ability of both cationic and anionic CS derivatives. In addition, a drastic decrease in metabolic activity and EPS production was observed. The superiority of SCS over WCS can be explained by its ability to form an impermeable layer surrounding the cell and block all the channels that are participating in transporting the solutes, thereby causing weakening of the cells and severe leakage of the cell constituents, leading to ultimate cell death.
Based on the antibacterial properties of CS, Liu's group researched the development of pH-adaptive nanocapsules containing CS nanoparticles (NPs) as the core to investigate the penetration ability and bactericidal effect on S. aureus biofilm.65 The nanocapsule PMPC-CS was constructed by decorating CS NPs with a thin layer of cross-linked zwitterionic phosphorylcholine-based copolymer (PMPC) synthesized via the in situ polymerization of glycerol dimethacrylate and 2-methacryloyloxyethyl phosphorylcholine (MPC). Subsequently, the surface of the structured nanocapsules was modified by partially replacing the MPC with different compositions of cationic N-(3-aminopropyl) methacrylamide (APM), resulting in the formation of other nanocapsules: P(MPC1-co-APM1)-CS and P(MPC1-co-APM4)-CS. Though the traditional cationic molecules facilitate the electrostatic attraction with bacterial membranes, the presence of an excessive amount of positive charge often decreases the biofilm penetration efficiency owing to their strong adhesion to the EPS matrix. Accordingly, the accumulation and penetration ability of the as-prepared nanocapsules was ranked as follows: P(MPC1-co-APM4)-CS < P(MPC1-co-APM1)-CS < PMPC-CS (Fig. 3). However, PMPC-CS did not have any intrinsic antibiofilm capability. Thus, the hydrophobic antimicrobial drug triclosan66,67 was encapsulated into these nanocapsules with a drug loading content (DLC) and drug loading efficiency (DLE) of 46% and 61%, respectively. All the loaded nanocapsules substantially prohibited S. aureus biofilm formation after 3 h of treatment and their activity was considerably higher at low pH 5.0 compared with physiological pH 7.4. Unlike free drugs, the triclosan-loaded CS nanocapsules have been shown to lower staphylococcal viabilities at pH 5.0 though they have similar MIC and MBC values. These results indicate that the release of triclosan in an acidic milieu is solely responsible for the biocidal activities of the nanocapsules.
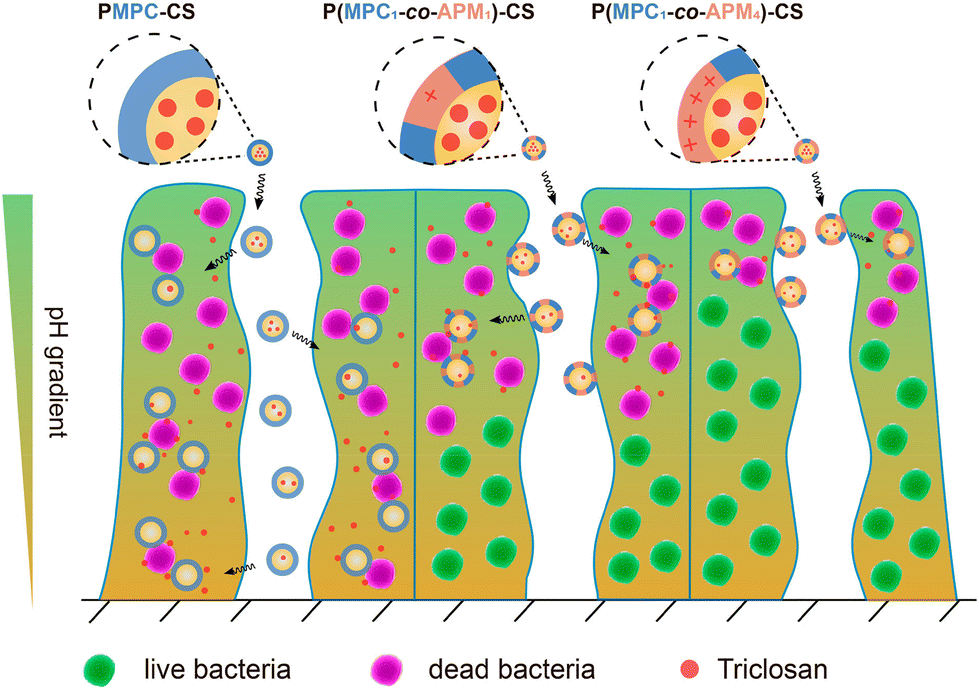 | ||
| Fig. 3 Schematic illustration of electrostatic attraction-induced S. aureus biofilm penetration by triclosan-loaded CS nanocapsules with variable APM to MPC ratios. “Reprinted from ref. 65 with permission from [American Chemical Society], Copyright [2019]”. | ||
In another report, CS together with hyaluronic acid (HA) was used as a coating material for antibiotic-loaded gelatine nanoparticles (GNPs). On account of the gelatinase- and hyaluronidase-promoted degradation, their antibiofilm properties were evaluated against Gram-negative wound-infecting bacteria Vibrio vulnificus (V. vulnificus) under biofilm mimetic conditions.68 Here, the FDA-approved tetracycline antibiotic doxycycline (Doxy),69 commonly used for the treatment of V. vulnificus-caused infections, was used as a model encapsulated drug. The GNPs were fabricated by the layer-by-layer (LBL) assembly of two oppositely charged coatings: polyanion HA and polycation CS, with the HA layer being exposed to the surroundings (Fig. 4A). To thoroughly understand their antibiofilm capacity, three types of Doxy-loaded GNP were produced, namely non-coated (Doxy-GNPs), CS-coated (CS-Doxy-GNPs), and CS-HA-coated GNPs (HA-CS-Doxy-GNPs). Amongst these, HA- and CS-coated NPs showed better biofilm penetration, visualized through CLSM imaging using the autofluorescence of GNPs (Fig. 4B). In comparison with free Doxy (MIC = 0.5 μg mL−1), the Doxy-loaded NP formulation showed a slightly lower MIC of 0.25 μg mL−1, indicating greater inhibitory power. When prefabricated biofilms were treated with HA-CS-Doxy-GNP, it was shown to furnish superior V. vulnificus biofilm penetration and eradication capabilities compared with free Doxy, with SEM demonstrating a considerable drop in EPS, and viability staining suggesting severe membrane damage (Fig. 4B). The authors hypothesized that the antibiofilm action of such GNPs follows two mechanisms: (a) degradation of the HA layer with bacterial hyaluronidases at the infection sites exposed the underlying cationic CS layer, allowing enhanced penetration and possible damage of the EPS matrix via electrostatic attraction;70 (b) swelling of the CS provided bacterial gelatinase with better access to the GNP core that eventually triggered the release of Doxy, leading to efficient bacterial cell death.71 Furthermore, the HA-CS-Doxy-GNPs were shown to be biocompatible with red blood cells (RBCs), fibroblasts, and endothelial cells.
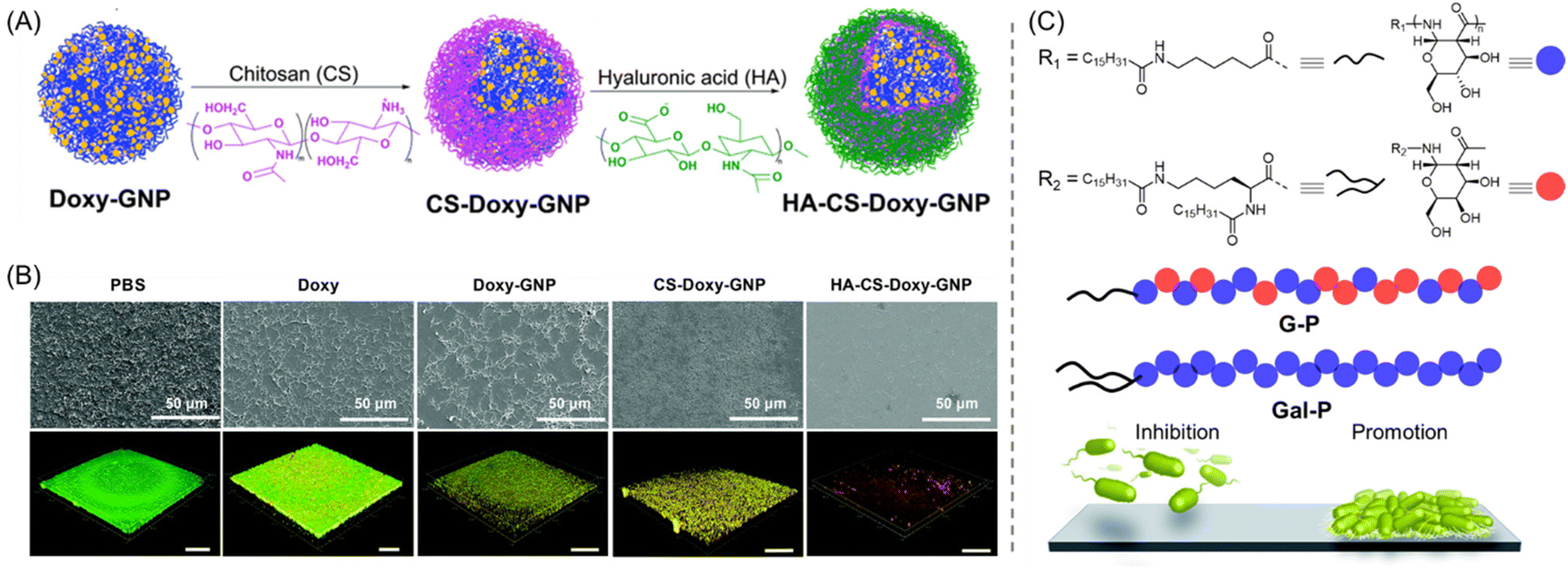 | ||
| Fig. 4 (A) Fabrication of HA-CS-Doxy-GNP via the layer-by-layer assembly of CS and HA. (B) SEM (top) and live/dead stained CLSM imaging (bottom) of V. vulnificus biofilms after being treated for 24 h with PBS, 50 μg mL−1 Doxy, and Doxy-loaded NP formulations at an equal Doxy concentration (live bacteria: green, whereas bacteria with broken membranes: red). “Reproduced from ref. 68 with permission from the [Royal Society of Chemistry], copyright [2022]”. (C) Chemical structures of glucose/galactose-derived PAS amphiphiles. G-P inhibits biofilm formation while Gal-P promotes it. “Reproduced with permission from ref. 76 from [Royal Society of Chemistry], Copyright [2014]”. | ||
Raj and co-workers developed chitosan–gum arabic embedded liposomes-alizarin nanocarriers (CGL-Alz NCs) to enhance drug accumulation, alizarin permeability and antibiofilm activities against single or combined species of Candida albicans (C. albicans) and S. aureus.72 A low-cost and environmentally friendly ionotropic gelation process was employed for the construction of the positive surface charged NCs. Different concentrations of synthesized NCs were incubated with C. albicans and S. aureus cultured bacteria for 24 h and almost 95% suppression of both the biofilms was observed at 50 μg mL−1 concentration. Notably, the CGL-Alz NCs were also successful in preventing the growth of mixed biofilm – C. albicans plus S. aureus – unlike free alizarin, which does not possess any potency to inhibit either bacteria or fungi. Surprisingly, the CGL-Alz NCs showed antimicrobial activity at a much higher concentration (>50 μg mL−1). The in vitro mechanistic studies revealed that the interaction between the cationic NCs and negatively charge bacterial and fungal membranes is mainly responsible for the effective permeabilization and damage of bacterial/fungi membranes.
In 2020, highly water-dispersed antibiofilm graphene/chitosan nanocomposites (GR/CS NCPs) were constructed by Maruthupandy et al. for the prevention of P. aeruginosa and Klebsiella pneumoniae (K. pneumoniae) biofilms.73 CS was homogeneously distributed into the GR nanosheets, and the antimicrobial potency of GR and GR/CS NCPs was separately evaluated. Both GR and GR/CS NCPs exhibited good antibiofilm activity; however, the latter outperformed the former. At a concentration of 40 μg mL−1, GR/CS NCPs displayed around a 92% and 94% reduction in biofilms of K. pneumoniae and P. aeruginosa, respectively, while similar reduction percentages were achieved with 70 μg mL−1 GR. The CLSM images indicated the severe degradation of K. pneumoniae and P. aeruginosa biofilm aggregations on treatment with GR and GR/CS NCPs via membrane distortions and damage. This fact was further corroborated by SEM observations that revealed irregular morphologies with compromised cellular death.
Recently, a new approach was introduced that boosts the potentiality of the membrane-active antimicrobial (MAA) chlorhexidine (CHX) by encapsulating CHX-liposomes into a CS hydrogel.74S. aureus and P. aeruginosa bacterial strains were chosen to evaluate the biofilm inhibition efficacy of free CHX and CHX-hydrogels. As disclosed by the biofilm inhibition assay, free CHX was moderately active against pre-formed biofilm, with an eradication rate of 29–43%, whereas the incorporation of CHX into the CS hydrogel significantly improved its potency to prevent biofilm development or disperse existing biofilm, with an abolition rate of 82–98% and 64% for S. aureus and P. aeruginosa, respectively.
Although the cationic small molecule biocides are being used as prospective antibiofilm agents, their non-specific targeting of bacteria/biofilm infection sites often limits their potential usage. To enhance the targeting efficiency, Hoque and Haldar loaded such biocides into dextran-derived hydrogels (Dex-5, Dex-10, and Dex-20), and noticed their remarkable antimicrobial and antibiofilm activity.75 The numbers after Dex represent the loaded weight% of biocide. These ideal delivery systems were constructed by combining dextran methacrylate (Dex-MA) and biocide with two cationic charges, and subsequent in situ photopolymerizations of the mixture. The loaded biocide exhibited outstanding stability with excellent distribution through the crosslinked network of dextran-based hydrogels. It has been demonstrated that the hydrogels progressively release the biocide over time and kill (100%) both Gram-positive (S. aureus) and Gram-negative (Escherichia coli (E. coli)) bacteria, along with methicillin-resistant S. aureus (MRSA). The effectiveness of the gels as antibiofilm agents was next assessed by examining how well-established bacterial biofilms could be removed upon the release of the biocide. After 2 days of treatment with Dex-10 hydrogel, negligible cells of both S. aureus and E. coli bacteria were observed, suggesting complete biofilm eradication. Furthermore, the in vivo efficacy of the hydrogels was evaluated using pre-formed biofilms in a murine model of superficial MRSA infections. In contrast to untreated skin samples, Dex-5-treated skin samples exhibited a 3.1 log (>99.9%) reduction, while Dex-10-treated samples showed a 4.8 log (>99.99%) reduction of MRSA after 4 days. An important finding was that the gels had great skin biocompatibility in a variety of animal models, and thus can be employed as secure and efficient antibacterial/antibiofilm agents to treat topical infections.
Apart from the CS and dextran, glucose/galactose-derived polymers have also been used for the inhibition/eradication of bacterial biofilms. In this context, Dane et al. synthesized a family of bioinspired poly-amido-saccharide (PAS) amphiphiles via the controlled anionic polymerization of galactose and/or glucose-derived β-lactam monomers using a pentafluorophenol-based ester initiator.76 The capability of this PAS to influence the growth of P. aeruginosa strain was evaluated. From the library of eight distinct amphiphiles, the authors revealed that two amphiphiles (G-P) prevented biofilm formation and one amphiphile (Gal-P) promoted biofilm growth at a concentration of ≥0.25 and ≥0.5 mg mL−1, respectively (Fig. 4C). More importantly, no such bacterial promotion or inhibition occurred with PAS homopolymer or random copolymer lacking hydrophobic end groups, thus suggesting the necessity of the amphiphilic structure for their bioactivity.
Very recently, the antimicrobial and antibiofilm properties of Ag(I) coordination polymer-doped hybrid biopolymer film made of potato starch (PS) or epoxidized soybean oil acrylate (SBO) were investigated. The biopolymer films containing 0.05–0.5 wt% doping with coordination polymer exhibited exceptional antibacterial efficacy against both Gram-negative (E. coli and P. aeruginosa) and Gram-positive (S. aureus and Staphylococcus epidermidis (S. epidermidis)) bacteria. Next, the biofilm inhibition capabilities of Ag-doped SBO films were assessed and revealed a 1–3 log reduction, i.e., 90.0–99.9% bacterial growth inhibition, of the aforesaid microbes.77 It was postulated that the ability of penetration with the release of Ag+ ions in the biofilm microenvironment is mainly attributed to its effective antibiofilm capacities.
4. Synthetic macromolecule-based antibiofilm agents
As discussed earlier, bacterial biofilms possess high resistance and tolerance toward conventional antibiotics and become more difficult to eradicate in the later stage. Therefore, a significant amount of attention has been paid to fewer prevailing approaches using tailored macromolecular species. These can directly target the fungi or biofilm-dwelling bacteria through a non-metabolic pathway or inhibit the formation of biofilms at an early stage. Moreover, a degradation of the preformed biofilms can be attained by mimicking the membrane-disruptive mechanism of commercial antibiotics in a polymeric system. Of note, the integration of physiological factors (pH, enzyme, ROS, hypoxia) responsible for biofilm inhibition can act synergistically with the polymeric species and produce lethal antibiofilm agents. In subsequent sub-sections, numerous synthetic macromolecular approaches for the inhibition, dispersion, and eradication of biofilms are discussed (Table 2).4.1 Cationic and charge-switchable polymers
Cationic polymers have emerged as one of the most effective macromolecular antibiofilm species. Their design was inspired by the bacterial and/or fungal inhibition features of antimicrobial peptides (AMPs).78,79 These peptides are composed of hydrophobic amino acids and cationic residues. Although AMPs were proved to eliminate biofilms, their implementation holds several shortcomings including high manufacturing costs due to multi-step solid-phase synthesis, low solubility owing to proteolytic degradation and rapid clearance in vivo.80,81 To overcome these bottlenecks, researchers have turned their investigation to the development of synthetic mimics of AMPs in the form of polymers (cationic or charge-switchable) which can offer high proteolytic stability, cost-effective preparation and excellent bacterial membrane rupturing properties. These polymers are chemically akin to AMPs and exhibit bacteriolysis via electrostatic interaction between the cationic unit of the polymer and the negatively charged bacterial outer membrane or cytoplasmic membrane, followed by insertion into phospholipid layers, that caused the killing of bacteria through disrupting the cell membrane and the leakage of cytoplasmic constituents.82,83 For this purpose, four types of cationic unit, namely amines (lysine amino acid mimic), quaternary amines, guanidine (arginine amino acid mimic), and phosphonium, have been used.Takahashi and co-workers investigated the antibacterial and antibiofilm effect of AMP-mimetic methacrylate homo (PE0) and amphiphilic copolymer (PE31) synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization against the cariogenic bacterium Streptococcus mutans (S. mutans) (Fig. 5A and B).84 The minimum biofilm inhibitory concentration (MBIC) was found to be 8.3 and 6.3 μg mL−1 for the PE0 and PE31, respectively, which are higher compared with the commercial antiseptic and disinfectant CHX (MBIC = 1.0 μg mL−1). Conversely, both the cationic homo- and copolymers exhibited an excellent eradication activity of 1-day matured S. mutans biofilm, while CHX and the cationic surfactant cetyltrimethylammonium bromide (CTAB) failed to do so. A reduction of 80–85% biofilm mass was attained within 2 h and 24 h at a polymer concentration of 1 mg mL−1 (Fig. 5C and D). Interestingly, there was no significant disparity in the biofilm eradication activity between PE31 and PE0 although the former showed higher antibacterial activity than the latter. This fact indicates that cationic functionality is the key factor in the eradication of S. mutans biofilm as it may cause physical disruption of biofilm by binding on the negatively charged extracellular biofilm matrix. Furthermore, the potentiality of these polymers was evaluated as a “liquid toothbrush” by performing the removal of biofilm through repeated rinsing and swishing.
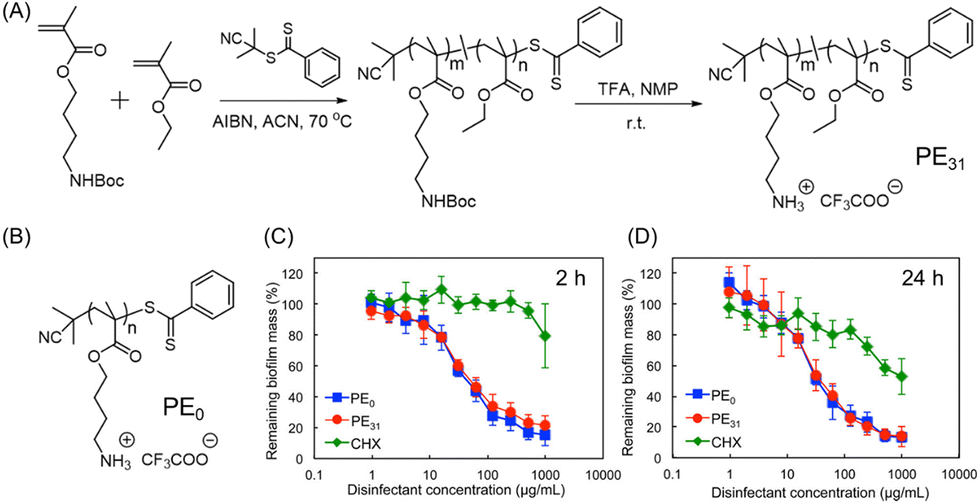 | ||
| Fig. 5 (A) Synthetic procedure of cationic amphiphilic copolymer PE31 by RAFT polymerization. (B) Chemical structure of PE0 homopolymer. (C and D) Influence of PE0, PE31 and CHX to reduce the established S. mutans biofilm biomass by CV staining after being treated for 2 h and 24 h. “Reprinted from ref. 84 with permission from [American Chemical Society], Copyright [2017]”. | ||
Joy and co-workers designed and developed a compositional variant of Tecoflex® polyurethane (PU) catheters by introducing a peptide-like cationic pendant to improve its antibiofilm activity and combat nosocomial infections.85 The cationic variant Tecoflex-NH3+ was synthesized via step-growth copolymerization of an amine-functionalized N-substituted diol monomer followed by acid-mediated deprotection. They chose E. coli bacteria to examine the antibiofilm activity, as it is known that catheter-associated urinary tract infections are often caused by fecal E. coli.86,87 In contrast to non-modified Tecoflex® coating, the cationic polyurethane coating has been shown to significantly slow down the biofilm build-up (even after 5 days of incubation) through the contact-killing mechanism, evidenced by bactericidal and electron microscopy assays. Interestingly, the cationic variant displayed excellent selective toxicity toward E. coli over mammalian cells due to the electrostatic interaction between the cationic PU and the negatively charged bacterial membrane.
Though the prevention of biofilm formation is feasible and has been widely explored, the eradication of surface-established biofilms remains one of the major challenges in this field. Recently, a synthetic, water-soluble AMP-mimetic PU was introduced that can inhibit the biofilm generation as well as disrupt matured biofilms of S. aureus, P. aeruginosa and E. coli, all of them showing resistance to the conventional antibiotics ciprofloxacin (CIP) and polymyxin B sulfate (Fig. 6A).88 The PUs composed of amino-acid-derived cationic and hydrophobic functionalities were synthesized by combining N-functionalized diethanolamide monomers with hexamethylene diisocyanate in a straightforward one-pot step-growth polymerization process. The cationic groups were engineered to be arginine-mimetic (mArg) or lysine-mimetic (mLys), while the hydrophobicity was adjusted by the incorporation of pendant phenylalanine (mPhe) and alanine (mAla) to ensure the substantial solubility of these polyurethanes in various growth media. For all the polymer compositions, the ratio of charge/hydrophobicity was fixed to 60/40 to mimic the structural aspects of AMPs.89,90 While these polyurethanes did not have antibacterial activity, they can prevent broad-spectrum biofilm inhibition at subinhibitory concentrations (S. aureus: MBIC = 4–16 μg mL−1; P. aeruginosa: MBIC = 4–8 μg mL−1; and E. coli: MBIC = 2–4 μg mL−1). As confocal microscopy suggested, mLys/mPhe and mLys/mAla were able to disrupt both the established P. aeruginosa biofilms and cell debris from the surface at a 4× concentration of MBIC (32 μg mL−1), higher than that of mArg/mPhe (16 μg mL−1) (Fig. 6B). Significantly, the conventional antibiotics were unable to disrupt the P. aeruginosa, which further supports the efficiency of these PUs for antibiofilm therapeutics without being poisonous to mammalian cells.
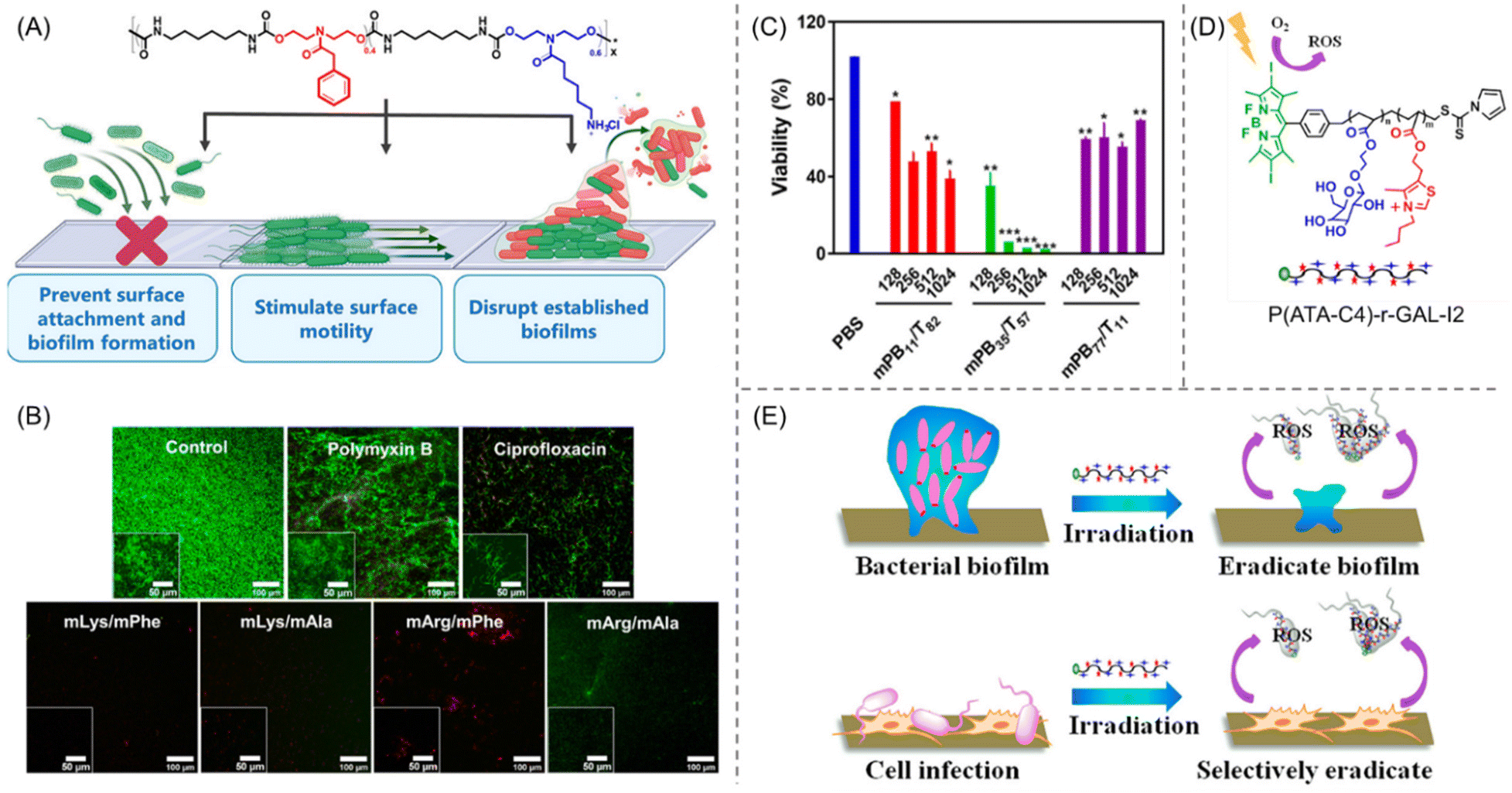 | ||
| Fig. 6 (A) The molecular structure of peptidomimetic PUs and a cartoon illustration of their biofilm inhibition and eradication activities. (B) CLSM photographs of prefabricated P. aeruginosa biofilms exposed to polymyxin B, ciprofloxacin, and diverse amino-acid-derived PUs. Red and green staining represent dead and live cells, respectively. “Reprinted with permission from ref. 88 [American Chemical Society], Copyright [2021]”. (C) Quantitative analysis of biofilm viability by a CFU reduction assay employing various PEGylated amphiphilic cationic polymers of different concentrations (μg mL−1). “Reprinted with permission from ref. 95 [American Chemical Society], Copyright [2020]”. (D) Molecular structure of BODIPY-tethered polymer P(ATA-C4)-r-GAL-I2. (E) ROS-stimulated eradication of bacteria and bacterial biofilm under visible light irradiation using P(ATA-C4)-r-GAL-I2. “Reprinted with permission from ref. 96 [American Chemical Society], Copyright [2018]”. | ||
Another class of PU-based alternating biomimetic polymers were examined for antibacterial and antibiofilm activity by Ghosh's research group.91 Several PUs were synthesized by integrating variable hydrophobic and cationic residues into the polymeric backbone. Amongst them, two PUs contain rigid cyclic hydrocarbons (C6, C13) and another five are composed of flexible linear hydrocarbons (C4, C6, C8). Unlike rigid PUs, flexible polymers showed better broad-spectrum antibacterial activity. Interestingly, the flexible PU with a C6 linker (F-PU-C6) appeared to be the most lethal one, with an MIC value of ∼2.0 μg mL−1, while containing guanidium or quaternary amines as cationic groups, due to the optimal balance between charge density and hydrophobicity. After treatment of a 3-day aged S. aureus/E. coli biofilm for 3 h, F-PU-C6 was successful in destroying the biofilm, with a concentration of 30 μg mL−1, which is comparable to the eradication efficiency of the common antibiotic levofloxacin or polymyxin-B. While these amphiphilic PUs were lethal against bacteria, they were identified to be non-toxic against mammalian cells, as evident by cytotoxic and hemolysis assays.
Chen and co-workers92 demonstrated the biofilm disassembly and inhibition ability of cationic D-type polypeptides, as D-type amino acids (DAAs) were found to prevent existing biofilm,93,94 though it is questioned by many scientists. The amphiphilic polypeptide, poly(L-lysine)33-block-poly(D-phenylalanine)14 was synthesized by N-carboxyanhydride (NCA) ring-opening polymerization using hexamethyldisilazane (HMDS) as an initiator. In comparison with the L-type polypeptide i.e., poly(L-lysine)33-block-poly(D-phenylalanine)14 (inhibition ratio: 24.6%), the D-type one showed much stronger surface biofilm eradication activity (inhibition ratio: 49.4%) of Bacillus subtilis (B. subtilis) at the same low concentration (24 μg mL−1), indicated by microbial experiments. This study suggests that D-type polypeptides are a much more effective biofilm removal agent than their opposite isomer, and represent a potential platform for DAA-embedded exterminator design.
To understand the structure–antibacterial activity relationship of AMP-mimetic polymers against E. coli and S. aureus, a series of PEGylated amphiphilic cationic macromolecules were synthesized by varying the structure, PEGylation, content and chain length of the hydrophobic segments, and the molecular weights.95 Quaternary ammonium groups were chosen as cationic residues owing to their ability to interact with anionic biocomponents, while PEGylation was used to modify the hydrophilic–hydrophobic balance in the polymer. Overall, the PEGylated random cationic copolymer (mPBx/Ty) has been shown to exhibit better antibiofilm activity and biocompatibility than the non-PEGylated random copolymer (PBx/Ty) containing similar compositions of cationic and hydrophobic residues. Amongst all the PEGylated polymers, mPB35/T57 has been proved to possess the highest antibacterial activity with the same MIC and MBC value of 8 μg mL−1 against E. coli. Next, the eradication activity against E. coli biofilm was examined by crystal violet (CV) staining and colony-forming unit (CFU) assays using levofloxacin and phosphate buffered saline (PBS) as a negative and positive control, respectively. As per the CV staining assay, mPB35/T57 showed a concentration sensitivity in biofilm removal efficacy, with 85% residual biofilm mass at 128 μg mL−1 that decreased to 25% at 1024 μg mL−1, implying that 75% biofilm could be eliminated at 1024 μg mL−1 (Fig. 6C). However, the CFU assay revealed that mPB35/T57 did not show significant bacterial killing activity (>30%) at a concentration of ≤128 μg mL−1 even if the treatment time was extended from 4 h to 16 h. Interestingly, more than 99% killing of bacteria was attained using mPB35/T57 at 1024 μg mL−1 in 4 h, which indicates that the PEGylated cationic copolymer does not enter the biofilm instantly but rather it penetrated slowly to kill bacteria, and a high polymer concentration could do the job efficiently.
Dai et al.96 designed and discussed the antibiofilm feature of RAFT-synthesized galactose-functionalized thiazole-based cationic copolymers embedded with a photodynamic therapy (PDT) agent, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY). BODIPY was used intentionally due to its excellent photostability and high quantum yield.97 Moreover, PDT agents can generate toxic reactive oxygen species (ROS) upon irradiation, which leads to killing of bacteria and, consequently, to removing biofilm without the rapid development of bacterial resistance.98 Four types of bacteria, E. coli, S. aureus, P. aeruginosa, and Bacillus amyloliquefaciens (B. amyloliquefaciens), have been used to examine the inhibition and eradication capability of the synthesized BODIPY-embedded copolymer P(ATA-C4)-r-GAL-I2 (Fig. 6D). The copolymer showed more than 80% and 90% inhibition activity at a concentration of 5.5 and 22 μg mL−1, confirmed by the CV staining method. Additionally, the biofilm removal efficacy of the polymer was evaluated against all these bacteria, which revealed >80% biofilm eradication at a concentration of 22 μg mL−1 that increases with increasing polymer concentration. The mechanism behind such eradication activity against both Gram-positive and Gram-negative bacteria is a combination of two factors: (1) penetration of the cell membrane through interacting with alginate (exists in EPS), affirmed by dynamic light scattering (DLS) measurement; and (2) BODIPY-mediated generation of the toxic ROS upon irradiating with visible light (Fig. 6E). Furthermore, the polymers are highly selective towards bacterial cells over mammalian cells and furnished no discernible cytotoxicity and hemolytic activity.
While most of the AMP-mimetic synthetic polymers were focused on the inhibition and/or removal of planktonic microbes, eradication of multidrug-resistant (MDR) “superbugs”, namely MRSA biofilms, was relatively overlooked though it possesses a serious threat to global health systems. In this regard, Rotello et al.99 engineered a series of poly(oxanorborneneimide) cationic polymeric nanoparticles (P1–P6 PNPs), synthesized via ring-opening metathesis polymerization (ROMP), that can simultaneously eradicate the preformed biofilms and maintain higher therapeutic indices against RBCs, critical for effective application in clinical settings (Fig. 7A).100 A 1000-fold increase in MIC value (0.064 μM to 64 μM) was observed with increasing the hydrophobic chain length bridging between the quaternary cationic moiety and the polymeric backbone (Fig. 7B). However, there was no such effect of cationic headgroup hydrophobicity on the inhibition performances. Amongst them, P5 PNP was demonstrated to be the most-effective nanoparticle that can eradicate 90% of biofilm mass (including S. aureus – MRSA) at a concentration of 1–3 μM with unprecedented therapeutic indices (TI) ranging from 60 to 165 (Fig. 7C). The successful biofilm removal ability of P5 PNP was further investigated in mammalian NIH 3T3 fibroblast cells at the same concentration, where no significant cytotoxicity was observed. Noteworthy, no bacterial resistance toward these PNP nanoparticles was noticed even after 20 serial passages as there was no change in MIC, contrary to clinically relevant antibiotics such as ciprofloxacin, ceftazidime and tetracycline that showed a 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 4200 and 256-fold increase in MIC value, respectively (Fig. 7D).
000, 4200 and 256-fold increase in MIC value, respectively (Fig. 7D).
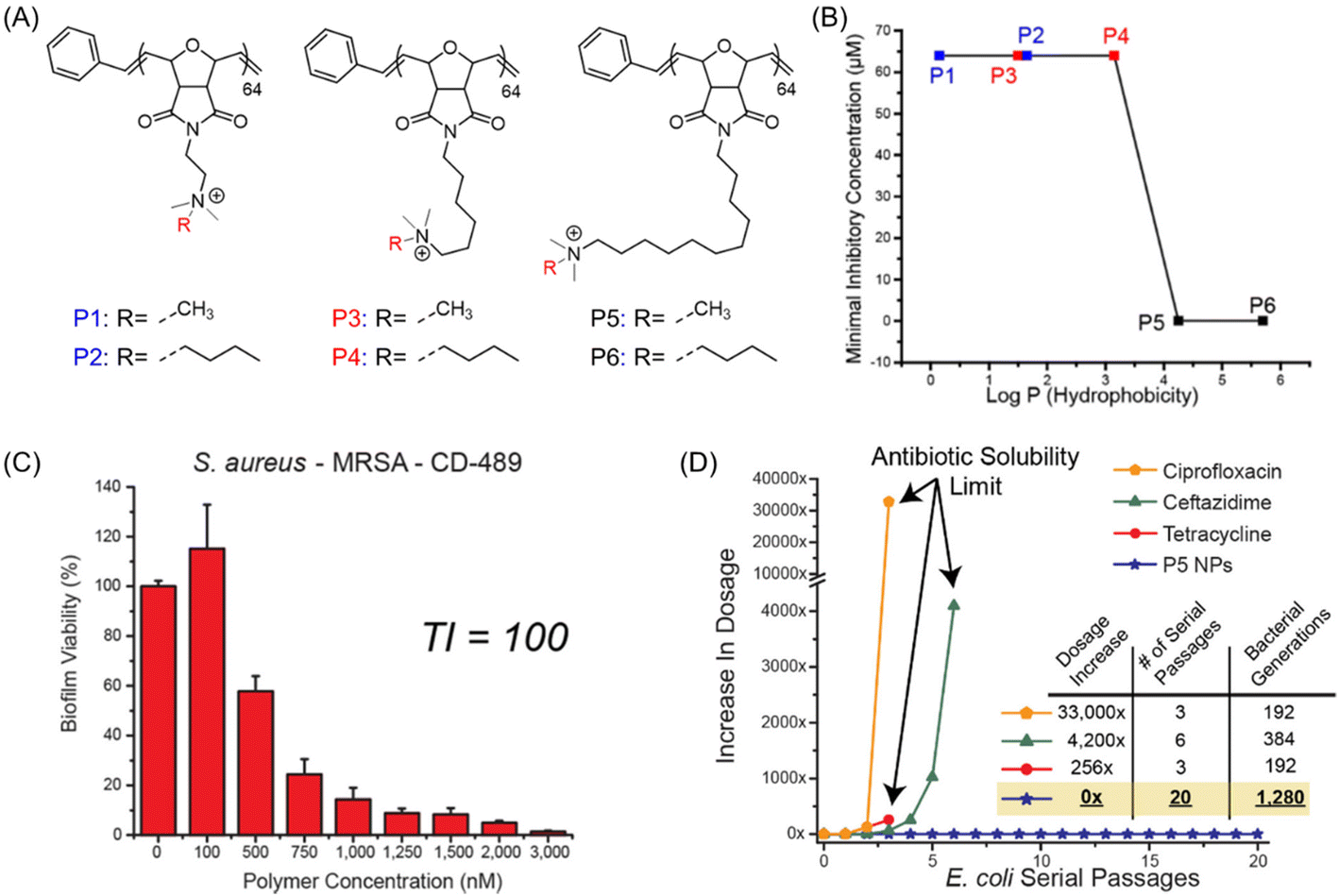 | ||
| Fig. 7 (A) Chemical structures of antibiofilm poly(oxanorborneneimides) (P1–P6). (B) Determined MICs of the polymers against their hydrophobicity (logP). (C) Viability of 24 h aged S. aureus biofilm after being exposed to P5 PNP for 3 h (TI = therapeutic index). (D) Resistant development of sub-MIC-level commercial antimicrobials along with P5 PNP during serial passages of E. coli bacteria. “Reproduced from ref. 99 with permission from [American Chemical Society], Copyright [2018]”. | ||
To further enhance the effectiveness of such a class of PNPs in combating MDR bacterial and biofilm infections, the same group has meticulously combined PNPs with an antibiotic (colistin) to achieve synergistic therapy.101 In comparison with colistin alone, a 16- to 32-fold decrease in colistin dosage was required for PNP–colistin conjugates to inhibit and/or remove MDR Gram-negative bacteria and biofilm matrix. This fact is most likely attributed to the enhanced accumulation of antibiotics inside the biofilms through the membrane disruption pathway, resulting in the synergistic effect of the PNP–colistin combination to eradicate biofilms.
In addition to the non-degradable cationic polymers, degradable polymers with proper functionalities have also been used to eradicate biofilms. Jeremy et al. reported polycarbonate-based antimicrobial polymers with varied chain lengths synthesized via organocatalytic ring-opening polymerization of an N-methyldiethanolamine-derived 8-membered ring followed by quaternization with methyl iodide.102 These polycarbonates have been tested for antimicrobial activity against different clinically relevant microbes including Gram-positive bacteria (S. aureus and Staphylococcus epidermidis (S. epidermidis)), Gram-negative bacteria (E. coli and P. aeruginosa) and fungus (C. albicans). All the quaternized polymers exhibited significant antimicrobial activity with an MIC value of 5–63 μg mL−1 and 63–250 μg mL−1 for Gram-positive and Gram-negative bacteria, respectively. The existence of an extra membrane in Gram-negative bacteria could be the reason for their higher MIC values. SEM results suggested that the polymers follow a similar membrane-lytic mechanism to AMPs as bacterial membrane disruption and cell debris were observed for both S. aureus and E. coli after 2 h treatment with 2× MIC. Also, the polymer exhibited a dose-dependent biofilm elimination of both S. aureus and E. coli. As disclosed by the CV staining method, the percentage of biomass left was around 85% at 2× MIC concentration, whereas it decreased to ∼25% upon increasing the concentration up to 8× MIC. More importantly, the polymer was found to remove biofilm from the bloodstream of a S. aureus-infected mouse model more rapidly than the antibiotic augmentin. Overall, this macromolecular approach has the potential to provide a strong platform for removing matured biofilms in both in vitro and in vivo conditions.
Shen and co-workers103 investigated antimicrobial and biofilm elimination activity using several quaternized polyethyleneimines (QPEI), prepared by treating commercially available PEI1200 with variable alkyl bromides in ethanol. Both PEI and QPEI displayed antimicrobial properties against broad-spectrum bacteria; especially, PEI1200-C6 (quaternized with 1-bromohexanal) exhibited the highest activity (low MIC value) among them all. Besides, it showed better biofilm eradication activity than PEI1200 alone, examined by an in vitro live/dead cell staining assay.
Until recently, quaternary ammonium group-containing polymers have been used to penetrate bacterial membranes. However, Wang et al. have introduced main chain sulfonium homopolymers (PS+), synthesized via a thiol–ene reaction and subsequent methylation to eradicate both fungal and bacterial biofilms (Fig. 8A).104 All the presented polysulfoniums showed good antimicrobial activity against pathogenic microbes – S. aureus, E. coli, C. albicans – with MICs in the range of 0.5–32 μg mL−1 that are comparable to a clinically used antibacterial (erythromycin) and an antifungal drug (amphotericin B). For biofilm eradication, a PS+-C6 system containing a C6H12 hydrophobic spacer was proved to be the most successful candidate because of its highest charge density among all the synthesized sulfonium-based polymers. The PS+-C6 polymer significantly reduced the biofilm masses and displayed excellent killing efficacies: 99% and 99.5% for E. coli and C. albicans, respectively, at 2× MIC. However, in the case of S. aureus biofilm, a relatively higher concentration (8× MIC) was required to attain a similar reduction in biofilm mass. Following treatment with the PS+-C6 polymer for 24 h, thick biofilms produced mostly red fluorescent signals that signified the existence of dead microorganisms; however, most of the cells in the control groups were still alive (Fig. 8B). The successful biofilm extermination via a membrane rupture process was also demonstrated by an alteration in the shape of the bacterial cells, monitored using SEM. The aforesaid results suggest that highly charged cationic polysulfoniums can permeate the negatively charged EPS matrix of biofilms via electrostatic interactions and induce their lethal antibiofilm activity at significantly low doses with minimal hemolytic toxicity.
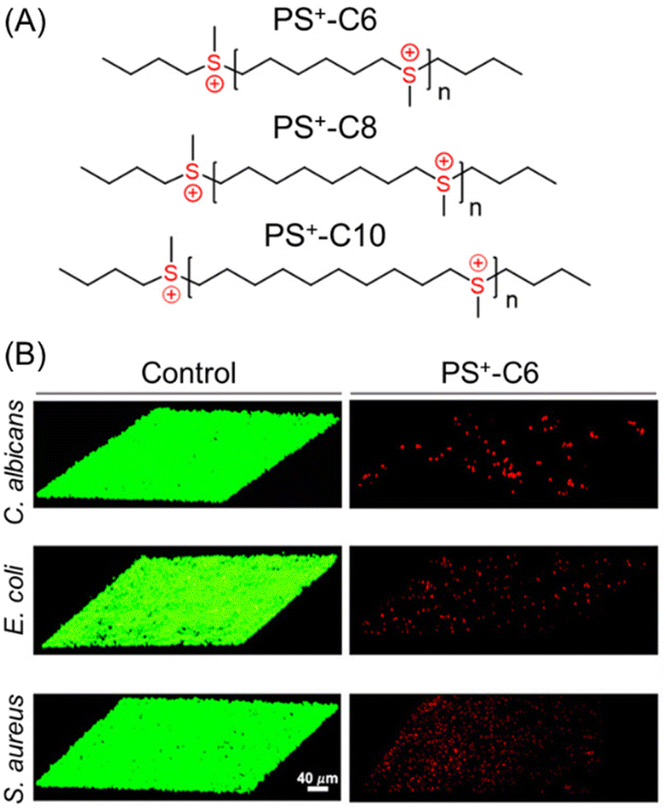 | ||
| Fig. 8 (A) Chemical structures of main chain sulfonium-containing homopolymers. (B) CLSM images of C. albicans-, E. coli- and S. aureus-constructed biofilms treated with the control solution and PS+-C6. “Reprinted from ref. 104 with permission from [American Chemical Society] Copyright [2021]”. | ||
Despite reasonable advantages of cationic polymers as antibiofilm materials, their activity has been somewhat restricted because of their high hemolysis ratio, lower selectivity and pronounced toxicity toward healthy mammalian cells. To overcome these issues, a new attractive approach has been recently developed for biofilm eradication harnessing the bacterial biofilm microenvironment. Due to bacterial metabolism and the consequent production of acidic by-products including lactic acid and acetic acid, microorganisms trapped in the biofilm create a confined acidic microenvironment (pH 4.5–6.5).105,106 Considering this acidic microenvironment, a handful of charge-switchable (neutral/−ve to +ve) polymeric materials have been developed with superior biofilm abolition performance. Noteworthy, combining antibiotics with such polymers can further broaden their antibacterial spectrum through synergistic effects.
Li et al. developed a biocompatible approach utilizing negatively charged polymeric nanocarriers for the targeted release of CHX in the cariogenic biofilm, based on the pH difference between the acidic biofilm (pH 5.5) and healthy oral cavity (pH 7.4).107 At first, a cationic block copolymer poly(ethylene glycol)-block-poly(2-(((2-aminoethyl)carbamoyl)oxy)ethyl methacrylate) (PEG-b-PAECOEMA) was synthesized via atom transfer radical polymerization (ATRP). To endow the cationic polymer with pH sensitivity in an acidic milieu, it was further treated with citraconic anhydride (CA) to generate negatively charged PEG-b-PAECOEMA/CA that can self-assemble into core–shell micellar structures (PICMs) while mixing with cationic CHX through electrostatic attraction. The resulting PICMs have been shown to exhibit good antibacterial capability against both planktonic bacteria (MIC and MBC = 16 μg mL−1) and biofilm of S. mutans. Relative to pH 7.4, a higher CHX release was achieved from PICMs at pH 5.5 due to acid-responsive cleavage of the citraconic amide linker.
Zhang's research group introduced a series of RAFT-synthesized amphiphilic ionizable glycomimetic block copolymers to explore the protonation–antibacterial activity relationship.108 Here, three types of tertiary amine-based hydrophobic block, namely poly(2-(diisopropylamino)ethyl methacrylate) (pDPA), poly(2-(hexamethyleneimino)ethyl methacrylate) (pHMEMA) and poly(2-(dibutylamino)ethyl methacrylate) (pDBA), with pKa values of 6.0, 6.3 and 4.7, respectively, have been used to examine their antibacterial and antibiofilm performances against P. aeruginosa. In fact, these blocks were incorporated as a comonomer partner to prepare the block copolymers p(GEMA-r-FEMA)-b-pR (R = DPA, DBA or HMEMA) using the macro chain transfer agent p(GEMA-r-FEMA) [poly(2-(β-D-galactosyloxy) ethyl methacrylate-random-2-fucose ethyl methacrylate)]. Galactose and fucose moieties were intended to specifically target the surface of P. aeruginosa.109 Subsequently, the block copolymers were subjected to encapsulation of the broad-spectrum antibiotic nitroxoline (NQ), resulting in the formation of NPs: NP-DPA@NQ, NP-HMEMA@NQ and NP-DBA@NQ. Amongst these, NP-DPA@NQ offered the highest bacterial killing and growth inhibition activity, with an MIC value of 15.6 μg mL−1 at pH 6.0, while no such activity was found at pH 7.4, implying a pH-mediated binding affinity of P. aeruginosa with cationic pDPA and a metal-chelation-dependent mechanism of NQ.110,111 Interestingly, the blank NPs (without NQ loading) showed ∼58–74% biofilm inhibition at a concentration of 500 μg mL−1; however, similar results were obtained at a much lower concentration (40 μg mL−1) with NQ-loaded NPs, as verified by CLSM imaging indicating the formation of red-stained dead cell aggregates (Fig. 9A). This overall protonation–activity relationship provides a paradigm for simultaneous antibacterial therapies via targeted drug release at the infectious sites.
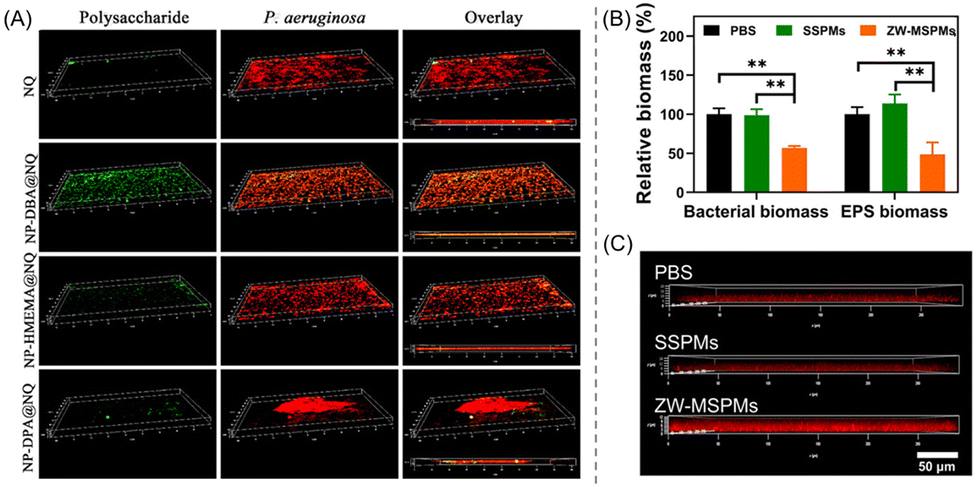 | ||
| Fig. 9 (A) CLSM 3D snapshots of NQ-loaded NPs treated P. aeruginosa biofilm after 24 h, showing superior biofilm inhibition activity of NP-DPA@NQ NPs among others. “Reproduced from ref. 108 with permission from [American Chemical Society], Copyright [2020]”. (B) PBS-, SSPM- and ZW-MSPM-treated relative biomass percentages of bacteria and EPS. (C) CLSM images of the penetration of red fluorescent CIP-containing PBS, SSPM and ZW-MSPM suspensions into S. aureus biofilm after a 2 h incubation. “Reproduced from ref. 114 with permission from [Science], Copyright [2020]”. | ||
Another charge-switchable nanocarrier combined with antibiotic azithromycin (AZM) was fabricated for the treatment of chronic lung infection caused by P. aeruginosa.112 The electrostatic complexation between a block copolymer, 2,3-dimethyl maleic anhydride (DA) modified PEG-block-polylysine (PEG-b-PLys), and a dendrimer, AZM conjugated amino-ended poly(amidoamine) (PAMAM), resulted in the formation of size (112 nm) and charge adaptive (−2.2 mV; sensitivity to acidic milieu) AZM-clustered NPs (AZM-DA NPs). As supported by the in vitro bactericidal assays, the AZM-DA NPs not only exhibited outstanding antibacterial ability (decrease of 99.994% bacterial colonies) but also showed a remarkable reduction (99.998%) in biofilm mass compared with only AZM and pH-insensitive NPs, AZM-SA NPs. This fact is ascribed to the size compression and charge reversal phenomena of the NPs upon arrival in the acidic biofilm environment, which concurrently triggered the release of cationic NPs and enhanced the penetration and accumulation of AZM inside the biofilm. Moreover, the excellent antibiofilm activity of the NPs was successfully executed on a P. aeruginosa infected in vivo mice model.
Similarly, Hong and co-workers113 developed AZM-encapsulated well-defined surface charge-switchable micelles (SCSMs) containing acid-sensitive acyl hydrazone linker and demonstrated its release behaviour to P. aeruginosa infected biofilm. To investigate the applicability of AZM-loaded SCSMs, two controls have been used, namely free AZM and AZM-loaded surface charge unswitchable micelles (AZM-SCUMs). The AZM-SCSMs showed an obvious lower MIC value than the others at pH 5.5 due to charge reversion in an acidic biofilm milieu. At a low concentration of 32 g mL−1 at pH 5.5, the benefit of AZM-SCSMs in terms of a decrease in the viability of P. aeruginosa biofilm became apparent. However, they virtually lost their antibiofilm effectiveness at pH 7.4 and needed more AZM (512 g mL−1) to completely remove the biofilm. Their antibiofilm activity was also examined under in vivo conditions.
An exciting approach featuring zwitterionic (ZW) micelles with dual functions – self-targeting and biofilm disruption – was rationally designed and constructed by Tian et al.114 The ZW micelles (ZW-MSPMs) were prepared through self-assembly of PEG-block-poly(caprolactone) (PEG-b-PCL) with PEG-block-poly(quaternary amino ester) (PEG-b-PQAE) containing pH-responsive carboxybetaine under physiological conditions. To showcase the influence of ZW in bacterial killing, PEG-b-PCL single-shell polymeric micelles (SSPMs) without ZW characteristics were synthesized as a control. As in vitro studies suggested, ZW-MSPMs were able to reduce by half the established biofilm thickness and biomass of both S. aureus and EPS, much higher than that with SSPMs and the PBS control at a concentration of 200 μg mL−1 (Fig. 9B). Interestingly, the remaining biofilm was easier to eradicate by using the common antibiotic CIP (Fig. 9C). Besides, the zwitterionic micelles with CIP have been shown to completely eradicate staphylococcal biofilms formed in mice underneath an abdominal imaging window (AIW) within 5 days, whereas SSPMs did not show any activity. The mechanism of action of ZW-MSPMs is associated with the structural conversion of the zwitterionic poly(quaternary-amino-ester) to a cationic lactone in a biofilm microenvironment at pH 5.0 that induces self-targeting, penetration, and collective accumulation inside staphylococcal biofilms.
In order to visualize real-time biofilm eradication, a pH-responsive green-fluorescent polymer–drug system composed of modified naphthalic anhydride (NA-COOH) was developed.115 The synthesis of the targeted branched polymer involved three steps: (i) proper functionalization of naphthalic anhydride to prepare NA-COOH; (ii) Michael addition polymerization of N,N′-methylenebisacrylamide (MBA) and 1-(2-aminoethyl)piperazine (AEPZ) to form poly(MBA-AEPZ)-AEPZ; and (iii) incorporation of NA-COOH into the polymer using EDC/NHS chemistry. The final polymer poly(MBA-AEPZ)-AEPZ-NA exhibited a weak emission intensity at pH 7.4, but emitted an intense green emission when the pH was decreased from 7.4 to 5.0 due to protonation of the embedded morpholine entities (Fig. 10A). Such a fluorescence switching phenomena was also observed in living zebrafish. However, the antibacterial performance of the polymer was poor against S. aureus and P. aeruginosa. Meanwhile, the biofilm ablation activity of the polymer was evaluated using the CV staining method. In the case of S. aureus, the biofilm mass reduced from 31.6% to 2.8% after treatment with polymer together with vancomycin. Similarly for P. aeruginosa, tobramycin and polymer + tobramycin exhibited a 16.1% and 2.7% reduction in biofilm, implying a higher antibiofilm activity of the combination of poly(MBA-AEPZ)-AEPZ-NA and antibiotics. Inspired by the pH-tunable fluorescence properties, the above polymer combination was implemented to visualize the real-time change in biofilm biomass through confocal laser scanning microscopy, which revealed a reduction in green emission intensity over time, signifying biofilm eradication (Fig. 10B).
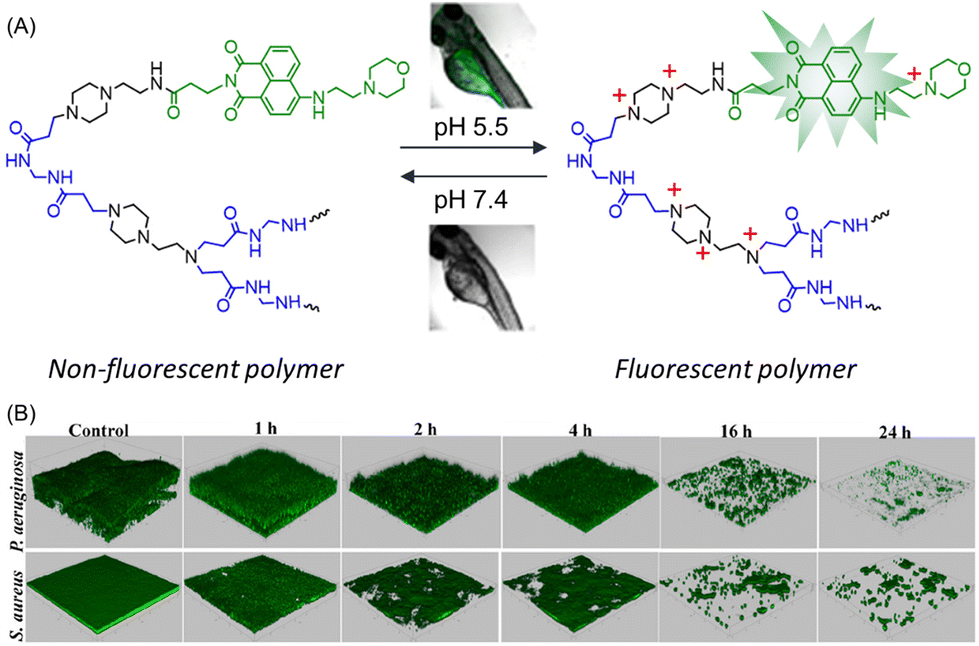 | ||
| Fig. 10 (A) Structural representation and pH-induced photophysical behaviour of poly(MBA-AEPZ)-AEPZ-NA. (B) Real-time detection/eradication of P. aeruginosa and S. aureus biofilms with green fluorescent poly(MBA-AEPZ)-AEPZ-NA + antibiotics. “Reproduced from ref. 115 with permission from [American Chemical Society], Copyright [2022]”. | ||
4.2 Conjugated polymers
Conjugated polymers (CPs) are composed of two major components: delocalized π-conjugated backbone and multifunctional pendants. The intrinsic light-amplified and light-harvested capabilities render CPs promising materials in a myriad of applications ranging from optoelectronic devices and chemical sensing to cell imaging. Notably, the binding of CPs on the surface of bacteria and their exerting antibacterial activity have been unveiled recently.116,117 To this end, the pendants of CPs were deliberately modified with cationic residues to make them water-soluble and concurrently enable the interaction with bacterial membranes. The antibacterial and/or antibiofilm activities of CPs mainly follow two pathways:(a) The pendant cationic moieties allow CPs to damage the bacterial membranes by reacting electrostatically with the negatively charged Gram-positive and Gram-negative bacteria; and (b) upon illuminating with proper light, the π-conjugated backbone can react with the surrounding O2 and produce ROS, leading to rapid cell deactivation. Yet, implementation of CPs as an antibiofilm agent is not much explored and only a very few studies are reported, which are being discussed in this section.
The development of water-soluble CPs was reported to inhibit the growth and eradicate the biofilm of S. aureus.118 Quaternary ammonium ion-functionalized blue-fluorescent poly-{[(9,9-bis(6′-N,N,N-trimethylammonium)hexyl)fluorenylenepenylene]dibromide} (PFP) was used as a model CP (Fig. 11A). Apparently, the PFP did not show any bactericidal or biofilm ablation activity up to 1–20 μM concentration. However, it was shown to furnish antibacterial activity when the concentration was raised to 50–100 μM, as indicated by the CV staining assay. The interaction between PFP and S. aureus was validated by zeta potential and CLSM studies, which revealed an alteration of surface charge from −ve to +ve and a formation of aggregates. Subsequently, the biofilm extermination of S. aureus was examined using PFP with and without irradiation with white light. As was obvious, PFP in the presence of white light exhibited pronounced biofilm removal activity within 25 min of irradiation, contrary to only PFP, suggesting ROS-promoted biofilm elimination (Fig. 11A).
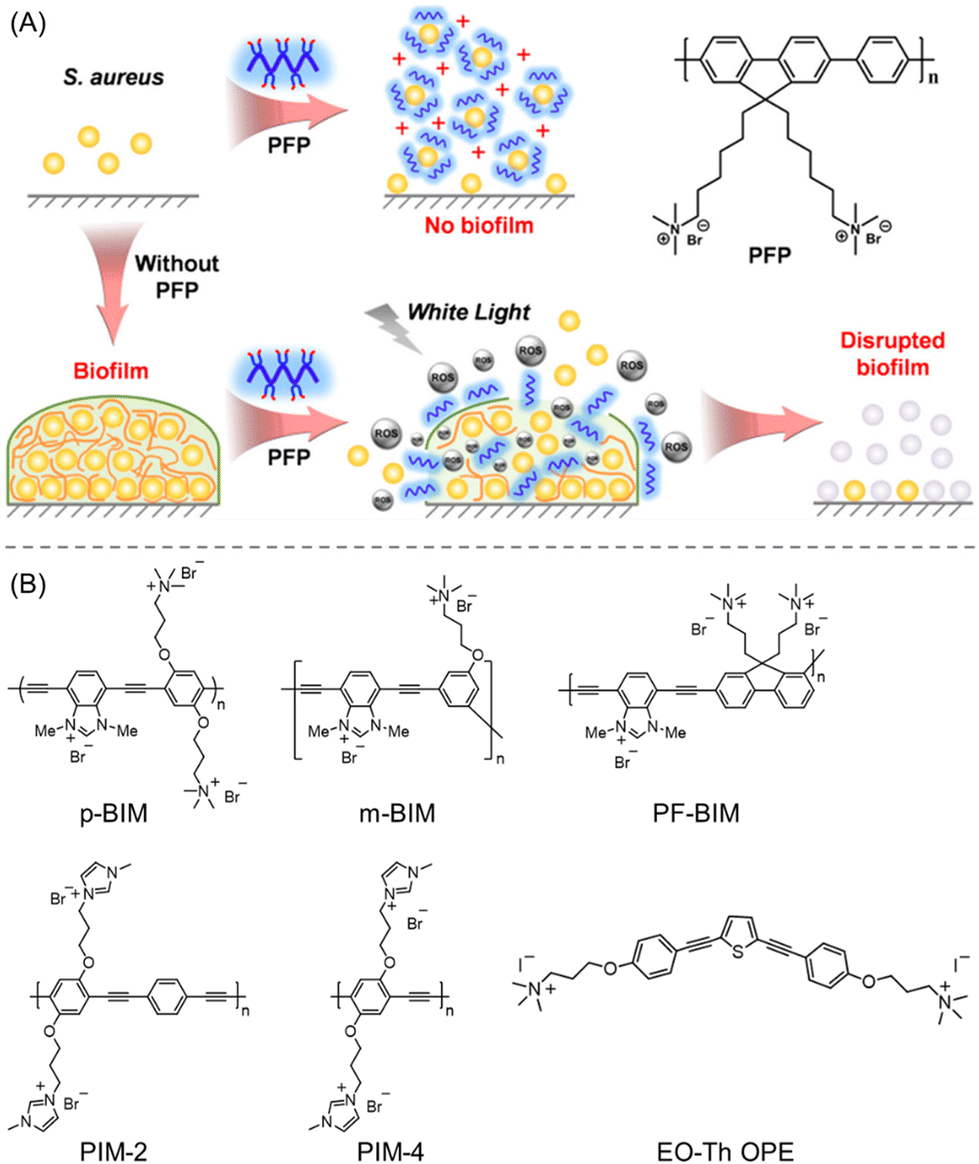 | ||
| Fig. 11 (A) Schematic interpretation of the PFP chemical structure and its mechanistic pathway for biofilm inhibition and the eradication of matured biofilms (under white light illumination). “Reproduced from ref. 118 with permission from [American Chemical Society] Copyright [2017]”. (B) Molecular structures of employed linear cationic CPs to treat C. albicans-caused infections. “Reproduced from ref. 120 with permission from [American Chemical Society] Copyright [2020]”. | ||
Nowadays one of the rising worldwide public health concerns is the spread of infections brought on by a pathogenic fungus, C. albicans. Although current antifungal medications are often used to treat Candida infections, they are harmful in terms of their high toxicity and multidrug-resistant behaviours.119 In this regard, Schanze et al.120 developed a facile strategy using linear cationic CPs to treat C. albicans-caused infections (Fig. 11B). They have rationally synthesized a new family of water-soluble CPs with imidazolium functionality including poly(m-phenylene ethynylene) (m-BIM), polyfluorene (PF-BIM) and poly(p-phenylene ethynylene) (p-BIM), and extensively investigated their inherent photophysical properties and potential antifungal/antibiofilm activity. Additionally, two other polymers of poly(p-phenylene ethynylene) backbone and quaternary ammonium-functionalized oligo(thiophene-ethynylene) (EO-Th OPE) were used for fungicidal activity comparison. Unlike m-BIM (MIC90 = 8 M), all other CPs exhibited MIC90 < 1 M after 60 min of photolysis; however, their antifungal activity was severely compromised under dark conditions. Similarly, the synthesized CPs displayed great potency towards C. albicans biofilm reduction when exposed to 420 nm light at a concentration of 128 μg mL−1. Notably, the cytotoxicities of EO-Th OPE and m-BIM were found to be 6× higher than other CPs, which thus restricted their in vivo applications. Hence, the linear polymers (PIM-2, PIM-4, p-BIM, and PF-BIM) were established to be an excellent class of antifungals with low toxicity to human cells. Though it is arguable, the authors suggested that the effective antifungal activity of such CPs mainly originated from the ROS production and interaction of the cationic imidazole moieties121 with the surface of the bacterial cell membrane, resulting in retention, deformation, and damage.
The utilization of CPs as a platform for antibiofilm application was further recognized by Alston and co-workers.122 They reported photothermal ablation (PTA)-active non-toxic hybrid donor–acceptor polymer particles (H-DAPPs-PTA) synthesized by a 1:1 combination of fluorescent poly(3-hexylthiophene-2,5 diyl) (P3HT) and heat-generation polymer poly[4,4-bis(2-ethylhexyl)-cyclopenta[2,1-b;3,4-b]dithiophene-2,6-diyl-alt-2,1,3-benzoselenadiazole-4,7 diyl] (PCPDTBSe) in 1 mL tetrahydrofuran followed by injecting into water containing Pluronic F-127. The H-DPPAs were evaluated alone and in combination with the antibiotic clindamycin, against planktonic Streptococcus pyogenes (S. pyogenes), S. aureus and its biofilm. With exposure to 60 seconds of 800 nm light, H-DPPAs-PTA generates temperature variations of 27.6–73.1 °C that led to complete elimination of planktonic S. aureus bacteria and drastically reduced bacterial viability inside biofilms by more than 4 log (>99%). These CPs were able to inactivate the bacteria within the biofilm, but could not significantly mitigate the biofilm polysaccharides, as suggested by confocal analysis. In contrast to H-DAPPs-PTA and clindamycin alone, the combined particle (H-DAPPs-PTA + clindamycin) showed higher S. aureus antibiofilm activity by reducing the bacterial viability by ∼3 log at an average temperature of 55.1 °C and 100 μg mL−1 concentration. The present results indicated that the near infrared (NIR)-active H-DAPPs-PTA has great potential to combat biofilm infection by generating rapid localized hyperthermia.
Recently, Ghosh and co-workers reported photocatalytic bacterial inactivation by employing two distinct conjugated polymer nanostructures (CPNs): polythiophene (PEDOT) and polyaniline (PANI).123 As supported by the in vitro bactericidal assay, both the CPNs were shown to exhibit antimicrobial activity against S. aureus and E. coli under ultraviolet A (UVA) irradiation. However, negligible inhibition of bacterial growth was observed with bulk PEDOT and PANI without light. After being treated with PANI nanofibers, the obtained CFU reduction was around 7 log and 4 log for S. aureus and E. coli, respectively, at 33 ng mL−1 concentration, whereas the same exposure concentration of PEDOT nanofibers led to a >6 log CFU reduction, and complete prevention of E. coli and S. aureus growth was reached at 333 and 883 ng mL−1, respectively. The antimicrobial effect of these CPNs could be ascribed to the photoactivated generation of ROS that induced cell membrane damage and eventually impeded their biofilm-forming ability.
4.3 Polymeric nanocomposites
The ability of highly effective antibiofilm agents including cationic macromolecules, proteins, and β-lactamase inhibitor is often severely encumbered by their low water solubility, high toxicity, and most importantly, lack of specificity toward the infected bacterial cell.124,125 Therefore, suitable antibiofilm agents are required to circumvent the aforesaid issues. Metallic nanomaterials featuring a huge surface area, small volume, and enriched multifunction integration could be possible disinfectants as they exhibit highly selective antibacterial activity against various microbes: fungi, bacteria, and viruses.126 However, the aggregation of metallic NPs in water and unwanted oxidation incited a loss of antimicrobial activity.127,128 Emerging strategies based on polymer-supported metal nanocomposites led to the design of stable yet effective nanomaterials for biofilm inhibition and/or eradication. As per the intended applications, the structural parameters of such nanocomposites can be tuned with stimulus-responsiveness, PDT or an alternating magnetic field effect (AMF) to form an appropriate antibiofilm agent. In the following section, a few examples of polymer composites are highlighted where metallic NPs result in an intrinsic change in antibiofilm activity.Li and co-workers prepared a series of membrane-targeting cationic polymer-supported silver nanoparticles (AgNPs@polymers) to explore the structure–antibiofilm activity relationship against both Gram-positive and Gram-negative bacteria.129 Cationic polymers with varied molecular weights, pendant lengths, and structural topologies (linear, 4-arm, 8-arm) were developed to modify AgNPs. In addition to the cationic polymer, AgNPs were chosen as an antimicrobial entity owing to their ultra-high surface area that is susceptible to oxidative dissolution, facilitating Ag+ ion release, and thus resulting in the killing of bacteria by interacting with the thiol groups of vital enzymes present in the bacteria.130 Synthesis of the cationic part includes ATRP block copolymerization of PEG with 2-(dimethylamino)ethyl methacrylate (DMAEMA) followed by a quaternization of tertiary amino pendants with butyl bromide. Next, the reduction of AgNO3 with NaBH4 in the polymer solution resulted in the formation of AgNPs@polymers where AgNPs adhered to the ammonium groups of the polymer. At a concentration of 5 μg mL−1, AgNPs@polymers sufficiently clogged the growth of P. aeruginosa and S. aureus. However, the antimicrobial and biofilm activity were shown to be increased with increasing arm length. Compared with nascent AgNPs, linear and 4-arm AgNPs@polymers, and 8-arm analogues (AgNPs@8-armPEG-b-DMAEMA-C4), exhibited a very low MIC value and 80% obliteration of P. aeruginosa and S. aureus biofilm at 64 μg mL−1 concentration due to the highest abundance of cationic charge. Notably, the nanocomposites were effective even after 30 generations without being faced with any resistance from the bacteria. A β-galactosidase assay strongly suggested that the AgNPs@8-armPEG-b-DMAEMA-C4 prevents the natural activity of bacterial intracellular enzymes, leading to bacterial cell death.
Also, carbohydrate-derived polymer poly(2-(acrylamido) glucopyranose) (PAGA) instead of PEG was developed to improve the antibacterial performance of such cationic-polymer embedded AgNPs.131 The synthesis of this polymer composite involved two steps: (1) the synthesis of two polymers (PAGA and quaternized PDMAEMA) through RAFT polymerization; and (2) the reduction of AgNO3 to AgNPs in the presence of these two polymer ligands. These nanocomposites displayed improved antibacterial activity with inhibition concentrations beyond 0.62, 0.16, and 0.16 μM for AgNPs@PAGA, AgNPs@PDAMEMA-C4, and AgNPs@PAGA/PDMAEMA-C4, respectively, against P. aeruginosa, E. coli, S. aureus and B. amyloliquefaciens. Also, AgNPs having both PAGA/PDAMEMA or PDAMEMA alone at the surface showed a higher percentage (∼80%) dispersion of biofilm masses of the four strains relative to the composite having only PAGA (∼40–60%), as implied by the CLSM images. The stronger bacterial inhibition and antibiofilm activity of AgNPs@PDAMEMA-C4 and AgNPs@PAGA/PDMAEMA-C4 was ascribed to the synergistic effect of the release of high-concentration Ag+ ions along with the glucosamine-mediated internalization into bacterial cells.132 The authors concluded that the introduction of PAGA not only made the system biocompatible but also induced no cytotoxicity to mammalian cells or red blood cell hemolysis.
Apart from cationic polymers, a pH-responsive charge-reversal polymer has also been introduced to fabricate the AgNPs’ surface. Wu et al. methodically developed biofilm-responsive silver nanoantibiotics (rAgNAs) composed of silver nanoclusters (AgNCs) and charge-reversal (+ve to −ve) polymer ligand, and investigated their bactericidal effect against the MRSA biofilm (Fig. 12A).133 The nanoantibiotic rAgNAs were constructed through the self-assembly of AgNCs with the pH-sensitive polymer ligand poly(ethylene glycol)-poly(aminopropyl imidazole-aspartate)-polyalanine (PEG-PSB-PALA) under physiological conditions. They exhibited excellent colloidal stability, prolonged blood circulation and high specificity at the biofilm infection site due to charge inversion of the imidazole moiety in an acidic biofilm microenvironment. Both in vitro and in vivo assays proclaimed rAgNAs as a lethal antibacterial agent against MRSA planktonic bacteria and biofilm with the same MIC and MBC value of ∼50 μg mL−1. To examine the pH-induced bactericidal effect, control nanoantibiotics (devoid of pH-responsive imidazole), uAgNAs containing AgNCs and poly(ethylene glycol)-poly(β-benzil-ι-aspartate)-polyalanine (PEG-PIB-PALA) were also prepared. At a 4-fold MIC-dosage, rAgNAs caused about 72.9% clearance of the MRSA biofilm, whereas uAgNAs or vancomycin instigated only an 8–25% reduction (Fig. 12B). The bactericidal effect of rAgNAs was also effective in a pyomyositis mice model. The superiority of rAgNAs over uAgNAs for antibiofilm was attributed to: (a) the acidic microenvironment of biofilm facilitating enhanced accumulation and penetration through a protonation of imidazole functionalities; and (b) the disassembly of rAgNAs into small nanoclusters resulting in dual accelerated Ag+ ion leaching, leading to the destruction of the bacterial cell membrane (Fig. 12A).
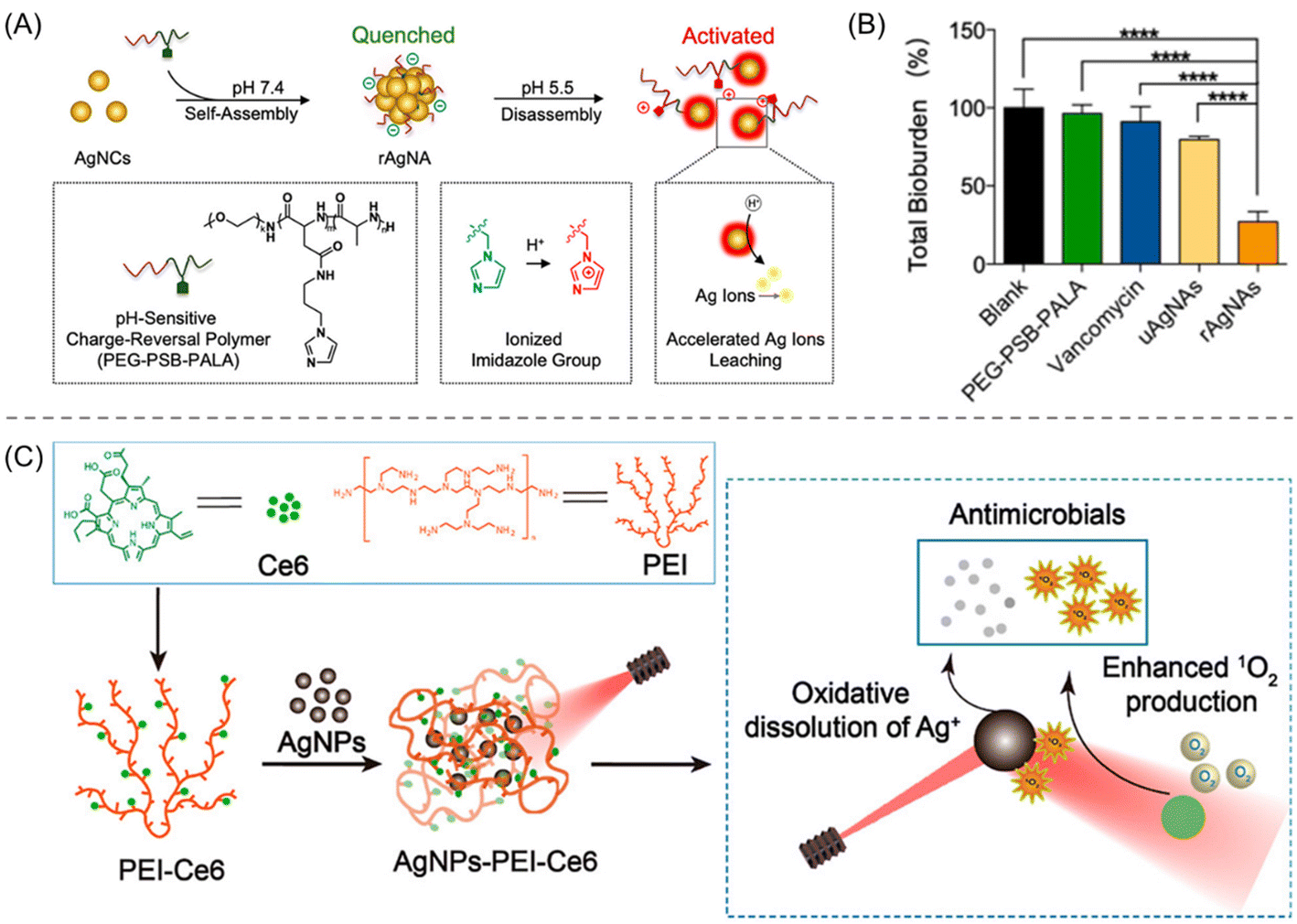 | ||
| Fig. 12 (A) Pictorial representation of the fabrication and mechanistic explanation of pH-reversal rAgNAs. (B) Quantitative analysis of the total bioburden reduction of MRSA biofilm after treatment with rAgNAs with different control compounds. “Reproduced from ref. 133 with permission from [American Chemical Society], Copyright [2019]”. (C) Procedure for the construction of AgNPs-PEI-Ce6. Higher ROS generation is promoted by the SPR effect of AgNPs that in turn release antibiofilm Ag+ ions. “Reproduced from ref. 134 with permission from [American Chemical Society], Copyright [2022]”. | ||
Zhou et al. exploited the combined approach of PDT and AgNPs to combat biofilm formation by broad-spectrum bacteria.134 Such a combinatorial approach led to higher ROS generation, promoted by the surface plasmon resonance (SPR) effect that can oxidize the AgNPs to release Ag+ ions, particularly in the biofilm microenvironment (Fig. 12C). Initially, a photosensitizer, chlorin-e6 (Ce6), was modified on cationic polyethyleneimine (PEI). Afterwards, it was used as a ligand to coat negatively charged AgNPs via electrostatic interaction to yield a hybrid nanocomposite, AgNPs-PEI-Ce6 (Fig. 12C). In conjunction with PDT, the cationic surface of the nanocomposite also facilitates the membrane penetration of bacterial cells; thus the killing of bacteria occurred. The AgNPs-PEI-Ce6 exhibited a better antibacterial effect against both E. coli and S. aureus bacteria than the other control materials used. Interestingly, the MIC and MBC values of the composites against E. coli (MIC = 0.001 mg mL−1; MBC = 0.003 mg mL−1) are much lower than against S. aureus (MIC = 0.2 mg mL−1; MBC = 1 mg mL−1) due to a higher susceptibility of Gram-negative bacteria to Ag+ ions, manifested by the standard plate count method with irradiation at a wavelength of 660 nm. As the live/dead assay data suggest, AgNPs-PEI-Ce6 can kill most of the bacteria (<105 CFU per mL) compared with AgNPs (>106 CFU per mL) and PEI-Ce6 (>106 CFU per mL) because of its combinatorial effect. However, the biofilm ablation effect was higher for S. aureus than E. coli as the latter has a protective outer membrane. Furthermore, they investigated the effect of AgNPs-PEI-Ce6 against the S. aureus infection model on the skin of mice, and a faster-wound healing process by reducing the number of bacteria in the infected site was observed.
Yin and co-workers demonstrated the utilization of gold nanorods (GNRs) instead of AgNPs to attain the MRSA biofilm obliteration effect.135 The surface of the mesoporous silica-coated GNRs was rationally decorated by in situ polymerizations of N-isopropyl acrylamide (NIPAM), acrylic acid (AA), and N-allylmethylamine (MAA) followed by CIP encapsulation (loading content = 9.8%) to produce the final nanocomposite GNR@P(NIPAM-AA-MAA) with the chemo-photothermal feature. While poly(N-isopropyl acrylamide) (PNIPAM) offers tunable thermosensitivity, poly(acrylic acid) (PAA) and poly(N-allylmethylamine) (PMAA) were introduced owing to their charge-switchable characteristics. The surface charge switches from negative to positive upon reaching the acidic bacterial infectious site (pH 5.5), which induced deep penetration and adhesion of the nanocomposites on the biofilm by electrostatic attraction. Importantly, intense local hyperthermia resulting from the photothermal effect of GNRs under NIR irradiation can kill bacteria and destroy cell integrity. Also, it causes rapid volume shrinkage of the nanocomposites followed by the release of CIP antibiotics, improving deeper penetration and enhanced biofilm eradication, both in vitro and in vivo (Fig. 13). Almost 90% of the MRSA bacteria were extinct in the presence of NIR irradiation, whereas ∼10% bacteria elimination was noticed without NIR radiation. Notably, the GNR@(NIPAM-AA-MAA)-CIP + NIR group appeared to be the most successful antibiofilm agent when compared with other control groups investigated in the present work and its activity was maximum at pH 5.5.
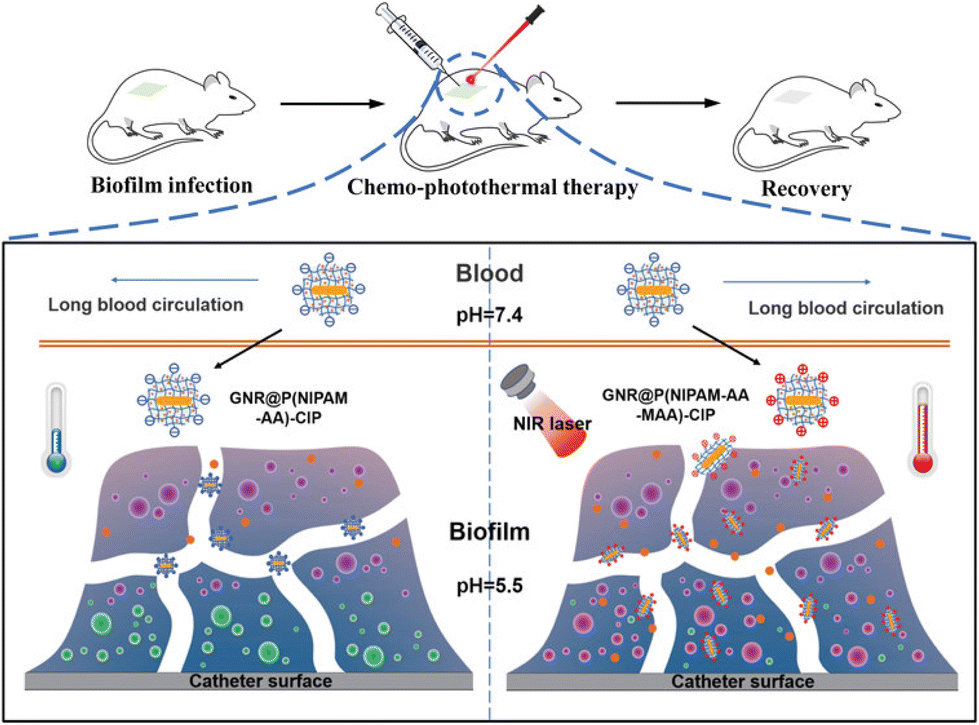 | ||
| Fig. 13 Targeted in vivo thermo-chemotherapy for MRSA biofilms using GNR@P(NIPAM-AA-MAA) in the presence of NIR irradiation. “Reproduced from ref. 135 with permission from [American Chemical Society], Copyright [2022]”. | ||
Along with silver and gold, magnetic iron NPs have also been embedded into a polymer matrix to accomplish a greener, cost-effective, and straightforward approach for bacterial removal and “on-demand” abolition activities. Positively charged poly(catechol-hexanediamine) decorated magnetic Fe3O4 NPs (Fe3O4@HDA) were prepared via mussel-inspired polymerization of hexanediamine and catechol in an aqueous medium for 1 h at room temperature. In comparison with nascent Fe3O4 NPs, Fe3O4@HDA displayed much higher (90.5%) bacteria-killing activity and bacteria-capturing ability in contaminated water. Meanwhile, the matured S. aureus biofilm can be potentially degraded within 10 min of NIR irradiation using Fe3O4@HDA via local hyperthermia. Combining the NIR activity and magnetic response property, the presented nanocomposites showed high antibacterial effects and in situ eradication.136
To understand the interaction between Fe3O4 NPs and biofilm matrix, two oppositely charged NPs were fabricated and their ability to penetrate biofilm was evaluated under AMF-triggered hyperthermia activation.137 A trisodium citrate-assisted solvothermal technique was used to make negatively charged Fe3O4 NPs and its subsequent surface-functionalized with PEI generated positively charged Fe3O4 NPs i.e., Fe3O4@PEI NPs (Fig. 14). The latter showed strong adherence to the negatively charged planktonic and sessile cells, leading to higher antibacterial activity and proficient biofilm eradication compared with the negatively charged former NPs. In response to AMF, Fe3O4@PEI NPs induced the formation of ROS in the bacterial cells by thermal damage and physical stress that eventually sabotaged the cell membrane, and disruption of biofilm took place (Fig. 14). Accordingly, both the Fe3O4 NPs and Fe3O4@PEI NPs exhibited biofilm ablation activity; however, their potency differs in the absence and presence of AMF. Without AMF, Fe3O4 NPs did not show any biofilm biomass reduction activity, whereas Fe3O4@PEI NPs reduced 15–20% of the biomass for both the E. coli and S. aureus strains. In the presence of AMF-triggering, the reduction value with Fe3O4 NPs and Fe3O4@PEI NPs was boosted up to ∼38–40% and ∼85–87%, respectively. These results unequivocally suggest a synergistic antibacterial approach using AMF along with the cationic polymer.
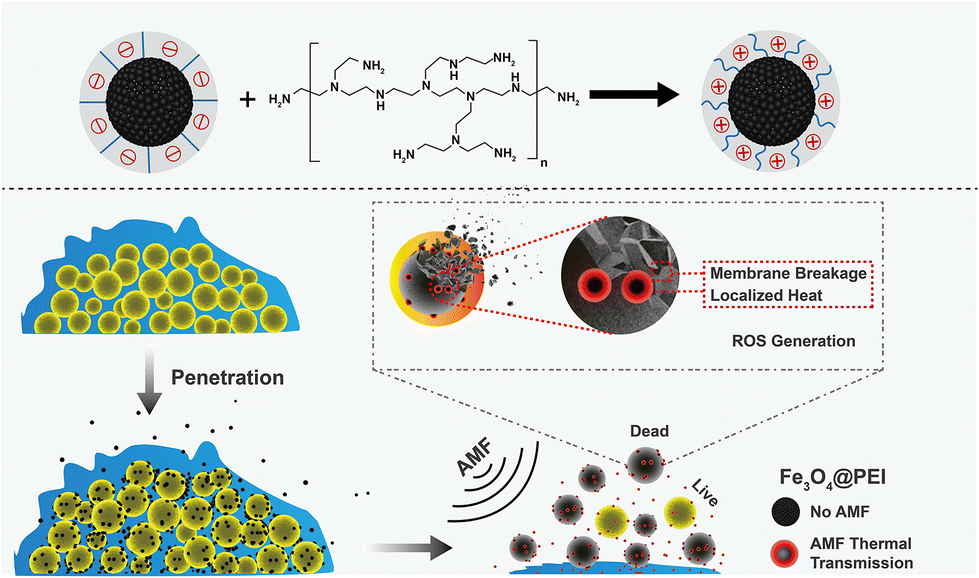 | ||
| Fig. 14 Cartoon illustration of the underlying antibiofilm mechanism of Fe3O4@PEI NPs via the enhanced penetration and potential membrane disruption of bacterial biofilm under AMF-triggering. “Reproduced from ref. 137 with permission from [American Chemical Society], Copyright [2022]”. | ||
Liu's group developed dual-functional polymer nanocomposites (PCBAA/ACP) having amorphous calcium phosphate (ACP) and zwitterionic poly(carboxybetaine acrylamide) (PCBAA) to withstand carcinogenic S. mutans adhesion and biofilm formation in enamel.138 As disclosed by SEM results, the inhibition percentage of S. mutans attachment on the enamel surface was 90.7% and 90.4% for the PCBAA and PCBAA/ACP, respectively, after 6 h treatment. Subsequently, the growth and biofilm formation of the S. mutans on the enamel surface was evaluated. Both PCBAA and PCBAA/ACP were found to show similar antibiofilm activity, suggesting no such effect of ACP on antibacterial properties. Besides, contrasted with fluoride, PCBAA/ACP demonstrated better effects in promoting the in vitro and in vivo remineralization of demineralized enamel and blockage of the dentinal tubules.
4.4 Polymeric hydrogels
An alternative tactic involving hydrogels with prerequisite biofilm-eradicating features has become increasingly popular in the recent past. The well-organized 3D structure of hydrogels, made with hydrophilic polymers, endows some inherent characteristics such as high water content, biocompatibility, excellent oxygen permeability and so forth.139 Moreover, their properties can be tuned with various stimuli – pH, redox, temperature, etc. – to construct a multifunctional platform.140,141 Because of the aforesaid aspects, polymeric hydrogels have been recently employed as antibacterial coatings,142,143 considering the electrostatic attraction between the oppositely charged hydrogel and the bacterial membranes. However, they were successful in the prophylactic treatment of bacteria but failed to exterminate bacterial biofilm. To address this problem, researchers came up with an approach that combines cationic functionalities with other antibiofilm factors like ROS, photothermal therapy (PTT), and metal ions (Ag+, Cu2+) to combat broad-spectrum biofilm formation and eradication along with the promotion of wound healing.Yeo and co-workers demonstrated a new convenient pathway for biofilm eradication using cationic hydrogels by virtue of non-leaching-based debridement, followed by ex situ contact-killing distant from the infected area.144 The cationic hydrogel PEI-PEGMA (Fig. 15A) was synthesized by the UV-mediated grafting of methacrylate-modified star polyethyleneimine (PEI) with poly(ethylene) glycol dimethacrylate (PEGDMA) using Irgacure 2959 initiator. In addition, another hydrogel with a second graft of decane (PEI-decane-PEGMA) was prepared to understand the effect of hydrophobicity (Fig. 15A). As in vitro studies suggested, the hydrogels with a lower content of PEGDMA and PEI-decane-PEGMA were not only able to completely eradicate the biofilms of S. aureus, E. coli, Acinetobacter baumannii (A. baumannii; AB), K. pneumoniae, and P. aeruginosa (PA), but also to eradicate MRSA and carbapenem-resistant AB (CR-AB) and PA (CR-PA) – pathogens of great concern worldwide as declared by the WHO.145,146 The PEI hydrogels were further implemented for in vivo biofilm elimination in a murine excisional wound infection model, which revealed that the hydrogel exhibited an >99.99% reduction in biofilms biomass of MRSA, CR-AB and CR-PA, whereas commercial silver-containing wound dressing showed almost no reduction in biofilm. The overall antibiofilm effect of the PEI hydrogels can be explained by two distinct bacterial killing modes: (a) debridement – the high pore size allows the hydrogels to absorb bacterial biofilm from the wound site through diffusion and slow fluid flow without clogging; subsequently, the absorbed bacteria undergo ex situ contact killing via PEI-induced membrane damage over time; and (b) triggered release – infection-triggered degradation of the hydrogels caused the release of bactericidal cationic PEI polymer that can kill bacteria both in the wound site and the dressing, which eventually leaves a cleaner wound throughout the healing process (Fig. 15B).
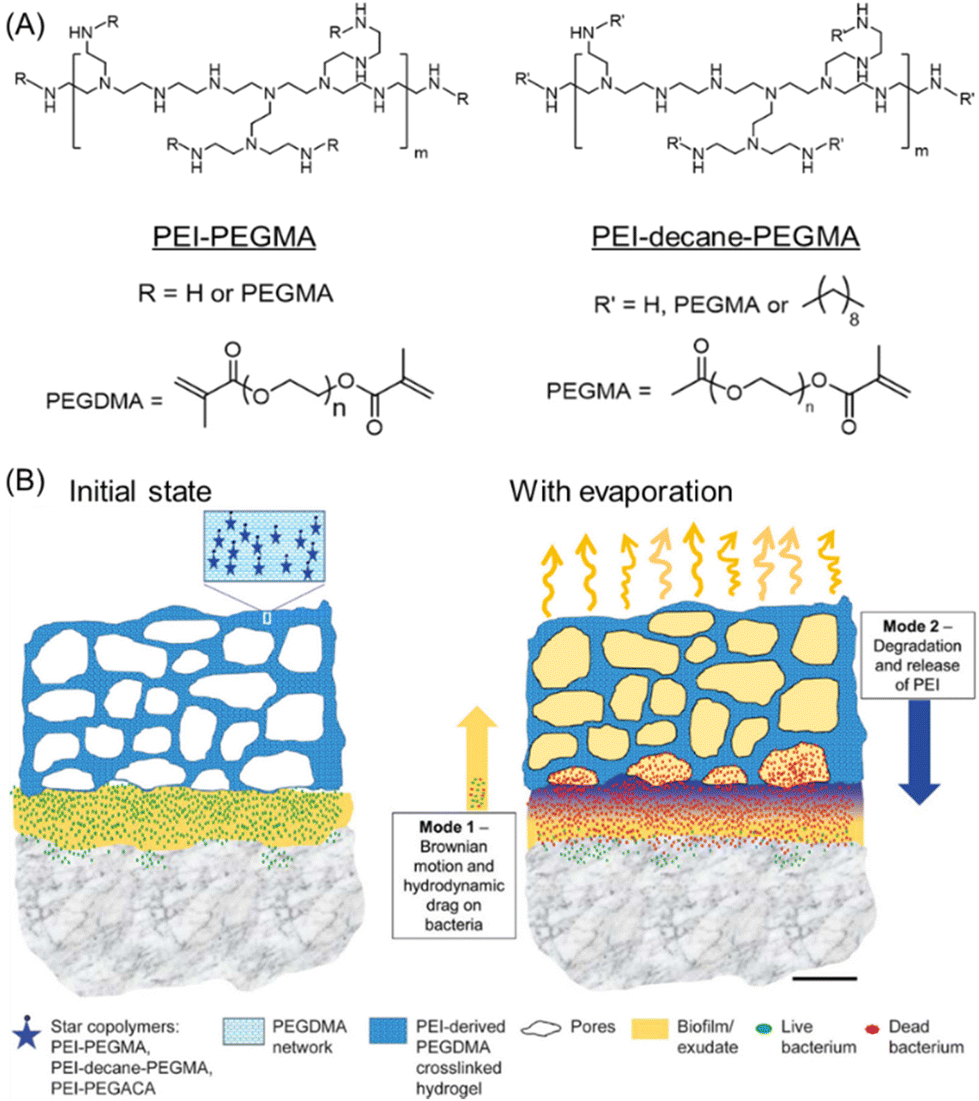 | ||
| Fig. 15 (A) Chemical structures of PEI-PEGMA and PEI-decane-PEGMA. (B) Illustration of the biofilm killing process of non-leaching biofilm-debridement and infection-degradable hydrogels in their initial state and with evaporation. “Reproduced from ref. 144 with permission from [American Chemical Society], Copyright [2018]”. | ||
Feng et al. designed and developed a dual-functional intelligent hydrogel for the naked-eye visual detection and photothermal treatment of bacterial biofilm.147 Self-assembly of bromothymol blue (BTB), β-glycerophosphate (β-GP), and NIR-absorbing conjugated polymer poly-{2,5-thiophen-co-[3,6-di(thiophen-2-yl)-2,5-bis(N,N,N-trimethylhexan-1-aminiu-m)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione]-bromide-co-4,7-(2,1,3-benzothia-diazole)} (PTDBD) and CS resulted in the formation of hybrid hydrogel BTB/PTDBD/CS. β-GP induced thermosensitivity in the hydrogels and displayed sol–gel transition behaviour as the temperature increased, facilitating a powerful local antibacterial action.
On the other hand, the pH-sensitive behaviour of BTB allows the in situ detection of bacterial biofilms via an emergent visible colour change from green to pale yellow in the acidic milieu of biofilms, whereas PTDBD empowered the hydrogel with NIR-mediated bacterial killing activity. The S. aureus constructed biofilm was used to examine the biofilm annihilation ability of this hydrogel. Even at the highest concentration of PTDBD, the hydrogel destroyed around 40% of the bacteria in the absence of laser irradiation. Nevertheless, it could effectively kill almost all the bacteria while being exposed to a laser beam (1.0 W cm−2, 808 nm) for 8 min at a PTDBD concentration of 40 μg mL−1. Interestingly, its killing efficiency was further enhanced when the concentration reached 60 μg mL−1. Hence, the antibacterial activity of this hydrogel mainly arises from the generation of NIR light-promoted local hyperthermia. Furthermore, the BTB/PTDBD/CS hydrogel was successfully used to treat and diagnose S. aureus-infected mice.
As mentioned earlier, the ultrasmall AgNPs are particularly vulnerable to quick oxidation that results in short-term action through an immediate release of silver ions. In this regard, Haidari et al.148 designed a novel hydrogel system with long-term stability, sustained release of Ag+ ions along with potent antibacterial activity (Fig. 16). The polymeric hydrogel was fabricated by incorporating different concentrations of mercaptosuccinic acid (MSA) coated AgNPs (AgNPs@MSA) into the biocompatible thermosensitive Pluronic F-127 hydrogel. The hydrogel showed a good bactericidal effect against both planktonic Gram-positive and Gram-negative bacteria as suggested by an in vitro CV assay. A concentration-dependent biofilm disruption study was also performed against individual S. epidermidis, S. aureus, and P. aeruginosa, and all these variants experienced considerable biofilm disruption up to ∼80% upon incubation with 50 μg g−1 AgNP-loaded hydrogel, which is much higher compared with control CIP. The persuasive antibacterial action of AgNP-loaded hydrogel could be illustrated by the continuous release of Ag+ ions from the hydrogel over an extended period that caused considerable bacterial cell damage and led to death (Fig. 16). Overall, the advantage of the present system lies in the combined use of ultrasmall (<3 nm) AgNPs and hydrogel, which serves as the reservoir for the controlled release of silver ions with an improved biofilm obliteration capacity. Interestingly, the effective antibacterial concentration of the formulation shows minimal toxicity to human fibroblasts and keratinocytes.
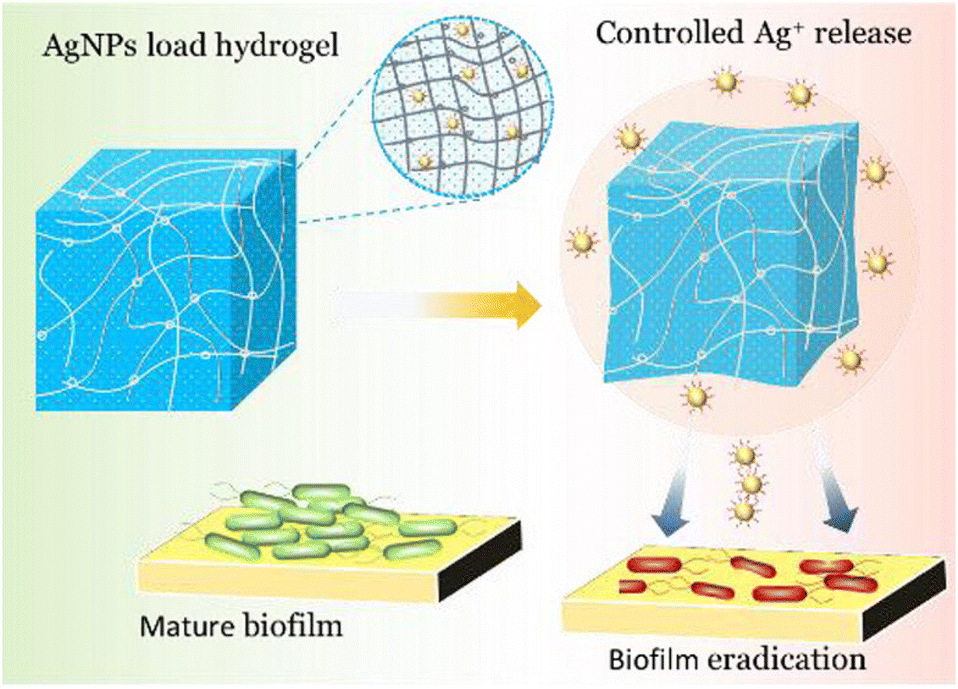 | ||
| Fig. 16 Controlled Ag+ release-promoted antibiofilm activity of AgNP-loaded hydrogel. “Reproduced from ref. 148 with permission from [American Chemical Society] Copyright [2020]”. | ||
Gharibi and Agarwal reported the synthesis of thermoplastic PU-based materials grafted with cationic polyhexamethylene guanidine (PHMG) featuring contact-active antibacterial activity against both Gram-positive and Gram-negative bacteria.149,150 These PHMG-grafted PU films or hydrogels have shown fast and efficient biofilm ablation capability. Moreover, the tunable physico-mechanical properties of such PUs can be exploited for wound dressings, and as an antibiofilm coating on biomedical devices.
Poly(vinyl alcohol) (PVA), an eco-friendly synthetic vinyl polymer is hydrophilic, water-soluble, non-carcinogenic, biocompatible and capable of forming films and gel under proper conditions. To date, it has been used as a primary material in various biomedical fields for contact lenses, vocal cord reconstruction, and artificial cartilages,151 but it has no intrinsic antibacterial and/or antifungal activity owing to its mild acidic nature that prevents the interaction between PVA and bacterial/fungal cell membranes.152 However, the use of non-toxic PVA integrating with an antibacterial agent (PTT or chitosan) is worth exploring.
Taking advantage of the photodynamic/photothermal effect of copper sulfide, a unique synergistic approach to inhibit and eradicate biofilms was established utilizing lignin-copper sulfide (LS-CuS) incorporated PVA hydrogel (LS-CuS@PVA).153 The preparation of such hydrogels involves two steps: (1) PVA solution was first combined with the LS-CuS nanocomposites and glutaraldehyde (GA) to generate a homogeneous mixture which was then (2) crosslinked to gel in the presence of HCl catalyst (Fig. 17A). Subsequently, the bactericidal impact of LS-CuS@PVA was thoroughly assessed against E. coli and S. aureus, considering its photothermal, photodynamic and peroxidase-like activity. The PVA hydrogel dramatically reduced CFUs by 4.8 log and 3.8 log for S. aureus and E. coli respectively, in the presence of H2O2 under NIR light irradiation for 10 min. Next, the S. aureus biofilm eradication efficiency of LS-CuS@PVA was investigated using different sets of experimental conditions: LS-CuS@PVA + H2O2, LS-CuS@PVA and H2O2 alone in the presence or absence of NIR light via a CV staining assay (Fig. 17B). In the absence of NIR irradiation, partial biofilm destruction was achieved with both LS-CuS@PVA and LS-CuS@PVA + H2O2, whereas almost complete removal of the S. aureus biofilm biomass was attained upon 10 min exposure to NIR light (Fig. 17B). These results unequivocally suggest the synergistic effect of CuS-promoted local hyperthermia and production of ˙OH radicals was mainly responsible for bacterial cell death radicals were mainly responsible for bacterial cell death.154,155 Such a superior antibacterial and antibiofilm capability makes this PVA-hydrogel a potential candidate for wound dressings. A series of antibiofilm PVA-derived hydrogels were prepared by incorporating antimicrobial polymer CS and AgNPs.156 Initially, CS and PVA were blended and subsequently crosslinked using trimellitic anhydride isothiocyanate. The CS content was varied during the formation of hydrogels, and AgNPs of different percentages were further loaded to reinforce the antimicrobial and antibiofilm performances. All the hydrogels with and without AgNPs displayed antimicrobial and antibiofilm formation activities against both Gram-negative and Gram-positive bacteria. However, the hydrogel loaded with a maximum percentage (3%) of AgNPs was more potent than other hydrogels, supported by a lower MBIC value against biofilms of A. baumannii (MBIC = 3.90 μg mL−1), B. subtilis (MBIC = 1.95 μg mL−1) and C. albicans (MBIC = 1.95 μg mL−1). Interestingly, the hydrogel also proved to be much more effective than the commercial antibiotics amphotericin B and vancomycin.
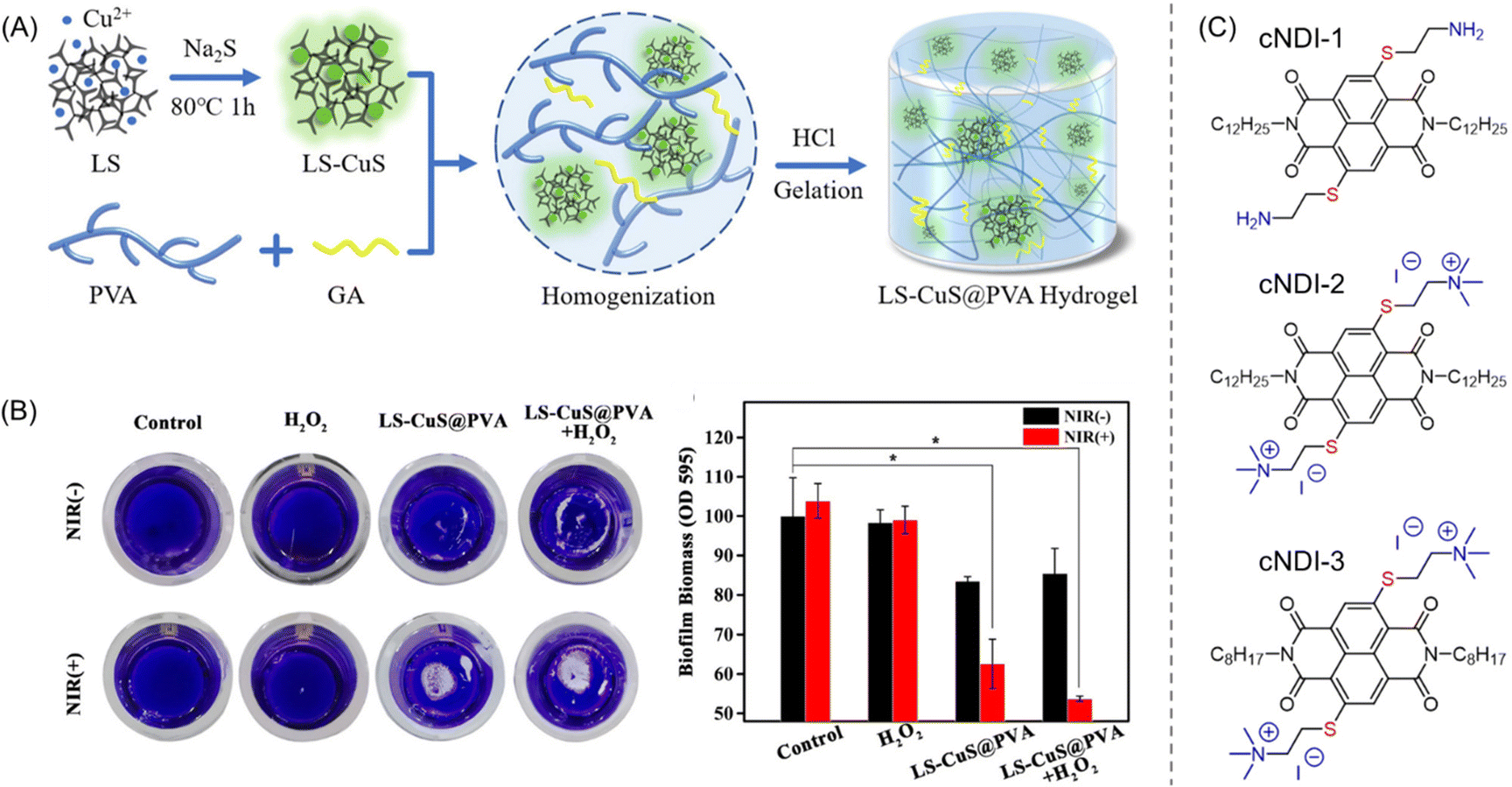 | ||
| Fig. 17 (A) Protocol employed for the preparation of LS-CuS@PVA hydrogels. “Reproduced from ref. 153 with permission from [American Chemical Society] Copyright [2022]”. (B) Photographs of CV-stained S. aureus biofilm, and corresponding biomass after being treated with H2O2, LS-CuS@PVA and LS-CuS@PVA + H2O2 in the presence and absence of NIR irradiation. (C) Molecular structure of tested cNDIs for antibiofilm activity. “Reproduced from ref. 162 with permission from [American Chemical Society] Copyright [2021]”. | ||
Recently, Tarawneh and co-workers developed a facile approach for the treatment of catheter-associated urinary tract infections (CAUTIs) by preparing a poly 2-hydroxyethyl methacrylate (p-HEMA) containing hydrogel composed of the antibiotics cefixime trihydrate (CFX), rifampicin (RIF) alone and a combination of RIF and CIF.157 To demonstrate the antibacterial activity, different ratios of antibiotics (CFX, RIF and the mixture of CIF and RIF) to p-HEMA hydrogel were used against S. aureus, E. coli, and P. aeruginosa. The p-HEMA hydrogel containing 3:1 RIF:CFX has shown the highest antibacterial resilience against S. aureus and E. coli, while the hydrogel loaded with equimolar ratios of RIF and CFX was effective for inhibiting the growth of P. aeruginosa. After establishing the bacterial growth inhibition performances of p-HEMA hydrogel, it was tested for the removal of mixed biofilm created with the microbes mentioned above. Amongst these, only the RIF- and RIF/CFX (3:1) loaded p-HEMA hydrogels appeared to be most successful. Moreover, the hydrogels showed negligible toxicity on HEK293 healthy cells, making them a prospective candidate for urinary biomaterials.
4.5 Other polymers
Chen and co-workers meticulously fabricated a degradable supramolecular photodynamic polymer (SPP) through metal ion coordination and cucurbit[8]uril (CB[8])-promoted host–guest interactions for the efficient elimination of S. aureus biofilms.158 Initially, a viologen pendant block copolymer was prepared via RAFT polymerization and subjected to undergoing host–guest complexation with a rationally designed cationic porphyrin photosensitizer in the presence of CB[8]. The biofilm elimination capability of SPP and only porphyrin was then assessed by a CV staining assay. In the dark, the CFU reduction was around 20% for both compounds. Meanwhile, a ∼70% and ∼100% reduction in CFU was noticed for the supramolecular polymer and free porphyrin, respectively, under white light illumination. This fact is ascribed to the local enrichment of the cationic photosensitizer around the biofilm through double-layer electrostatic attraction159,160 and the generation of ROS leading to bacterial cell death, further verified by CLSM imaging.Zink's group researched the development of antibiotic-loaded mesoporous silica NPs (MSNs) combined with magnetic NPs for the synergistic annihilation of P. aeruginosa-constructed bacterial biofilm.161 Here, a multi-stimulus-responsive nanoplatform was prepared by the supramolecular co-assembly of a magnetic core containing ofloxacin-encapsulated CB[6]-capped MSNs and melittin-loaded adamantane-decorated MSNs. Under AMF heating, both melittin and ofloxacin were shown to be effectively released from the supramolecular co-assemblies. These co-assemblies could efficiently kill and completely remove the biofilm biomass after being incubated with P. aeruginosa bacteria, suggesting their outstanding antibiofilm efficacy. Moreover, they exerted negligible cytotoxicity on the mammalian cells. It is worth mentioning that the as-prepared MSNs supramolecular assemblies played an important role in biofilm removal as they could easily penetrate the biofilm and facilitate prolonged interaction between the loaded antibiotics and the biofilms.
Recently, Ghosh et al. demonstrated the antibacterial and biofilm eradication ability of ultrathin two-dimensional (2D) nanosheets with amine functionalities.162 Here, three types of 2D nanosheets (cNDI-1, cNDI-2, and cNDI-3) with a height of <1.5 nm were prepared by the aqueous self-assembly of core-substituted naphthalene diimide (cNDI)-based π-amphiphiles (Fig. 17C). These 2D sheets have been shown to exhibit broad-spectrum antibacterial activity against both E. coli and S. aureus bacterial strains with an MIC of less than 100 μg mL−1. Of the three, the cNDI with quaternary ammonium entities and a C12 hydrophobic chain (cNDI-2) was identified as the most lethal one, presenting the lowest MIC value (30 μg mL−1). Consequently, the antibiofilm performances of these nanosheets were evaluated by CV staining and CFU reduction assays using different aged E. coli/S. aureus biofilms. In the case of E. coli, >80% biofilm (24 h aged) was destroyed with 2× MIC (62.5 μg mL−1), whereas almost complete abolition of 96 h aged biofilm was achieved at a concentration of 125 μg mL−1. Comparable efficacies were observed for S. aureus biofilm as well. In-depth mechanistic studies using SEM and CLSM proposed leakage in the bacterial membranes leading to cell death within the biofilm. Furthermore, the cNDI 2D nanosheets showed excellent hemocompatibility (<5% hemolysis of RBCs) and non-cytotoxicity to HeLa cells.
5. Conclusions and outlook
Polymeric materials have emerged as a state-of-the-art alternative to traditional antibiotics/antimicrobials for the successful suppression and destruction of bacterial assembly-driven mature biofilms. Utilization of these materials is very advantageous because of their excellent biocompatibility, simple structural representation and synthetic protocols, easy functionalization/post-polymerization modifications, stimulus-responsiveness and more importantly their outstanding cytocompatibility and hemocompatibility. Therefore, a variety of polymer-based materials have been fabricated as potential antibiofilm agents so far. In this review, we have thoroughly explicated the tailor-made synthesis of those systems and their multiple modes of action for the eradication and/or inhibition of various types of bacterial biofilm under in vitro and/or in vivo conditions. Biopolymers, viz. chitosan and dextran, were emphasized as excellent candidates for destroying and dispersing established biofilms, thanks to their biodegradability. Modern strategies employing several synthetic macromolecules with inherent or externally induced antibiofilm features were also highlighted. Firstly, the role of cationic and charge-switchable polymers in the prevention of planktonic bacteria and obliteration of biofilms (and MDR biofilms) was demonstrated, followed by the antibiofilm activity of π-conjugated polymers, polymer nanocomposites with metal NPs, and biocompatible polymer-derived hydrogels. Additionally, a few examples of supramolecular polymers were examined for antibacterial and biofilm annihilation. These various forms of polymeric material have distinct approaches to the eradication or growth inhibition process. However, the basic antibiofilm mechanism mostly deals with the damage/rupturing of the bacterial membrane, which can be accelerated by synergistic therapy using a myriad of external factors: pH, PDT, PTT, ROS, AMF, etc. Unlike off-targeting with conventional antibiotics, these materials can specifically target bacterial cells and release loaded drugs at the infection site.Though polymer chemistry's adaptability confers novel weapons against microbial diseases163,164 there are still numerous issues exists that need to be solved before implementing them in clinical settings. According to our predictions, more sophisticated approaches will likely be used in the future, such as anti-infective therapeutic materials that combine mucoadhesion and simultaneous biofilm-targeting capability, and multi-modal killing efficacy. The bottlenecks associated with the current strategies include: (a) unsolicited interactions of positively charged polymeric materials with the cellular proteins, inorganic cations (Mg2+ and Ca2+) and healthy mammalian cells that hampered in vivo antibacterial performances; (b) accumulation on the surface of the biofilm preventing the further enrichment of antimicrobials, leading to incomplete excision of the biofilm-residing bacteria; and (c) a short circulation time of the materials in the physiological fluid due to the existence of the EPS, resulting in weak antibiofilm activity. Even though some of these problems have been moderately resolved by exploiting the stimulus-responsive facets of polymeric materials, the cytotoxicity of these systems remains a great concern. In summary, it should be stressed that certain systems have both multiple positive impacts and some unavoidable negative consequences, and polymeric materials are no exception. We envisioned that the polymeric materials will be a realistic choice for the treatment of bacterial biofilm-caused infections in the forthcoming years if those unwelcome issues are resolved. Also, we anticipate that this review will provide a new horizon for modern antibiofilm techniques, and further open the door for the development of new sophisticated polymeric materials for the inhibition and eradication of bacterial biofilms.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Science and Engineering Research Board (SERB), Government of India [file no.: CRG/2021/000095].References
- [Internet] WHO. Résistance aux antimicrobiens: les enjeux de la réunion des Nations Unies. Revised 27 September 2020. https://www.who.int/bulletin/volumes/94/9/16-020916/fr/.
- Y. Liu, L. Shi, L. Su, H. C. van der Mei, P. C. Jutte, Y. J. Ren and H. J. Busscher, Chem. Soc. Rev., 2019, 48, 428–446 RSC.
- H. C. Flemming, P. Baveye, T. R. Neu, P. Stoodley, U. Szewzyk, J. Wingender and S. Wuertz, npj Biofilms Microbiomes, 2021, 7, 10 CrossRef PubMed.
- X. K. Ding, S. Duan, X. J. Ding, R. H. Liu and F. J. Xu, Adv. Funct. Mater., 2018, 28, 1802140 CrossRef.
- J. N. Wilking, V. Zaburdaev, M. D. Volder, R. Losick, M. P. Brenner and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2012, 110, 848–852 CrossRef.
- Z. W. Wang and S. Chen, Appl. Microbiol. Biotechnol., 2009, 83, 1–18 CrossRef CAS PubMed.
- S. J. Edwards and B. V. Kjellerup, Appl. Microbiol. Biotechnol., 2013, 97, 9909–9921 CrossRef CAS.
- T. Parween, P. Bhandari, Z. H. Siddiqui, S. Jan, T. Fatma and P. K. Patanjali, Mycoremediation Environ. Sustainability, 2017, 1, 39–51 Search PubMed.
- B. M. Peters, M. A. Jabra-Rizk, G. A. O'May, J. W. Costerton and M. E. Shirtliff, Clin. Microbiol. Rev., 2012, 25, 193–213 CrossRef PubMed.
- T. S. Murali, S. Kavitha, J. Spoorthi, D. V. Bhat, A. S. B. Prasad, Z. Upton, L. Ramachandra, R. V. Acharya and K. Satyamoorthy, J. Med. Microbiol., 2014, 63, 1377–1385 CrossRef CAS PubMed.
- J. J. Swartjes, P. K. Sharma, T. G. van Kooten, H. C. van der Mei, M. Mahmoudi, H. J. Busscher and E. T. J. Rochford, Curr. Med. Chem., 2015, 22, 2116–2129 CrossRef CAS.
- Y. Zhao, Q. Guo, X. Dai, X. Wei, Y. Yu, X. L. Chen, C. Li, Z. Cao and X. Zhang, Adv. Mater., 2018, 31, 1806024 CrossRef PubMed.
- K. Hede, Nature, 2014, 509, S2–S3 CrossRef PubMed.
- P. E. Kolenbrander, R. J. Palmer, S. Periasamy and N. S. Jakubovics, Nat. Rev. Microbiol., 2010, 8, 471–480 CrossRef CAS.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS PubMed.
- H. J. Busscher, H. C. van der Mei, G. Subbiahdoss, P. C. Jutte, J. J. A. M. van den Dungen, S. A. J. Zaat, M. J. Schultz and D. W. Grainger, Sci. Transl. Med., 2012, 4, 153 Search PubMed.
- A. D. Verderosa, M. Totsika and K. E. Fairfull-Smith, Front. Chem., 2019, 7, 824 CrossRef CAS PubMed.
- B. Parrino, D. Schillaci, I. Carnevale, E. Giovannetti, P. Diana, G. Cirrincione and S. Cascioferro, Eur. J. Med. Chem., 2019, 161, 154–178 CrossRef CAS PubMed.
- A. Al-Jumaili, S. Alancherry, K. Bazaka and M. V. Jacob, Materials, 2017, 10, 1066 CrossRef.
- K. P. Miller, L. Wang, B. C. Benicewicz and A. W. Decho, Chem. Soc. Rev., 2015, 44, 7787–7807 RSC.
- M.-H. Kim, IEEE Trans. Nanobiosci., 2016, 15, 294–304 Search PubMed.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, Bioconjugate Chem., 2019, 30, 218–230 CrossRef CAS PubMed.
- L. D. Blackman, Y. Qu, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2021, 50, 1587–1616 RSC.
- D. S. W. Benoit, K. R. Sims Jr. and D. Fraser, ACS Nano, 2019, 13, 4869–4875 CrossRef CAS PubMed.
- C. Li, E. J. Cornel and J. Du, ACS Appl. Polym. Mater., 2021, 3, 2218–2232 CrossRef CAS.
- C. Sahli, S. E. Moya, J. S. Lomas, C. Gravier-Pelletier, R. Briandet and M. Hémadi, Theranostics, 2022, 12, 2383–2405 CrossRef CAS PubMed.
- M. E. Davey and G. A. O'Toole, Microbiol. Mol. Biol. Rev., 2000, 64, 847–867 CrossRef CAS PubMed.
- J. Palmer, S. Flint and J. Brooks, J. Ind. Microbiol. Biotechnol., 2007, 34, 577–588 CrossRef CAS PubMed.
- Y. Qu, Y. Li, D. R. Cameron, C. D. Easton, X. Zhu, M. Zhu, M. Salwiczek, B. W. Muir, H. Thissen, A. Daley, J. S. Forsythe, A. Y. Peleg and T. Lithgow, Front. Microbiol., 2020, 11, 920 CrossRef PubMed.
- R. Bos, FEMS Microbiol. Rev., 1999, 23, 179–229 CrossRef CAS.
- M. Otto, Gram-positive Pathogens, 2019, pp. 699–711, DOI:10.1128/9781683670131.ch43.
- H. C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS.
- H. C. Flemming, T. R. Neu and D. J. Wozniak, J. Bacteriol., 2007, 189, 7945–7947 CrossRef CAS.
- C. Solano, M. Echeverz and I. Lasa, Curr. Opin. Microbiol., 2014, 18, 96–104 CrossRef CAS.
- M. R. Parsek and E. P. Greenberg, Trends Microbiol., 2005, 13, 27–33 CrossRef CAS PubMed.
- M. B. Miller and B. L. Bassler, Annu. Rev. Microbiol., 2001, 55, 165–199 CrossRef CAS.
- P. Albuquerque and A. Casadevall, Med. Mycol., 2012, 50, 337–345 CrossRef CAS PubMed.
- G. O'Toole, H. B. Kaplan and R. Kolter, Annu. Rev. Microbiol., 2000, 54, 49–79 CrossRef.
- P. S. Stewart and J. William-Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS.
- M. Moscoso, E. Garcıa and R. Lopez, J. Bacteriol., 2006, 188, 7785–7795 CrossRef CAS PubMed.
- K. Lewis, Biochemistry, 2005, 70, 267–274 CAS.
- K. Lewis, in Bacterial Biofilms, ed. T. Romeo, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 107–131, DOI:10.1007/978-3-540-75418-3.
- S. S. Ozcan, M. Dieser, A. E. Parker, N. Balasubramanian and C. M. Foreman, J. Ind. Microbiol. Biotechnol., 2019, 46, 1103–1111 CrossRef CAS.
- A. A. Saqr, M. F. Aldawsari, E.-S. Khafagy, M. A. Shaldam, W. A. H. Hegazy and H. A. Abbas, Antibiotics, 2021, 10, 1385 CrossRef CAS.
- N. H. Mahdally, R. F. George, M. T. Kashef, M. Al-Ghobashy, F. E. Murad and A. S. Attia, Front. Microbiol., 2021, 12, 700494 CrossRef.
- J. Liu, J.-S. Hou, Y.-Q. Chang, L.-J. Peng, X.-Y. Zhang, Z.-Y. Miao, P.-H. Sun, J. Lin and W.-M. Chen, J. Med. Chem., 2022, 65, 688–709 CrossRef CAS.
- M. Harmsen, M. Lappann, S. Knøchel and S. Molin, Appl. Environ. Microbiol., 2010, 76, 2271–2279 CrossRef CAS PubMed.
- M. J. Huseby, A. C. Kruse, J. Digre, P. L. Kohler, J. A. Vocke, E. E. Mann, K. W. Bayles, G. A. Bohach, P. M. Schlievert, D. H. Ohlendorf and C. A. Earhart, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14407–14412 CrossRef CAS.
- C. B. Whitchurch, T. Tolker-Nielsen, P. C. Ragas and J. S. Mattick, Science, 2002, 295, 1487 CrossRef CAS.
- J. J. T. M. Swartjes, T. Das, S. Sharifi, G. Subbiahdoss, P. K. Sharma, B. P. Krom, H. J. Busscher and H. C. V. D. Mei, Adv. Funct. Mater., 2013, 23, 2843–2849 CrossRef CAS.
- W. Tasia, C. Lei, Y. Cao, Q. Ye, Y. He and C. Xu, Nanoscale, 2020, 12, 2328–2332 RSC.
- C. Liu, Y. Zhao, W. Su, J. Chai, L. Xu, J. Cao and Y. Liu, J. Mater. Chem. B, 2020, 8, 4395–4401 RSC.
- T. Böttcher, I. Kolodkin-Gal, R. Kolter, R. Losick and J. Clardy, J. Am. Chem. Soc., 2013, 135, 2927–2930 CrossRef.
- M. S. Ganewatta, K. P. Miller, S. P. Singleton, P. Mehrpouya-Bahrami, Y. P. Chen, Y. Yan, M. Nagarkatti, P. Nagarkatti, A. W. Decho and C. Tang, Biomacromolecules, 2015, 16, 3336–3344 CrossRef CAS PubMed.
- D. Freebairn, D. Linton, E. Harkin-Jones, D. S. Jones, B. F. Gilmore and S. P. Gorman, Expert Rev. Med. Devices, 2013, 10, 85–103 CrossRef CAS PubMed.
- R. E. W. Hancock and A. Rozek, FEMS Microbiol. Lett., 2002, 206, 143–149 CrossRef CAS PubMed.
- B. P. Conlon, S. E. Rowe and K. Lewis, Adv. Exp. Med. Biol., 2015, 831, 1–9 CrossRef PubMed.
- K. Forier, K. Raemdonck, S. C. De Smedt, J. Demeester, T. Coenye and K. Braeckmans, J. Controlled Release, 2014, 190, 607–623 CrossRef CAS.
- Z. Rukavina and Z. Vanic, Pharmaceutics, 2016, 8, 18 CrossRef PubMed.
- A. Zhang, H. Mu, W. Zhang, G. Cui, J. Zhu and J. Duan, Sci. Rep., 2013, 3, 3364 CrossRef PubMed.
- B. Orgaz, M. M. Lobete, C. H. Puga and C. S. Jose, Int. J. Mol. Sci., 2011, 12, 817–828 CrossRef CAS PubMed.
- P. Sahariah, M. Másson and R. L. Meyer, Biomacromolecules, 2018, 19, 3649–3658 CrossRef CAS PubMed.
- P. Sahariah, V. S. Gaware, R. Lieder, S. Jónsdóttir, M. Á. Hjálmarsdóttir, O. E. Sigurjonsson and M. Másson, Mar. Drugs, 2014, 12, 4635–4658 CrossRef PubMed.
- Y. Liu, Y. Jiang, J. Zhu, J. Huang and H. Zhang, Carbohydr. Polym., 2019, 206, 412–419 CrossRef CAS.
- J. Cao, Y. Zhao, Y. Liu, S. Tian, C. Zheng, C. Liu, Y. Zhai, Y. An, H. J. Busscher, L. Shi and Y. Liu, ACS Macro Lett., 2019, 8, 651–657 CrossRef.
- L. Pavez, N. Tobar, C. Chacón, R. Arancibia, C. Martinez, C. Tapia, A. Pastor, M. González, J. Martínez and P. C. Smith, J. Periodontal Res., 2018, 53, 232–239 CrossRef CAS.
- X. C. Chen, K. F. Ren, J. H. Zhang, D. D. Li, E. Zhao, Z. J. Zhao, Z. K. Xu and J. Ji, Adv. Funct. Mater., 2015, 25, 7470–7477 CrossRef.
- Y. Wang and A. Shukla, Biomater. Sci., 2022, 10, 2831–2843 RSC.
- S. A. Trinh, H. E. Gavin and K. J. F. Satchell, Antimicrob. Agents Chemother., 2017, 61, e01106–e01117 CrossRef CAS PubMed.
- Z. V. Feng, I. L. Gunsolus, T. A. Qiu, K. R. Hurley, L. H. Nyberg, H. Frew, K. P. Johnson, A. M. Vartanian, L. M. Jacob, S. E. Lohse, M. D. Torelli, R. J. Hamers, C. J. Murphy and C. L. Haynes, Chem. Sci., 2015, 6, 5186–5196 RSC.
- W. Wang, X. Hao, S. Chen, Z. Yang, C. Wang, R. Yan, X. Zhang, H. Liu, Q. Shao and Z. Guo, Polymer, 2018, 158, 223–230 CrossRef CAS.
- V. Raj, Y. Kim, Y.-G. Kim, J.-H. Lee and J. Lee, Carbohydr. Polym., 2022, 284, 118959 CrossRef CAS.
- M. Maruthupandy, G. Rajivgandhi, S. Kadaikunnan, T. Veeramani, N. S. Alharbi, T. Muneeswaran, J. M. Khaled, W. J. Li and K. F. Alanzi, Carbohydr. Polym., 2020, 230, 115646 CrossRef PubMed.
- L. M. Hemmingsen, B. Giordani, A. K. Pettersen, B. Vitali, P. Basnet and N. S. Basnet, Carbohydr. Polym., 2021, 262, 117939 CrossRef CAS PubMed.
- J. Hoque and J. Haldar, ACS Appl. Mater. Interfaces, 2017, 9, 15975–15985 CrossRef CAS.
- E. L. Dane, A. E. Ballok, G. A. O'Toole and M. W. Grinstaff, Chem. Sci., 2014, 5, 551–557 RSC.
- T. A. Fernandes, I. F. M. Costa, P. Jorge, A. C. Sousa, V. André, R. G. Cabral, N. Cerca and A. M. Kirillov, ACS Appl. Mater. Interfaces, 2022, 14, 25104–25114 CrossRef CAS.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS.
- A. K. Marr, W. J. Gooderham and R. E. W. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- M. Sieprawska-Lupa, P. Mydel, K. Krawczyk, K. Wójcik, M. Puklo, B. Lupa, P. Suder, J. Silberring, M. Reed, J. Pohl, W. Shafer, F. McAleese, T. Foster, J. Travis and J. Potempa, Antimicrob. Agents Chemother., 2004, 48, 4673–4679 CrossRef CAS PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS.
- S. Yan, S. Chen, X. Gou, J. Yang, J. An, X. Jin, Y. W. Yang, L. Chen and H. Gao, Adv. Funct. Mater., 2019, 29, 1904683 CrossRef.
- H. Takahashi, E. T. Nadres and K. Kuroda, Biomacromolecules, 2017, 18, 257–265 CrossRef CAS.
- C. Peng, A. Vishwakarma, Z. Li, T. Miyoshi, H. A. Barton and A. Joy, Polym. Chem., 2018, 9, 3195–3198 RSC.
- S. M. Jacobsen, D. J. Stickler, H. L. T. Mobley and M. E. Shirtliff, Clin. Microbiol. Rev., 2008, 21, 26–59 CrossRef CAS PubMed.
- L. E. Nicolle, Antimicrob. Resist. Infect. Control, 2014, 3, 23 CrossRef PubMed.
- A. Vishwakarma, F. Dang, A. Ferrell, H. A. Barton and A. Joy, J. Am. Chem. Soc., 2021, 143, 9440–9449 CrossRef CAS PubMed.
- C. de la Fuente-Nunez, F. Reffuveille, S. C. Mansour, S. L. Reckseidler-Zenteno, D. Hernandez, G. Brackman, T. Coenye and R. E. W. Hancock, Chem. Biol., 2015, 22, 196–205 CrossRef CAS.
- E. F. Haney, Y. Brito-Sánchez, M. J. Trimble, S. C. Mansour, A. Cherkasov and R. E. W. Hancock, Sci. Rep., 2018, 8, 1871 CrossRef.
- R. Barman, D. Ray, V. K. Aswal and S. Ghosh, Polym. Chem., 2022, 13, 4384–4394 RSC.
- M. Chen, S. Zhang and Z. He, ACS Appl. Bio Mater., 2020, 3, 6343–6350 CrossRef PubMed.
- I. Kolodkin-Gal, D. Romero, S. Cao, J. Clardy, R. Kolter and R. Losick, Science, 2010, 328, 627–629 CrossRef CAS PubMed.
- E. C. Abenojar, S. Wickramasinghe, M. Ju, S. Uppaluri, A. Klika, J. George, W. Barsoum, S. J. Frangiamore, C. A. Higuera-Rueda and A. C. S. Samia, ACS Infect. Dis., 2018, 4, 1246–1256 CrossRef CAS PubMed.
- S. Zhao, W. Huang, C. Wang, Y. Wang, Y. Zhang, Z. Ye, J. Zhang, L. Deng and A. Dong, Biomacromolecules, 2020, 21, 5269–5281 CrossRef CAS PubMed.
- X. Dai, X. Chen, Y. Zhao, Y. Yu, X. Wei, X. Zhang and C. Li, Biomacromolecules, 2018, 19, 141–149 CrossRef CAS PubMed.
- A. Kamkaew and K. Burgess, J. Med. Chem., 2013, 56, 7608–7614 CrossRef CAS PubMed.
- M. B. Ballatore, J. Durantini, N. S. Gsponer, M. B. Suarez, M. Gervaldo, L. Otero, M. B. Spesia, M. E. Milanesio and E. N. Durantini, Environ. Sci. Technol., 2015, 49, 7456–7463 CrossRef CAS PubMed.
- A. Gupta, R. F. Landis, C. H. Li, M. Schnurr, R. Das, Y. W. Lee, M. Yazdani, Y. Liu, A. Kozlova and V. M. Rotello, J. Am. Chem. Soc., 2018, 140, 12137–12143 CrossRef CAS PubMed.
- S. Huo, Y. Jiang, A. Gupta, Z. Jiang, R. F. Landis, S. Hou, X. J. Liang and V. M. Rotello, ACS Nano, 2016, 10, 8732–8737 CrossRef CAS PubMed.
- A. Gupta, J. M. V. Makabenta, F. Schlüter, R. F. Landis, R. Das, M. Cuppels and V. M. Rotello, Adv. Ther., 2020, 3, 2000005 CrossRef CAS PubMed.
- P. K. T. Jeremy, D. J. Coady, H. Sardon, A. Yuen, S. Gao, S. W. Lim, Z. C. Liang, E. W. Tan, S. Venkataraman, A. C. Engler, M. Fevre, R. Ono, Y. Y. Yang and J. L. Hedrick, Adv. Healthcare Mater., 2017, 6, 1601420 CrossRef PubMed.
- T. Lan, Q. Guo and X. Shen, J. Biomater. Sci., Polym. Ed., 2019, 30, 1243–1259 CrossRef CAS PubMed.
- X. Wang, G. Wang, J. Zhao, Z. Zhu and J. Rao, ACS Macro Lett., 2021, 10, 1643–1649 CrossRef CAS.
- J. Xiao, M. I. Klein, M. L. Falsetta, B. Lu, C. M. Delahunty, J. R. Yates, A. Heydorn and H. Koo, PLoS Pathog., 2012, 8, 002623 Search PubMed.
- S. Piwat, H. Hassan, T. Kjeang, J. Lindehag, H. Wedin, R. Teanpaisan and G. Dahlén, Clin. Oral Investig., 2015, 19, 2179–2186 CrossRef CAS PubMed.
- Z. Zhao, C. Ding, Y. Wang, H. Tan and J. Li, Biomater. Sci., 2019, 7, 1643–1651 RSC.
- X. Dai, Y. Bai, Y. Zhang, Z. Ma, J. Li, H. Sun and X. Zhang, ACS Appl. Bio Mater., 2020, 3, 3868–3879 CrossRef CAS PubMed.
- A. Imberty, M. Wimmerova, E. P. Mitchell and N. G. Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- A. F. Radovic-Moreno, T. K. Lu, V. A. Puscasu, C. J. Yoon, R. Langer and O. C. Farokhzad, ACS Nano, 2012, 6, 4279–4287 CrossRef CAS.
- Y. Abouelhassan, Q. Yang, H. Yousaf, M. T. Nguyen, M. Rolfe, G. S. Schultz and R. W. Huigens 3rd, Int. J. Antimicrob. Agents, 2017, 49, 247–251 CrossRef CAS.
- Y. Gao, J. Wang, M. Chai, X. Li, Y. Deng, Q. Jin and J. Ji, ACS Nano, 2020, 14, 5686–5699 CrossRef CAS PubMed.
- X. Chen, R. Guo, C. Wang, K. Li, X. Jiang, H. He and W. Hong, J. Nanobiotechnol., 2021, 19, 99 CrossRef CAS.
- S. Tian, L. Su, Y. Liu, J. Cao, G. Yang, Y. Ren, F. Huang, J. Liu, Y. An, H. C. van der Mei, H. J. Busscher and L. Shi, Sci. Adv., 2020, 6, eabb1112 CrossRef CAS PubMed.
- X. Dai, Q. Xu, L. Yang, J. Ma and F. Gao, ACS Biomater. Sci. Eng., 2022, 8, 893–902 CrossRef CAS PubMed.
- S. Chemburu, T. S. Corbitt, L. K. Ista, E. Ji, J. Fulghum, G. P. Lopez, K. Ogawa, K. S. Schanze and D. G. Whitten, Langmuir, 2008, 24, 11053–11062 CrossRef CAS PubMed.
- T. S. Corbitt, J. R. Sommer, S. Chemburu, K. Ogawa, L. K. Ista, G. P. Lopez, D. G. Whitten and K. S. Schanze, ACS Appl. Mater. Interfaces, 2009, 1, 48–52 CrossRef CAS PubMed.
- P. Zhang, S. Li, H. Chen, X. Wang, L. Liu, F. Lv and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16933–16938 CrossRef CAS PubMed.
- R. J. Hamill, Drugs, 2013, 73, 919–934 CrossRef CAS PubMed.
- P. Jagadesan, Z. Yu, I. Barboza-Ramos, H. H. Lara, R. Vazquez-Munoz, J. L. López-Ribot and K. S. Schanze, Chem. Mater., 2020, 32, 6186–6196 CrossRef CAS.
- J. Guo, J. Qin, Y. Ren, B. Wang, H. Cui, Y. Ding, H. Mao and F. Yan, Polym. Chem., 2018, 9, 4611–4616 RSC.
- S. Y. Alston, S. Sarkar, M. Cochran, N. Kuthirummal and N. Levi, J. Microbiol. Methods, 2021, 190, 106328 CrossRef.
- S. Ghosh, G. Amariei, M. E. G. Mosquera and R. Rosal, J. Mater. Chem. B, 2021, 9, 4390–4399 RSC.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- J. Zhang, Y. P. Chen, K. P. Miller, M. S. Ganewatta, M. Bam, Y. Yan, M. Nagarkatti, A. W. Decho and C. Tang, J. Am. Chem. Soc., 2014, 136, 4873–4876 CrossRef CAS PubMed.
- Y. Wang, H.-B. Fang, Y.-Z. Zheng, R. Ye, X. Tao and J. F. Chen, Nanoscale, 2015, 7, 19118–19128 RSC.
- R. Kumar and H. Münstedt, Biomaterials, 2005, 26, 2081–2088 CrossRef CAS PubMed.
- S. Chernousova and M. Epple, Angew. Chem., Int. Ed., 2013, 52, 1636–1653 CrossRef CAS PubMed.
- X. Dai, X. Chen, J. Zhao, Y. Zhao, Q. Guo, T. Zhang, C. Chu, X. Zhang and C. Li, ACS Appl. Mater. Interfaces, 2017, 9, 13837–13848 CrossRef CAS PubMed.
- Q. L. Feng, J. Wu, G. Q. Chen, F. Z. Cui, T. N. Kim and J. O. Kim, Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS.
- Q. Guo, Y. Zhao, X. Dai, T. Zhang, Y. Yu, X. Zhang and C. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16834–16847 CrossRef CAS PubMed.
- A. Panáček, L. Kvítek, R. Prucek, M. Kolář, R. Večeřová, N. Pizúrová, V. K. Sharma, T. Nevěčná and R. Zbořil, J. Phys. Chem. B, 2006, 110, 16248–16253 CrossRef PubMed.
- J. Wu, F. Li, X. Hu, J. Lu, X. Sun, J. Gao and D. Ling, ACS Cent. Sci., 2019, 5, 1366–1376 CrossRef CAS PubMed.
- P. Sun, L. Ye, X. Tan, J. Peng, L. Zhao and Y. Zhou, ACS Appl. Nano Mater., 2022, 5, 8251–8259 CrossRef CAS.
- M. Yin, M. Yang, D. Yan, L. Yang, X. Wan, J. Xiao, Y. Yao and J. Luo, ACS Appl. Mater. Interfaces, 2022, 14, 8847–8864 CrossRef PubMed.
- Z. Shi, Y. Zhang, R. Dai, S. Chen, M. Zhang, L. Jin, J. Wang, W. Zhao and C. Zhao, Colloids Surf., B, 2020, 186, 1107282 CrossRef PubMed.
- W. Liu, W. Pei, M. Moradi, D. Zhao, Z. Li, M. Zhang, D. Xu and F. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 18794–18805 CrossRef CAS.
- J. He, J. Yang, M. Li, Y. Li, Y. Pang, J. Deng, X. Zhang and W. Liu, ACS Nano, 2022, 16, 3119–3134 CrossRef CAS PubMed.
- D. Gan, T. Xu, W. Xing, X. Ge, L. Fang, K. Wang, F. Ren and X. Lu, Adv. Funct. Mater., 2019, 29, 1805964 CrossRef.
- Z. Güngör and H. Ozay, J. Macromol. Sci., Part A: Pure Appl.Chem., 2022, 59, 295–306 CrossRef.
- T. Y. Zhan, S. Wang, Z. Y. Guo and Y. F. Hu, J. Macromol. Sci., Part A: Pure Appl.Chem., 2022, 59, 72–82 CrossRef CAS.
- P. Li, Y. F. Poon, W. Li, H.-Y. Zhu, S. H. Yeap, Y. Cao, X. Qi, C. Zhou, M. Lamrani, R. W. Beuerman, E.-T. Kang, Y. Mu, C. M. Li, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Nat. Mater., 2011, 10, 149–156 CrossRef CAS PubMed.
- C. Zhou, Y. Wu, K. R. V. Thappeta, J. T. L. Subramanian, D. Pranantyo, E.-T. Kang, H. Duan, K. Kline and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2017, 9, 36269–36280 CrossRef CAS PubMed.
- C. K. Yeo, Y. S. Vikhe, P. Li, Z. Guo, P. Greenberg, H. Duan, N. S. Tan and M. B. Chan-Park, ACS Appl. Mater. Interfaces, 2018, 10, 20356–20367 CrossRef CAS PubMed.
- H. Irawan, H. Yasa and K. P. Bali, Med. J., 2017, 6, S93–S96 Search PubMed.
- World Health Organization. WHO published a list of bacteria for which new antibiotics are urgently needed.
- H. Wang, S. Zhou, L. Guo, Y. Wang and L. Feng, ACS Appl. Mater. Interfaces, 2020, 12, 39685–39694 CrossRef CAS PubMed.
- H. Haidari, Z. Kopecki, R. Bright, A. J. Cowin, S. Garg, N. Goswami and K. Vasilev, ACS Appl. Mater. Interfaces, 2020, 12, 41011–41025 CrossRef CAS PubMed.
- R. Gharibi and S. Agarwal, ACS Appl. Polym. Mater., 2021, 3, 4695–4707 CrossRef CAS.
- R. Gharibi and S. Agarwal, ACS Appl. Bio Mater., 2021, 4, 4629–4640 CrossRef CAS PubMed.
- J. S. Gonzalez, A. S. Maiolo, C. E. Hoppe and V. A. Alvarez, Procedia Mater. Sci., 2012, 1, 483–490 CrossRef CAS.
- D. Hu and L. J. Wang, J. Appl. Polym. Sci., 2016, 133, 43552–43559 CrossRef.
- Y. Xie, C. Gan, Z. Li, W. Liu, D. Yang and X. Qiu, ACS Biomater. Sci. Eng., 2022, 8, 560–569 CrossRef CAS PubMed.
- S. Wang, A. Riedinger, H. Li, C. Fu, H. Liu, L. Li, T. Liu, L. Tan, M. J. Barthel, G. Pugliese, F. D. Donato, M. S. D'Abbusco, X. Meng, L. Manna, H. Meng and T. Pellegrino, ACS Nano, 2015, 9, 1788–1800 CrossRef CAS PubMed.
- W. Wang, B. Li, H. Yang, Z. Lin, L. Chen, Z. Li, J. Ge, T. Zhang, H. Xia, L. Li and Y. Lu, Nano Res., 2020, 13, 2156–2164 CrossRef CAS.
- R. T. Alfuraydi, F. M. Alminderej and N. A. Mohamed, Polymers, 2022, 14, 1619 CrossRef CAS PubMed.
- O. Tarawneh, H. A. Mahfouz, L. Hamadneh, A. A. Deeb, I. Al-Sheikh, W. Alwahsh and A. F. Abed, Sci. Rep., 2022, 12, 3900 CrossRef CAS PubMed.
- L. Chen, Y. Yang, P. Zhang, S. Wang, J. F. Xu and X. Zhang, ACS Appl. Bio Mater., 2019, 2, 2920–2926 CrossRef CAS PubMed.
- G. L. Y. Woo, M. W. Mittelman and J. P. Santerre, Biomaterials, 2000, 21, 1235–1246 CrossRef CAS PubMed.
- A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- Q. Yu, T. Deng, F.-C. Lin, B. Zhang and J. I. Zink, ACS Nano, 2020, 14, 5926–5937 CrossRef CAS PubMed.
- A. Mukherjee, R. Barman, B. Das and S. Ghosh, Chem. Mater., 2021, 33, 8656–8665 CrossRef CAS.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, J. Mater. Chem. B, 2019, 7, 3007–3018 RSC.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2018, 3, 769–780 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |