
PDAC-on-chip for in vitro modeling of stromal and pancreatic cancer cell crosstalk†
Viola
Sgarminato
ab,
Simone Luigi
Marasso
 cd,
Matteo
Cocuzza
cde,
Giorgio
Scordo‡
c,
Alberto
Ballesio
c,
Gianluca
Ciardelli
aef and
Chiara
Tonda-Turo
*ae
cd,
Matteo
Cocuzza
cde,
Giorgio
Scordo‡
c,
Alberto
Ballesio
c,
Gianluca
Ciardelli
aef and
Chiara
Tonda-Turo
*ae
aDepartment of Mechanical and Aerospace Engineering, Politecnico di Torino, Turin, Italy. E-mail: chiara.tondaturo@polito.it
bDepartment of Surgical Sciences, Università degli Studi di Torino, Corso Dogliotti, 14, 10126 Torino, Italy
cChilab – Materials and Microsystems Laboratory, Department of Applied Science and Technology (DISAT), Politecnico di Torino, Chivasso, TO, Italy
dCNR-IMEM, Parco Area delle Scienze, Parma, Italy
ePOLITO BIOMedLAB, Politecnico di Torino, Turin, Italy
fDepartment for Materials and Devices of the National Research Council, Institute for the Chemical and Physical Processes (CNR-IPCF UOS), Pisa, Italy
First published on 9th November 2022
Abstract
Pancreatic ductal adenocarcinoma (PDAC) mainly develops in the head of the pancreas, within the acino-ductal unit composed of acinar and ductal cells surrounded by pancreatic stellate cells (PSCs). PSCs strongly influence the tumor microenvironment by triggering an intense stromal deposition, which plays a key role in tumor progression and limits drug perfusion. We have developed a microfluidic in vitro model recreating the in vivo tumor-stroma crosstalk to replicate the steps of PDAC evolution towards the establishment of an efficient in vitro platform for innovative therapy validation. The multilayer PDAC-on-chip was designed to culture the PDAC cells and the PSCs embedded in a type I collagen gel in the top and bottom layers, respectively. The presence of a biomimetic nanofibrous membrane in the middle of the chip permits the control of interactions between the two cell lines and the easy analysis of the effects of the crosstalk on cell behavior. First, the PDAC-stromal cell relationship was evaluated under co-culture conditions on 24-well inserts including the PCL/Gel electrospun membrane. This simplified model shows that human fibroblasts change their morphology and secrete larger amounts of IL-6 cytokines in the presence of tumor cells, confirming the activation of stromal cells under co-culture. Then, the PDAC-on-chip system was validated by demonstrating that human fibroblasts seeded in a 3D collagen matrix in the bottom microchannel also change to a myofibroblast-like shape with increased expression of α-SMA and secrete larger amounts of IL-6 cytokines. This microfluidic system is suitable for the evaluation of drug efficacy and serves as a powerful tool for understanding the early evolution steps of PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC), commonly known as pancreatic cancer, is the most frequent type of exocrine pancreas tumor and one of the leading causes of cancer-related death worldwide with a five-year survival rate of below 9%.1,2The main reason for considering PDAC as a notably aggressive disease is its rapid evolution without clear symptoms in the early stages, resulting in a late diagnosis and poor clinical prognosis. Only 10% of the patients have resectable tumors since the majority of cases present extended metastases and advanced lesions at diagnosis.3 In addition, the genetic complexity and the heterogeneity of the pancreatic tumor microenvironment weaken the effectiveness of current treatments, despite improvements in the discovery of new therapeutical strategies.3–5 In particular, the PDAC microenvironment is mainly composed of a desmoplastic stroma, which strongly affects the tumor's evolution and represents a barrier against chemotherapy and radiotherapy.6–9 The stroma is mainly composed of collagens, with type I and III fibrillar collagens accounting for >90% of all collagen mass,10 and it is generated by the excessive extracellular matrix (ECM) deposition, principally from the pancreatic stellate cells (PSCs) that trigger an intense desmoplastic reaction within the tissue surrounding the cancer cells.9,11 More specifically, PSCs are normally located in the periacinar space around the acinar and ductal cells constituting the pancreatic adenomere, which represents the fundamental unit of the exocrine pancreas and the region where the pancreatic intraepithelial neoplasia (PanIN) occurs.3,12 During tumorigenesis, PSCs that are in a quiescent state become active and change their spindle-shaped morphology, exhibiting a myofibroblast-like phenotype. Their primary activation is the result of an inflammatory stimulus that causes the recruitment of the immune system cells into the tumor site (monocytes, T cells, neutrophils, macrophages, and mast cells). The inflammatory cells produce cytokines such as interleukin 6 (IL-6), which causes the mutation of the oncogene KRAS and the progression from PanIN to PDAC.6,13 Although important risk factors can contribute to the evolution of pancreatic cancer (such as smoking, obesity, type 2 diabetes, chronic pancreatitis, and alcoholism7) and mechanisms of maturation from the neoplasia are well documented,14,15 the alterations that give rise to the early lesions remain unclear.16 For these reasons, current research is focusing on the development of in vitro PDAC models for a deeper understanding of this pathology, the identification of new biomarkers and the establishment of screening tests, in order to enable the earlier detection of pancreatic cancer and improve the prognosis.17–22 However, one of the major limitations of PDAC in vitro models is the difficulty in replicating the tumor microenvironment surrounding the gland and the PDAC-stroma crosstalk, which constitutes an important feature that significantly affects tumor evolution and drug resistance.22–26 Among the existing in vitro models, organ-on-a-chip microfluidic systems represent a powerful tool to reproduce the cancer cell-stroma interactions in a controllable environment and under real-time monitoring.19,27–29 In addition, these devices make drug tests and cell analysis accessible and immediate due to the presence of typical micrometer-sized channels that allow the controlled patterning of stromal and cancer cells.30 Recent studies have demonstrated the possibility to include the stromal component on microfluidic platforms to better replicate the PDAC microenvironment by integrating hallmarks of pancreatic cancer desmoplastic tissue.20,31–34 However, the research is still far from a complete understanding of this disease and the mechanisms involved in the PDAC-stroma relationship. Further improvements are therefore needed in the design of microfluidic systems. In this study, a novel biomimetic PDAC-on-chip was designed to model pancreatic cancer and its interaction with the surrounding environment. In particular, PSCs were embedded in a type I collagen gel and cultured in bottom microchambers of the multilayered device, designed to both confine the gel (the central microchannel) and to guarantee the transport of nutrients in automated and dynamic conditions (the two lateral microchannels). The ductal cancer cells, on the other hand, were seeded in the top layer of the platform and they were allowed to grow on a nanofibrous polycaprolactone/gelatin (PCL/gel) membrane, which represents a biomimetic substrate since the ECM-like architecture typical of the electrospun matrices can support cell adhesion, spreading and functions.35–38 The blend of a synthetic thermoplastic polymer (PCL) and a natural polymer (gel) has been widely adopted in the fabrication of nanofibrous scaffolds as it allows the achievement of a combination of suitable mechanical and physicochemical properties with improved biocompatibility.39–42 Moreover, the presence of the electrospun membrane in the middle of the chip permits the control of interactions between the two cell lines and the easy analysis of the effects of the crosstalk on cell behavior. This aspect was also evaluated by implementing the co-culture conditions on 24-well inserts before proceeding with the experiments in the microfluidic system. In particular, preliminary assays were performed on commercial inserts modified by replacing the polyethylene terephthalate (PET) membrane with the PCL/gel nanofibrous mat and used to investigate the activity of human fibroblasts and human ductal cells (wild type or KRAS-mutated) seeded in mono- or co-culture.
Experimental section
Cell culture
Human pancreatic ductal epithelial cells (HPDE) stably expressing activated KRAS (HPDE-KRAS) and wild-type HPDE (HPDE-wt) were kindly provided by Prof. F. Bussolino and cultured in RPMI-1640 medium (Gibco, Thermo Fisher Scientific, Waltham, USA) supplemented with 1% penicillin–streptomycin (Gibco), 1% L-glutamine (Gibco) and 10% fetal bovine serum (FBS) (Gibco). Human foreskin fibroblasts (HFF1) cells were obtained from ATCC® and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 1% penicillin–streptomycin (Gibco), 2% L-glutamine (Gibco) and 15% FBS (Gibco). PSCs were purchased from iXCells Biotechnologies (San Diego, USA) and cultured in a stellate cell growth medium kit (iXCells Biotechnologies, San Diego, USA) composed by stellate cell basal medium supplemented with 0.2% stellate cell growth supplement, 1% penicillin–streptomycin–amphotericin B solution 100× and 10% FBS. Cell lines were maintained in a humidified CO2 incubator at 37 °C and 5% CO2.Preparation of modified transwell inserts using the PCL/gel electrospun membrane
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 w/w) in a mixture of acetic acid and formic acid in the ratio of 1:1 v/v. GPTMS (3-glycidoxypropyltrimethoxysilane) was added as a gelatin crosslinker to the final solution in a concentration of 3.68% v/v.38,43 The electrospinning process was performed by employing the NovaSpider instrument (CIC nanoGUNE, San Sebastian, Spain) and the parameters were set, in accordance with earlier tests, at 12 kV voltage, 0.5 ml h−1 flow rate and 12 cm distance.
:20 w/w) in a mixture of acetic acid and formic acid in the ratio of 1:1 v/v. GPTMS (3-glycidoxypropyltrimethoxysilane) was added as a gelatin crosslinker to the final solution in a concentration of 3.68% v/v.38,43 The electrospinning process was performed by employing the NovaSpider instrument (CIC nanoGUNE, San Sebastian, Spain) and the parameters were set, in accordance with earlier tests, at 12 kV voltage, 0.5 ml h−1 flow rate and 12 cm distance.
PCL/gel nanofibers were integrated into the commercial 24-well inserts. Specifically, each insert was modified by removing the polyethylene terephthalate (PET) membrane and leaving only the polystyrene support. The electrospun membranes were then cut in small square pieces and applied to the plastic inserts by employing poly-dimethylsiloxane (PDMS; Sylgard 184, Dow Chemical, Midland, USA) to favor the adhesion. In order to avoid the excessive impregnation of membranes with PDMS, a thin layer of PDMS was brushed on the insert's outer walls and a first pre-crosslinking was performed by maintaining the supports at 90 °C for about 5 min. After the positioning of the PCL/gel fibers on the polystyrene supports, an additional layer of PDMS was applied on the edges of the membranes and they were then kept at 70 °C for about 2 min to complete the curing of PDMS.
Cell seeding in transwell inserts
The transwell inserts modified with PCL/gel electrospun membranes were sterilized via UV light for 1 h before cell seeding. Cells were cultured both on the PCL/gel electrospun membrane-modified supports (TW_PCL/gel) and on the commercial 24-well PET inserts (TW_PET) with 8.0 μm pore size, 0.1 × 106 per cm2 pore density and 0.3 cm2 growth area. HFF1, HPDE-wt and HPDE-KRAS were seeded in mono- and co-culture. The co-culture medium was prepared in accordance with preliminary tests performed to individuate the appropriate concentration of supplements (Fig. S1†). In particular, they were maintained in DMEM/F-12 supplemented with 15% FBS (Gibco), 1% penicillin–streptomycin (Gibco) and 2% L-glutamine (Gibco). The 1:3 ratio of HPDE-wt and HPDE-KRAS to HFF1 cells was adopted for the co-culture conditions. The human fibroblasts and the ductal cells were seeded at densities of 3 × 104 cells per transwell and 1 × 104 cells per transwell, respectively. In particular, 35 μL of medium containing HFF1 and 20 μL of medium containing HPDE-KRAS or HPDE-wt were respectively pipetted on the upper and lower sides of the membrane. After seeding, the cells were allowed to adhere for approximately 1 h at 37 °C and 5% CO2 before adding the medium both in the inner and the outer compartments of the modified inserts. The medium was refreshed every 2 days during the culture.
Interleukin 6 (IL-6) cytokine release
Cytokine concentration was determined in cell supernatants that were collected from the upper side of the TW_PET and TW_PCL/gel membranes after 24 h, 48 h, 72 h and 7d of culture using the IL-6 human ELISA kit (Thermo Fisher Scientific, BMS213-2). The concentrations were calculated using the standard curve generated by plotting the absorbance values of each standard sample on the ordinate and the human IL-6 standard concentrations on the abscissa. This assay has allowed the quantification of the IL-6 cytokines released by cells seeded both on TW_PET and TW_PCL/gel in mono- and co-culture.Morphological analysis of HFF1 seeded on TW_PET inserts
Fluorescence imaging was carried out to analyze the changes in the cell morphology of human fibroblasts seeded in co-culture with HPDE-KRAS on the commercial PET transwell inserts (TW_PET). The inserts were washed once with 1× phosphate-buffered saline (PBS; Gibco) and fixed with 4% paraformaldehyde (PFA) (Sigma Aldrich) for 30 min at room temperature, after 24 h, 48 h and 72 h of culture. They were then washed twice with 1× PBS, permeabilized in 0.5% Triton X-100 (Sigma Aldrich) in 1× PBS for 10 min and incubated with 1% bovine serum albumin (BSA; Thermo Fisher Scientific) for 30 min to improve the staining. The cytoskeletons of HPDE-KRAS and HFF1 were stained with Alexa Fluor™ 488 Phalloidin (Thermo Fisher Scientific, A12379) at a 1:60 concentration in 1× PBS with 1% BSA solution, while the nuclei were visualized with DAPI reagent (4′,6-diamidino-2-phenylindole dihydrochloride; Thermo Fisher Scientific, D1306) at 1:1000 concentration in 1× PBS solution. All samples were then imaged by confocal microscopy (Eclipse Ti2, Nikon). The aspect ratios of HFF1 cells both in mono- and co-culture were also measured using Fiji (version 1.53c).
Embedding stromal cells into the collagen gel
Collagen gel solution (10 mg mL−1) was prepared by mixing sterile collagen type I from bovine hides (FibriCol®, Advanced Biomatrix, Carlsbad, USA) with 10× phosphate-buffered saline (PBS; Gibco) and sterile distilled water. The pH was adjusted to 7.4 using 0.1 M NaOH. Cells were embedded into the collagen gel at densities of 1 × 106 cells per ml (D1), 3 × 106 cells per ml (D2) and 5 × 106 cells per ml (D3). To compare the viability of human fibroblasts seeded at D1, D2 and D3 inside the collagen gel, the CellTiter-Glo® 3D cell viability assay (Promega, Milano, Italy) was performed according to the manufacturer's protocols. In addition, the LIVE/DEAD™ assay (Thermo Fisher Scientific) was carried out on the cells loaded within the collagen gel at the optimal density to further confirm the quantitative test (metabolic activity assay) results. Images were acquired with a fluorescent cell imager (ZOE™; Bio-Rad, Hercules, USA) and analyzed using Fiji (version 1.53c). The HFF1 cells were harvested and embedded in the type I collagen solution at a final density of 3 × 106 cells per ml. The same procedure was adopted to embed the PSCs in the collagen gel.Design and fabrication of the multilayer microfluidic device
The microfluidic device, composed of a bottom layer and a top layer connected by an electrospun membrane, was designed by Rhinoceros® (Robert McNeel & Associates) CAD software and then fabricated by replica molding. Briefly, poly-dimethylsiloxane (PDMS; Sylgard 184, Dow Chemical, Midland, USA) base and crosslinking agent were mixed thoroughly at a ratio of 10:1 w/w and poured onto the master mold and cured for 3 h at 60 °C. An additional layer was successively introduced to allocate the medium reservoirs, whose mold was fabricated through a laser cutter (Microla slider) on a polymethylmethacrylate (PMMA) substrate.
The chip was assembled through a plasma oxygen treatment of the PDMS surfaces to decrease the hydrophobicity and therefore facilitate the bond between the different layers. The electrospun PCL/gel membrane was inserted between the top and bottom layers to create an ECM-like substrate for epithelial cell growth.
Fluidic characterization of the bottom layer
Liquid insertion and diffusivity tests were performed to evaluate the influence of the distance between the pillars on the retention of the gel within the middle microchannel and the diffusivity of the medium from the lateral to the central channels, respectively. In particular, gaps of 50 μm and 75 μm between the pillars were analyzed.Cell seeding in the microfluidic device
The platform was treated with UV light for 1 h as a sterilization process before cell culture.:3 ratio of cancer cells to stromal cells was used since it has been reported as a commonly adopted value due to the relevancy that ratios of 1:1 to 1:3 have in vivo.44–46 Particularly, densities of 3.5 × 103 cells per channel and 10 × 103 cells per channel were used for cancer and stromal cells respectively. Cells were co-cultured and maintained in DMEM/F-12 supplemented with 15% FBS (Gibco), 1% penicillin–streptomycin (Gibco) and 2% L-glutamine (Gibco) since previous tests demonstrated the efficacy of this culture medium composition in promoting the cell viability (Fig. S1†). Stromal cells were embedded in the type I collagen gel as previously described and loaded in the central chamber of the bottom layer by using the inlet on the top layer corresponding to the central channel in the bottom layer. HPDE-KRAS cells were seeded by injecting 5 μL of medium with cells inside the inlet corresponding to the chamber in the top layer. Reservoirs were filled with culture medium and the medium was changed every day during the culture period.
The viability of cells inside the platform was analyzed by LIVE/DEAD assay at different time points. In particular, cells seeded within the microfluidic platforms and on the common polystyrene multiwell cell culture plate (controls) were incubated with LIVE/DEAD™ reagents (Thermo Fisher Scientific) according to the manufacturer's protocol. The PSCs embedded in the type I collagen gel and seeded in a plastic multiwell plate were also stained and used as control. Subsequently, the cultures were imaged by employing the ZOE™ fluorescent cell imager. Fluorescence staining and imaging were also performed to evaluate the morphology and the spatial distribution of HPDE-KRAS and PSCs seeded in the microfluidic platforms, both in mono- and co-culture, at different time steps. Each compartment of the device was washed once with 1× PBS (Gibco) and fixed with 4% PFA (Sigma Aldrich) for 30 min at room temperature. After washing twice with 1× PBS, the chambers were filled with 0.5% solution of Triton X-100 (Sigma Aldrich) in 1× PBS and maintained for 10 min at room temperature to allow cell membrane permeabilization. Then, samples were incubated with 1% BSA (Thermo Fisher Scientific) for 30 min to improve the staining. The cytoskeletons of cells in monoculture were visualized using the green-fluorescent Alexa FluorTM 488 Phalloidin (Thermo Fisher Scientific, A12379) while the actin filaments of cells seeded in co-culture were stained with Rhodamine Phalloidin (Thermo Fisher Scientific, R415) at 1:60 and 1:400 concentrations in solutions of 1× PBS with 1% BSA, respectively. Nuclei were observed with DAPI reagent (Thermo Fisher Scientific, D1306) as previously described. Images were acquired by confocal microscopy (Eclipse Ti2, Nikon) and analyzed using Fiji (version 1.53c).
Administration of the tumor supernatant to human fibroblasts seeded in the bottom layer
HFF1 cells embedded in type I collagen gel and seeded in the bottom layer of the microfluidic device were treated with supernatant collected from TW_PCL/gel seeded with HPDE-KRAS in monoculture after 72 h. The tumor supernatant was administered to HFF1 after 48 h of culture in the bottom layer. The same procedure was applied to HFF1 embedded in the type I collagen gel and seeded in a plastic multiwell plate, as a control. The expression of alpha-smooth muscle actin (α-SMA) was determined by immunofluorescence staining. Briefly, cultures in microchannels and in the controls were fixed after 48 h of incubation with HPDE-KRAS supernatant using 4% PFA for 30 min, and further treated with 0.5% Triton X-100 for 10 min. After blocking non-specific binding with 1% BSA, the primary antibody against α-SMA (1:100, Sigma Aldrich, A7607) was applied for overnight incubation at 4 °C. After incubation with goat anti-mouse IgG1 secondary antibody (1:1000, Thermo Fisher Scientific, SA5-10264), Alexa FluorTM 488 Phalloidin (Thermo Fisher Scientific, A12379) and DAPI (1:1000, Thermo Fisher Scientific, D1306), samples were analyzed by confocal microscopy (Eclipse Ti2, Nikon). The images were then analyzed and the fluorescence intensity (mean gray values) distribution was plotted using the Fiji software. In addition, the concentration of IL-6 cytokines released by HFF1 seeded in the bottom layer of the microfluidic chip was quantified using the IL-6 human ELISA kit (Thermo Fisher Scientific, BMS213-2) after 48 h of incubation with the HPDE-KRAS supernatant.
Testing the drug efficacy to induce HPDE-KRAS cell death using PDAC-on-chip
HPDE-KRAS were cultured in the top layer as previously described. To test the effect of anti-cancer agents on cell viability, 100 nM bortezomib (SelleckChem, Houston, USA) and 1 μM gemcitabine (SelleckChem, Houston, USA) in the HPDE growth medium were injected in the inlet corresponding to the microchamber of the top layer. HPDE-KRAS grown for 48 h were treated with the drug for 24 h and 96 h and they were successively stained using LIVE/DEAD™ reagents (Thermo Fisher Scientific) in accordance with the manufacturer's instructions.Statistical analysis
All bar graph data are presented as the mean ± standard deviation (SD) for three independent experiments per condition or time point. Significance was measured as indicated for each experiment, with two-way or one-way ANOVA followed by pairwise comparison with Tukey's multiple comparisons test using GraphPad Prism 9.3.1; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.Results
Increased IL-6 cytokines release by human fibroblasts under co-culture conditions
In order to evaluate the effect of the interaction between the human fibroblasts and the ductal cells, the concentration of IL-6 cytokines was measured through the ELISA test (Fig. 1) by analyzing the supernatants of HFF1, HPDE-KRAS and HPDE-wt seeded in mono- and co-culture on TW_PCL/gel and TW_PET inserts. The largest amount of IL-6 released by cells under monoculture (Fig. 1a) was quantified at 24 h after seeding on the TW_PET membranes by HFF1 cells. After 24 h, 48 h and 72 h of seeding, the IL-6 concentration analyzed in the monoculture HFF1 medium on the TW_PCL/gel membranes was significantly lower as compared to the TW_PET. This behavior could be ascribed to the more biomimetic architecture (Fig. S2†) and composition of electrospun nanofibers compared to PET, which reduced the fibroblast activation after cell culturing. The amounts of IL-6 determined in the media of HPDE-KRAS and HPDE-wt in monoculture were minimal (Fig. 1a) for all the tested conditions. In general, the most consistent release of cytokines was quantified in the supernatant of HFF1 in co-culture with the HPDE-KRAS cells (Fig. 1b). The same trend was observed on the TW_PCL/gel membranes, although the concentrations were lower than those measured on the TW_PET membranes.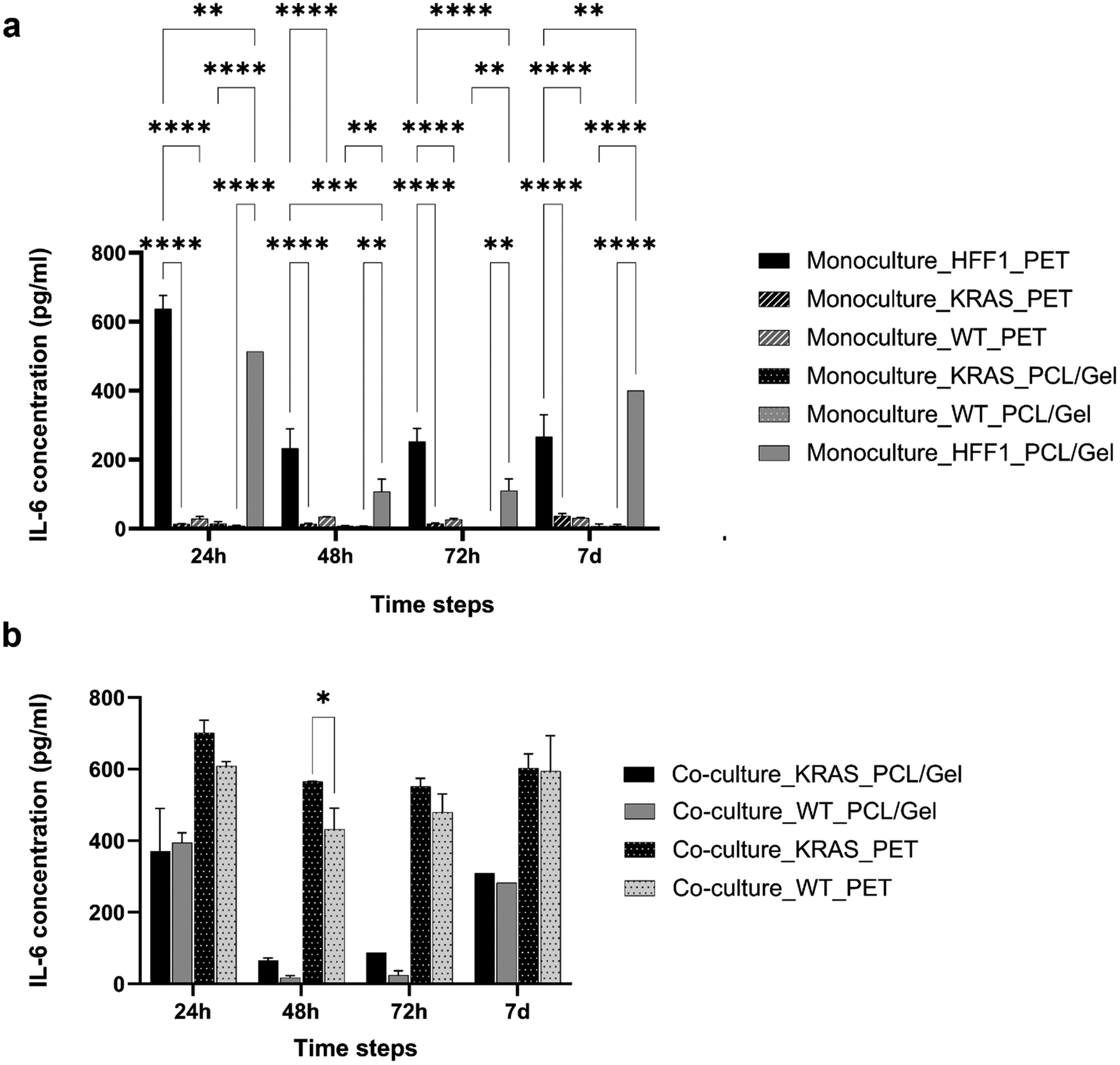 | ||
| Fig. 1 IL-6 cytokine concentration in supernatants collected from mono- and co-cultures in the transwell inserts. Bar plots of the data obtained from ELISA test IL-6 analysis for each culture condition on TW_PET (a) and TW_PCL/gel (b) grouped per time step (n = 3). Each condition has been assayed in duplicate following the manufacturer's instructions. Tukey's multiple comparisons test: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Morphological changes in human fibroblasts co-cultured with HPDE cells on the transwell inserts
Confocal imaging at different magnifications was performed to analyze the effects of tumor-stroma crosstalk on fibroblast morphology. As expected, HFF1 seeded on the TW_PET inserts showed changes in cytoskeletal organization when cultured with the HPDE-KRAS cells. The human fibroblasts assumed a more spindle-shaped conformation from 24 h to 72 h after seeding and in comparison with the monocultures, as confirmed by the aspect ratio measurements (Fig. S3†). Representative images of cell morphology changes are reported in Fig. 2.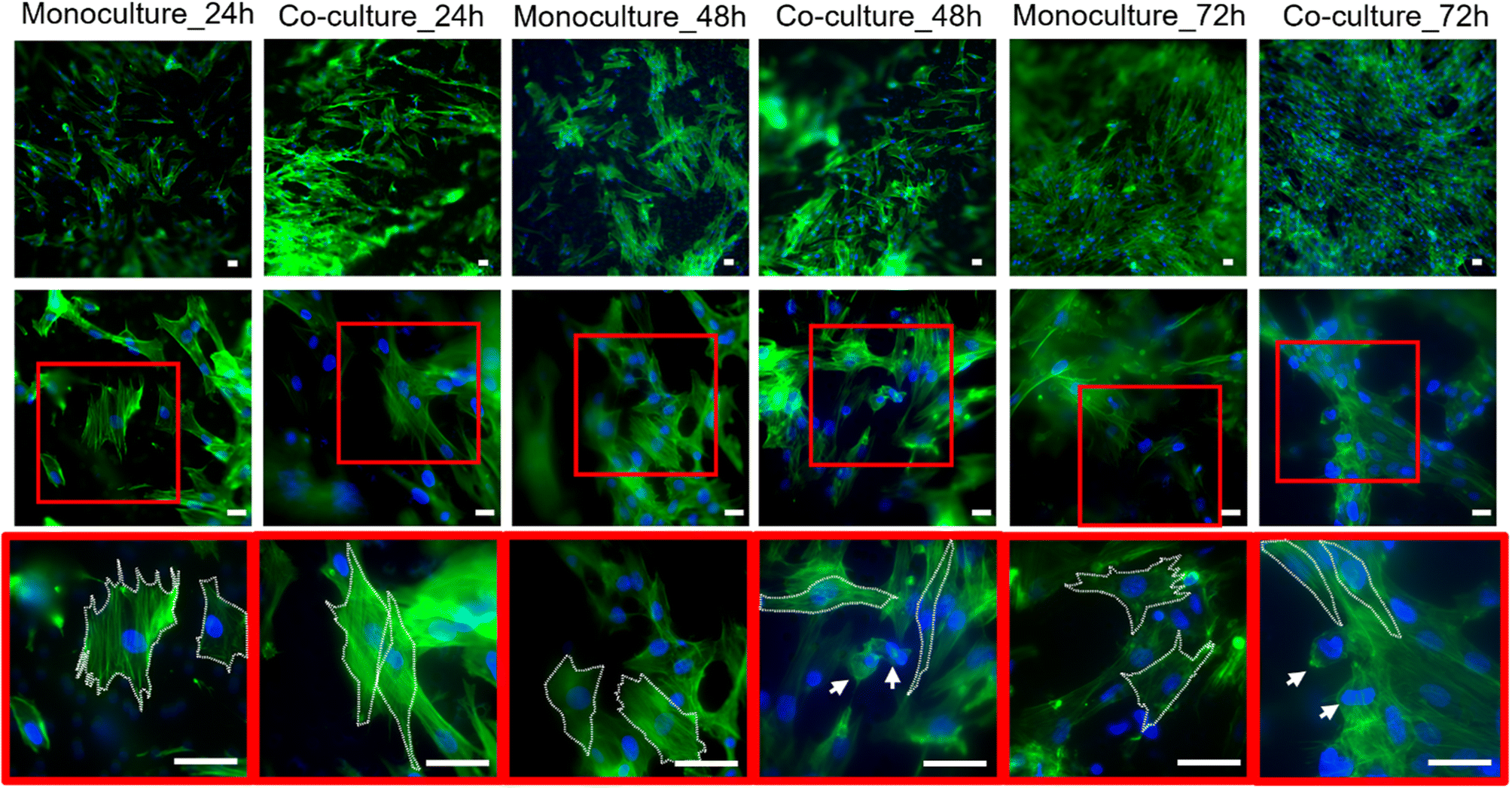 | ||
| Fig. 2 Effects of tumor-stroma crosstalk on human fibroblast morphology. Representative confocal images at different magnifications (20×, 60× and 100×) obtained upon the HFF1 and HPDE-KRAS staining of nuclei (DAPI) and F-actin (Alexa Fluor™ 488 Phalloidin) in monocoltures (HFF1) and co-cultures (HFF1 + HPDE-KRAS) 24 h, 48 h and 72 h after seeding (n = 3). The white arrows in the images at 100× indicate the HPDE-KRAS cells in co-culture with the human fibroblasts. Scale bars 50 μm. | ||
Viability of stromal cells embedded in type I collagen gel
CellTiterGlo 3D assay and LIVE/DEAD assay were performed to evaluate the viability of fibroblasts loaded within the type I collagen gel and cultured for one week (Fig. 3). Fig. 3a shows that cells seeded at a density of 3 × 106 cells per ml (D2) were more viable and active as compared to those seeded at a lower density of 1 × 106 cells per ml (D1). In addition, the D2 density was better as compared to a higher density D3 (5 × 106 cells per ml) as the cell viability increased with time, resulting in optimal fibroblast survival and proliferation within the gel. LIVE/DEAD images were acquired at 3 × 106 cells per ml (Fig. 3b–d), showing an increment in cell number during the culture period, confirming the optimal proliferative capability of fibroblasts embedded in the collagen gel at a density of 3 × 106 cells per ml (Fig. 3b–d). In particular, in accordance with the viability assay (Fig. 3a), cells were able to grow throughout the culture period, creating fibrillar and compact structures within the tridimensional (3D) framework (Fig. 3d).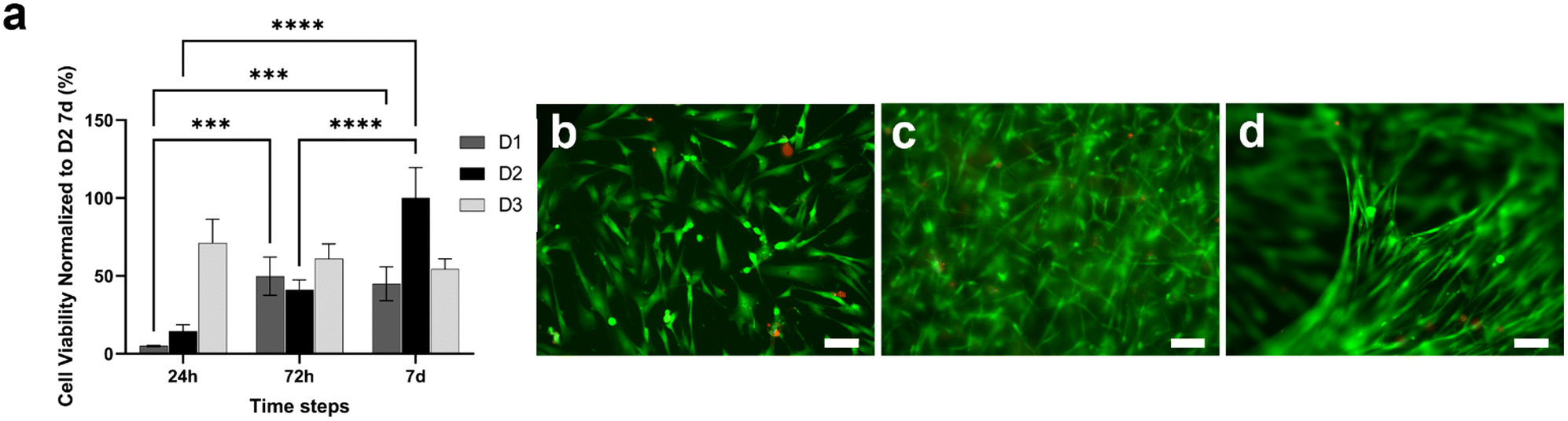 | ||
| Fig. 3 Human fibroblast viability in type I collagen gel. (a) Comparison of the viability of HFF1 seeded at D1 (1 × 106 cells per ml), D2 (3 × 106 cells per ml) and D3 (5 × 106 cells per ml) densities (n = 3). Tukey's multiple comparisons test: ***p < 0.001, ****p < 0.0001. (b–d) LIVE/DEAD images of HFF1 (3 × 106 cells per ml) embedded in the gel at 24 h (b), 72 h (c) and 7 d (d) after seeding. Cells were stained using calcein-AM for live cells and ethidium homodimer-1 for dead cells. Images are representative of at least six individual regions of the gels (n = 3). Scale bars 100 μm. | ||
Development of a functional multilayer device
The fabrication of the PDMS microfluidic system was carried out as previously illustrated. The platform consists of a top and a bottom layer divided by an electrospun PCL/gel membrane (Fig. 4). | ||
| Fig. 4 PDAC-on-chip: multilayer microfluidic device. (a) Top layer with the inlets, the outlet and the microchannel. (b) Bottom layer with the central microchannel delimited by two rows of micropillars (i–iii) and two lateral microchannels. (c) Assembled chip configuration showing the electrospun PCL/gel membrane inside the device. In the cross view, the nanofibrous matrix can be observed between the top layer (green) and the bottom layer (yellow). | ||
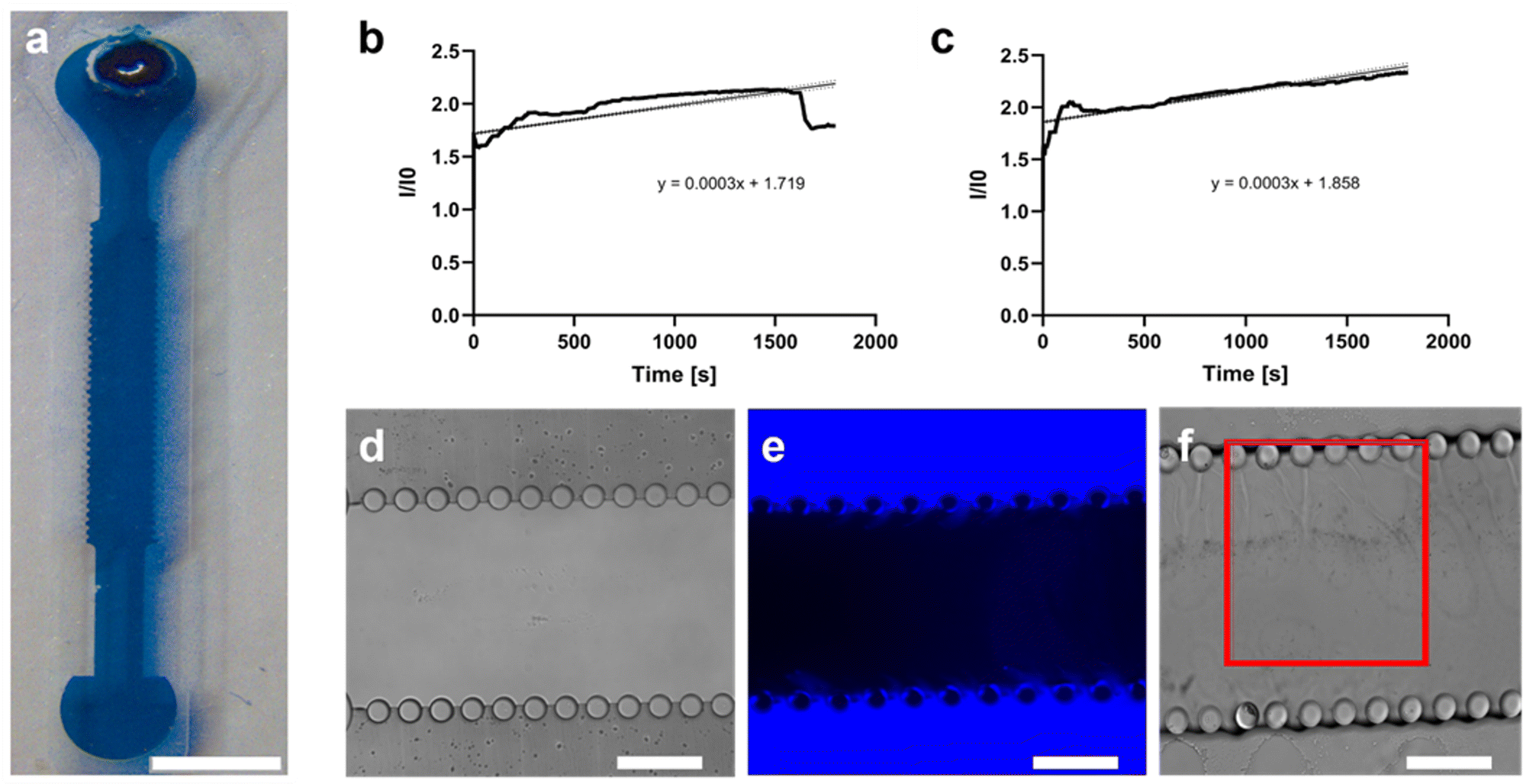 | ||
| Fig. 5 Fluidic characterization of the bottom layer. (a) Insertion test. Monitoring of the blue colored solution confinement inside the central microchannel. Scale bar 2 mm. (b–f) Diffusivity test. The fluorescence intensity of a stained solution was measured at different time intervals, for two pillar gaps, 75 μm (b) and 50 μm (c). The central chamber was filled with type I collagen gel (d) and the fluorescent solution was injected in the lateral microchannels (e) at t0 = 0 s. The average intensity was calculated considering the values registered in different ROIs (f). Scale bars 200 μm. | ||
Cell culture in the microfluidic device
The responses of PSCs and HPDE-KRAS cells, seeded in the bottom and top layers, respectively, were evaluated in terms of cell viability, proliferative capability and cell morphology. The PSCs embedded in the type I collagen gel and seeded in the central microchannel of the bottom layer (Fig. 6a) were able to colonize the chamber by modelling the hydrogel and forming dense tissue throughout the duration of the experiment. The LIVE/DEAD images (Fig. 6b) revealed that the stromal cells remained viable and active despite a few dead cells in the initial phase of seeding. This is probably related to the embedding procedure, which could be aggressive to the cells, causing a high number of dead cells, which was remarkable, from 24 h to 48 h and greatly decreased from 48 h to 72 h, after seeding. The morphological analyses demonstrated a uniform spatial distribution of PSCs that exhibited a high degree of cell spreading, developing interconnected multicellular networks (Fig. 6c). Furthermore, the fluorescence images showed the migratory ability of stromal cells, which crossed over the central channel 4 days after seeding. The ductal cells stably expressing activated KRAS (HPDE-KRAS) were seeded in the top layer of the microfluidic device (Fig. 7a) to evaluate their biological response in monoculture conditions. In particular, the PCL/gel electrospun membrane and the overhead PDMS layer were bonded to a glass coverslip in order to better monitor the cell behavior. The LIVE/DEAD assay revealed good cell viability on PCL/gelatin nanofibers, thus confirming the cytocompatibility of the membrane, which represents an optimal culture substrate for cell adhesion and proliferation (Fig. 7b). In addition, the cytoskeletal organization of the ductal cells was evaluated by fluorescence microscopy, which allowed the observation of the formation of the characteristic cobblestone morphology proving that cells were functionally active47,48 (Fig. 7c). The co-culture conditions were implemented in the assembled chip, where the different layers were joined together through a plasma oxygen treatment of the PDMS surfaces (Fig. 8a). Fig. 8b shows fluorescence images at higher magnifications where the PSCs (white arrows) display an elongated spindle shape, suggesting their possible activation. However, the PSCs are hardly visible and the quality of images reported in Fig. 8b is considerably inferior to those of monocultures. The hydrogel tends to impregnate the nanofibrous membrane, causing autofluorescence and noise effects in confocal images, limiting the detection of cell distribution.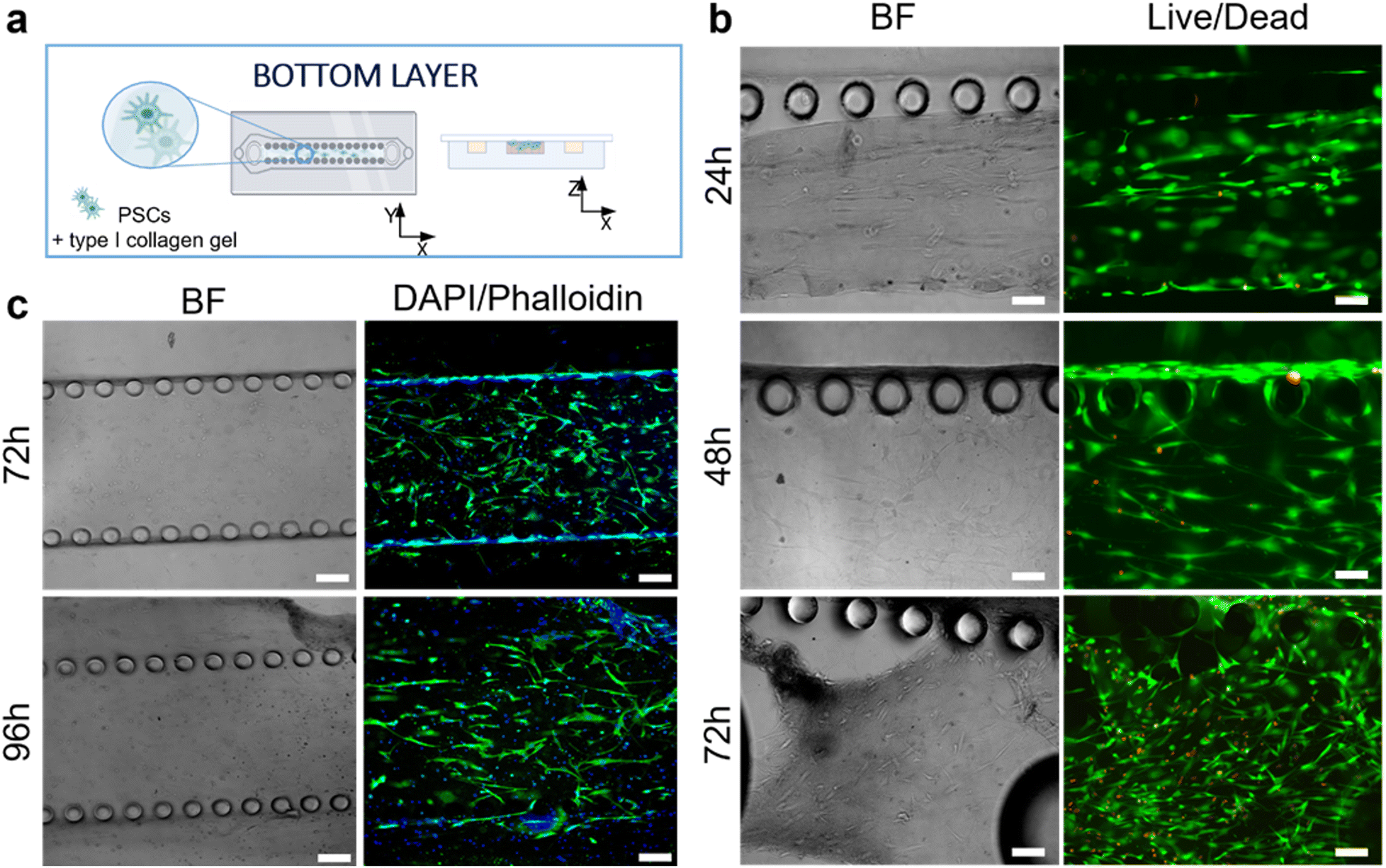 | ||
| Fig. 6 Seeding of human pancreatic stellate cells (PSCs) within the type I collagen gel in the bottom layer. (a) Schematic illustration of the bottom layer with the central microchannel containing the PSCs embedded in the collagen gel. (b) LIVE/DEAD images showing the viability of stromal cells at 24 h, 48 h and 72 h after seeding (n = 3). (c) Representative confocal images of PSCs in brightfield and upon staining with DAPI (nuclei) and Alexa Fluor™ 488 Phalloidin (cytoskeletons) at 72 h and 96 h after seeding. Scale bars 100 μm. | ||
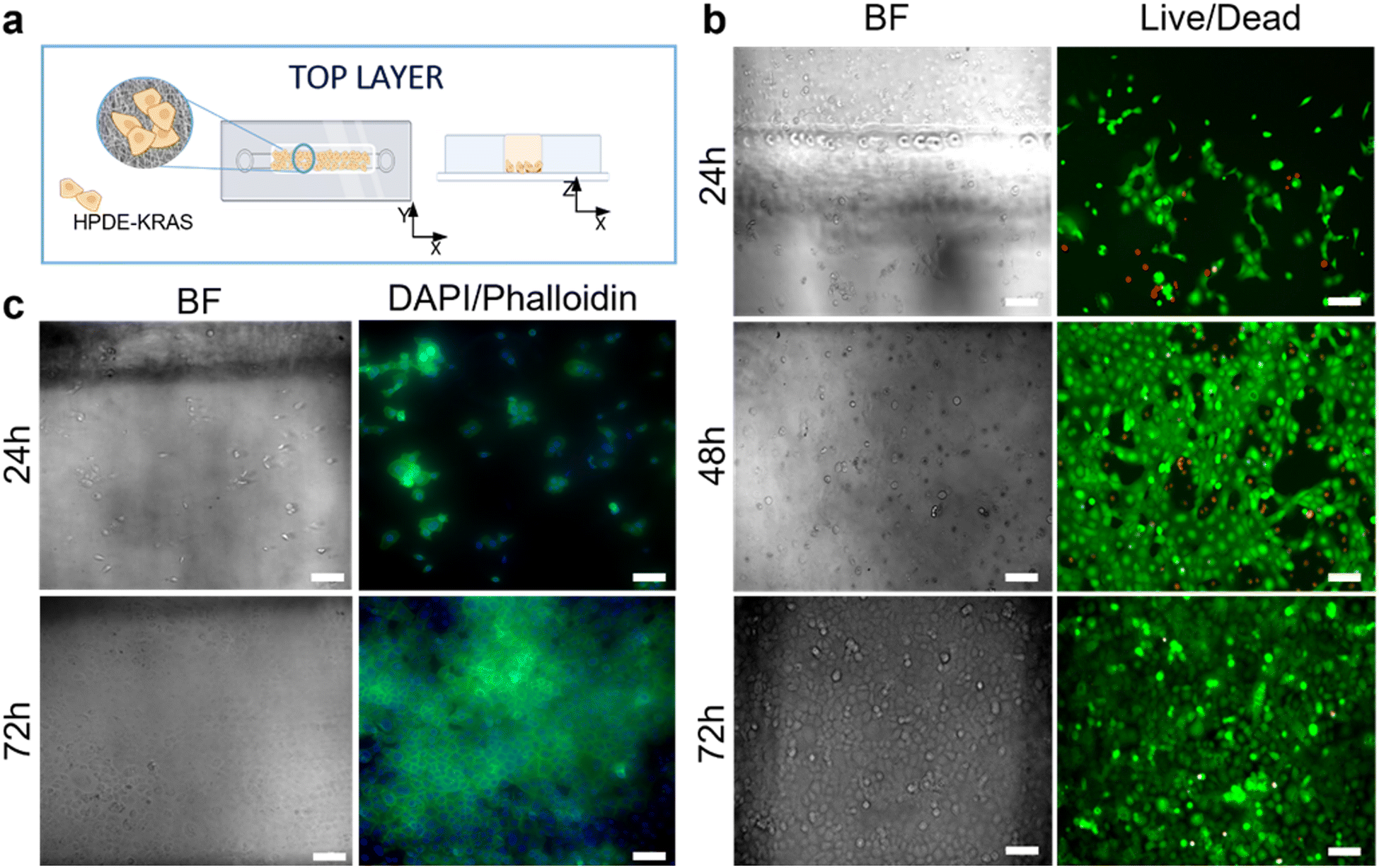 | ||
| Fig. 7 Seeding of PDAC cells in the top layer. (a) Schematic illustration of the top layer with the HPDE-KRAS cells seeded in the microchannel on the electrospun membrane. (b) LIVE/DEAD images showing the viability of HPDE-KRAS on the nanofibrous matrix in the chip at 24 h, 48 h and 72 h after seeding (n = 3). (c) Representative confocal images of HPDE-KRAS grown on the electrospun membrane inside the device upon staining with DAPI (nuclei) and Alexa Fluor™ 488 Phalloidin (cytoskeletons) at 24 h and 72 h after seeding. Scale bars 100 μm. | ||
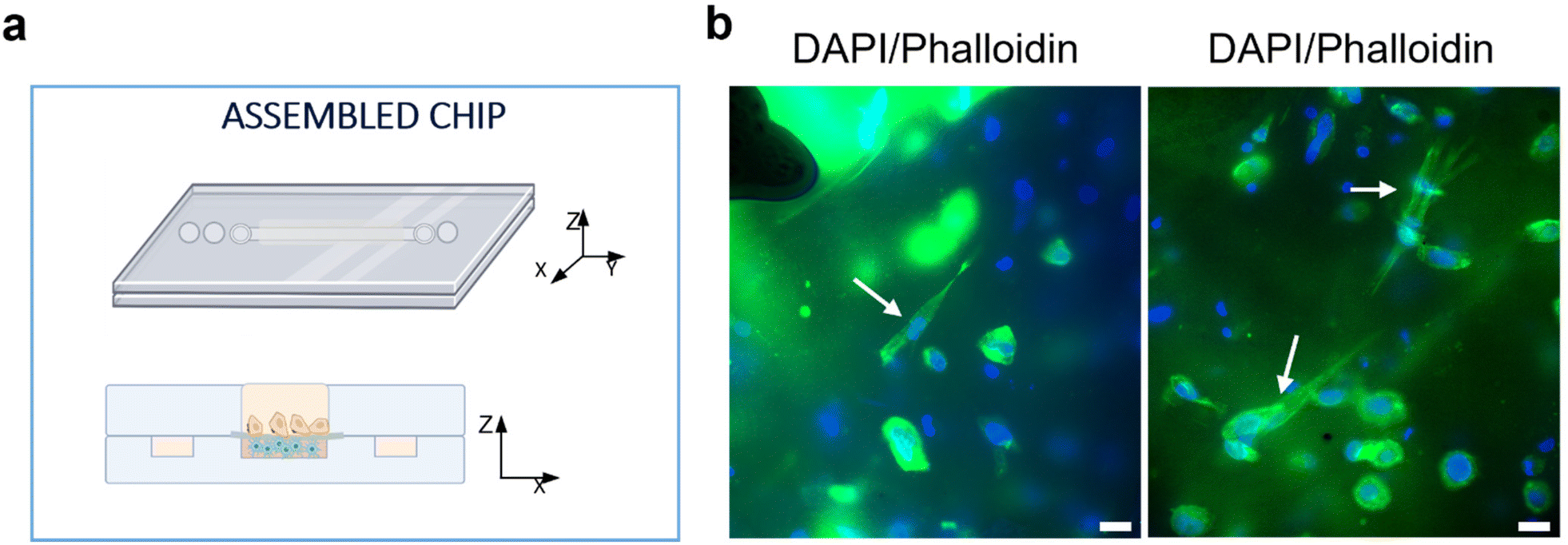 | ||
| Fig. 8 Seeding of PSCs and HPDE-KRAS in the assembled chip under co-culture conditions. (a) Schematic illustration of the assembled chip with the HPDE-KRAS cells seeded in the top and the PSCs in the bottom layers, respectively. (b) Representative confocal images at high magnifications showing the PSCs with elongated spindle shapes, indicated by the white arrows. Scale bars 20 μm. | ||
Activation of human fibroblasts cultured with HPDE-KRAS supernatant
The capability of exploiting the chip setup to monitor morphological changes of cells seeded in the bottom layer was demonstrated. HFF1 cells embedded in the collagen gel were biochemically stimulated using supernatant collected from TW_PCL/gel seeded with HPDE-KRAS in monoculture (Fig. 9). The cell morphology was evaluated and compared with data collected from the TW configurations previously analyzed. As shown in Fig. 9 and Fig. S3,† HFF1 assumed an elongated spindle shape, indicating their activation when cultured with the HPDE-KRAS supernatant for 48 h and upon staining with F-actin. The changes in cell morphology can be observed in Fig. 9a and b, which show the cytoskeletons of HFF1 in the microchamber of the bottom layers, cultured with (Fig. 9a) and without (Fig. 9b) the HPDE-KRAS supernatant. This evidence was further confirmed by the increment in α-SMA expression in fibroblasts embedded within the collagen gel and seeded both in the bottom layer and in the 24-wells used as controls (Fig. 9d and f). The amount of α-SMA was considerably higher in samples incubated with the tumor-derived supernatant (Fig. 9e and f) as compared to the “untreated” samples (Fig. 9c and d), confirming the activation of fibroblasts towards a myofibroblast phenotype. In addition, the graphs plotting the gray value measured on fluorescence images showed a more homogeneous distribution of the intensity values for the untreated conditions (Fig. S5ai–iii and bi–iii†) in comparison to the distributions of the treated samples (Fig. S5aiv–vi and biv–vi†). The peaks visible in Fig. S5avi and S5bvi† were correlated with a difference in the fluorescence intensity emitted by cells and background respectively, indicating a higher expression of α-SMA. Moreover, a higher level of IL-6 cytokines was measured in the medium of HFF1 seeded in PDAC-on-chip after the incubation with the pancreatic cancer cell supernatant. A 25% increment in the IL-6 cytokine amount was detected through the ELISA kit when HFF1 were treated with HPDE-KRAS supernatants as compared to HPDE (Fig. S5c†).
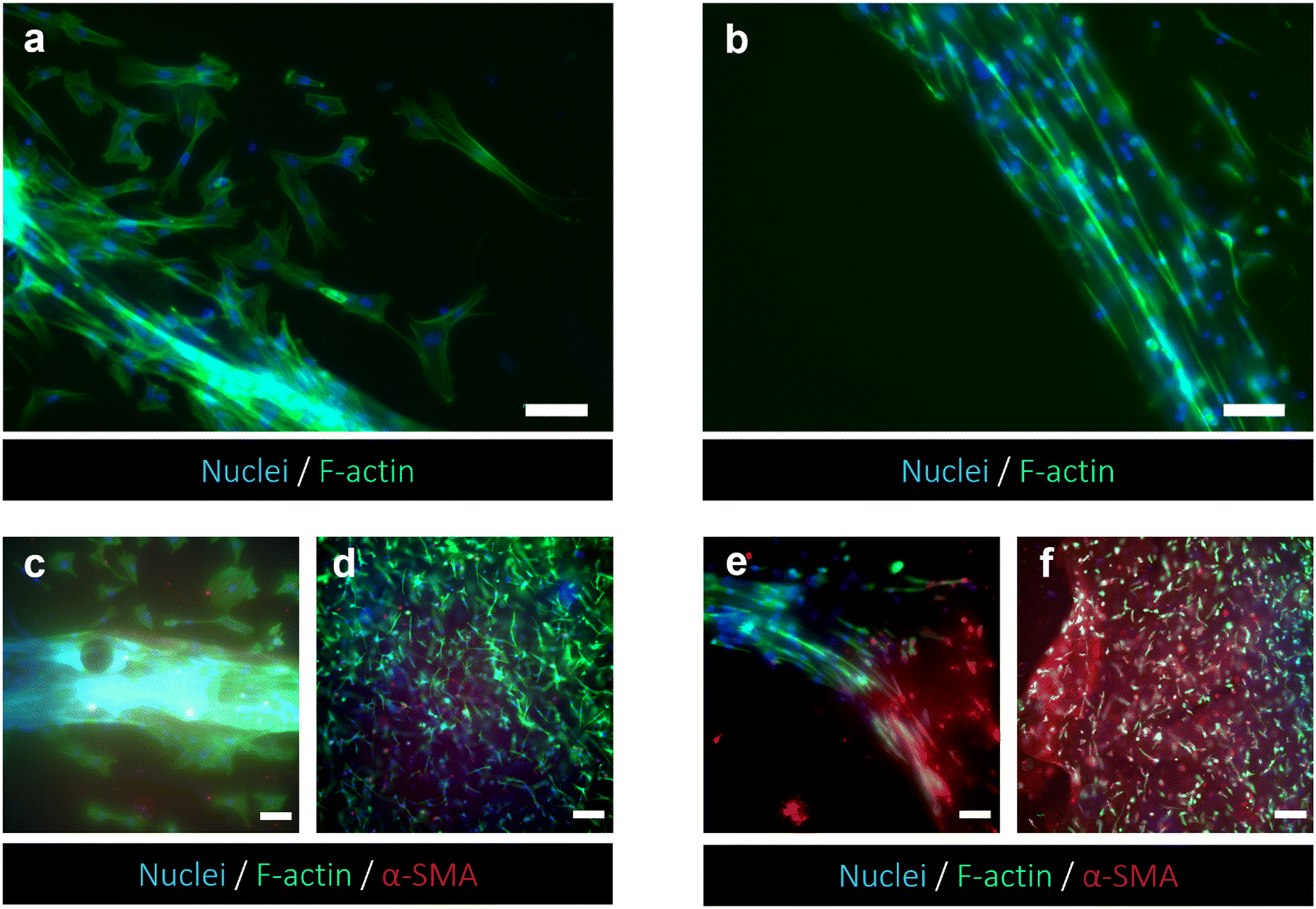 | ||
| Fig. 9 Activation of human fibroblasts upon culturing with the HPDE-KRAS supernatant. (a and b) Fluorescent images of HFF1 seeded in the bottom layer of PDAC-on-chip and stained with Alexa Fluor™ 488 Phalloidin (F-actin) and DAPI (nuclei) to highlight the differences between the fibroblasts before (a) and after (b) the incubation with tumor supernatant. Cells were grown for 48 h before staining. Images are representative of at least three images per individual experiment (n = 3). Scale bars 100 μm. (c–f) Representative confocal images showing the changes in morphology and α-SMA expression of HFF1 before (c and d) and after (e and f) the treatment with HPDE-KRAS supernatant. (c and e) HFF1 embedded in collagen hydrogel and seeded in the bottom layer of the microfluidic device. Scale bars 50 μm. (d and f) HFF1 grown within the type I collagen gel in the 24-wells and used as controls. Scale bars 100 μm. | ||
Drug sensitivity of HPDE-KRAS cultured in the microfluidic device
The HPDE-KRAS response to anti-cancer chemotherapeutic agents was analyzed and the validity of the PDAC-on-chip device in evaluating the efficacy of the drugs was demonstrated. The LIVE/DEAD images (Fig. 10) showed significant changes in the viability of cells exposed to bortezomib (Fig. 10a) and gemcitabine (Fig. 10b) after treatment periods of 24 h and 96 h. The decrease in cell number after the drug treatment was due to cell detachment following the cell apoptosis of pancreatic cancer cells induced by the chemotherapeutic treatment.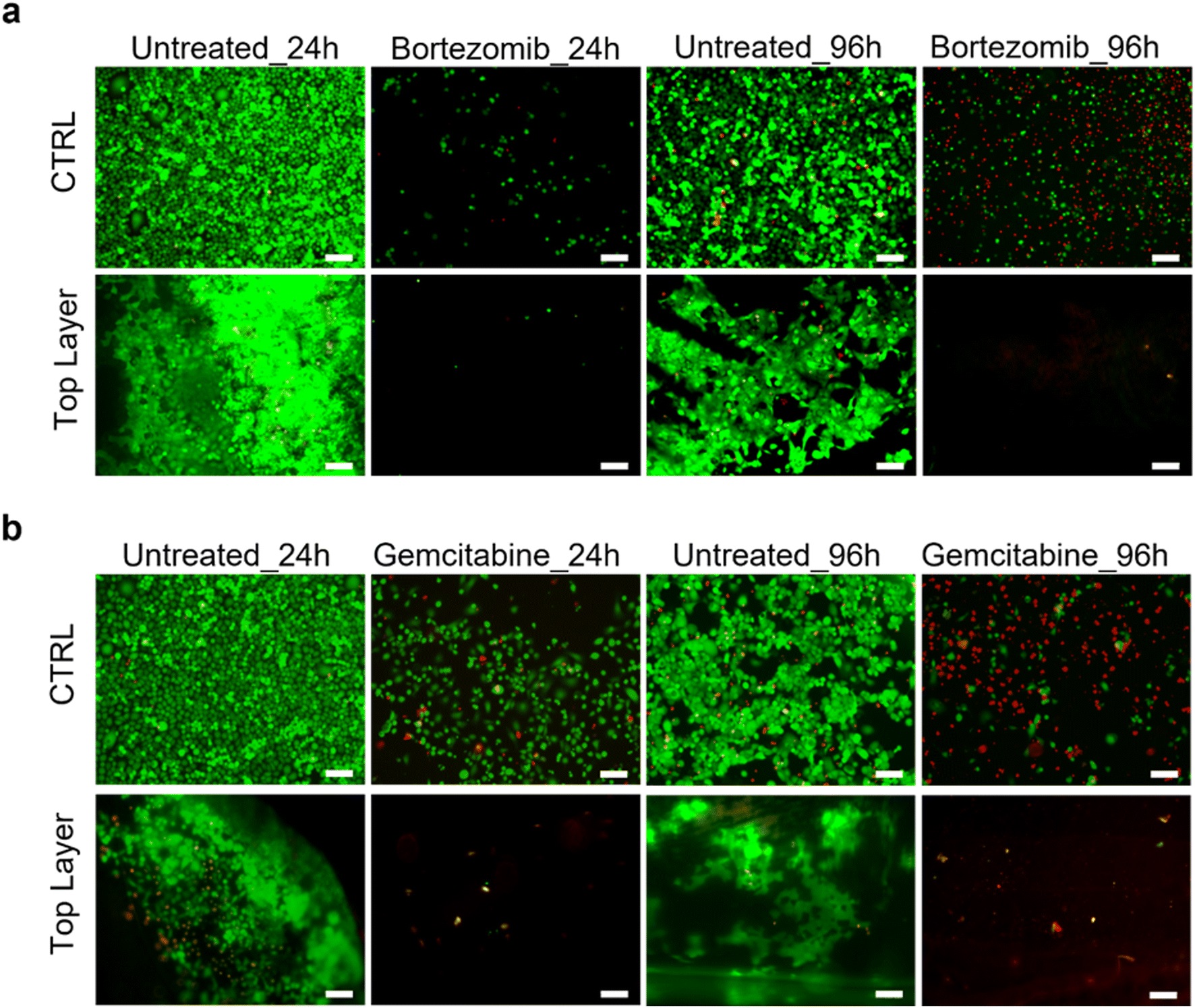 | ||
| Fig. 10 Sensitivity of HPDE-KRAS cells to anti-cancer drugs. (a and b) Cell viability analyses of HPDE-KRAS grown for 48 h and stained with calcein-AM (live cells) and ethidium homodimer-1 (dead cells) after 24 h and 96 h exposure to gemcitabine (a) and bortezonib (b). Images are representative of at least three images per individual experiment (n = 3). Scale bars 100 μm. | ||
Discussion
The interaction between pancreatic cancer and the surrounding microenvironment represents a key aspect that has to be investigated to enhance the knowledge of this malignant pathology and improve the efficacy of drug screening.49 As demonstrated,50–55 the stroma represents a histopathological hallmark of pancreatic ductal adenocarcinoma and plays a fundamental role in tumor progression.56,57 Although recent studies have shown the possibility of modeling the PDAC microenvironment in vitro,17,20,28,33,34,58–61 the tumor-stroma crosstalk remains extremely challenging to reproduce and monitor in functionally effective models.62–64We report herein the design, development and characterization of a multilayer PDAC-on-chip device to mimic the tumor-stroma relationship through the culture of human pancreatic ductal epithelial cells expressing the activated KRAS and human pancreatic stellate cells, on the same miniaturized platform. In particular, the HPDE-KRAS cells and the PSCs were respectively seeded in the top and in the bottom layers, separated by an electrospun PCL/gel membrane. The effects of the PDAC-stroma crosstalk on cellular behavior were analyzed in terms of fibroblast activation, which typically occurs during the inflammatory response characterizing the tumor development.65–68 The HFF1 cell line was used to perform these preliminary tests since the differences compared to the PSCs are minimal,69 with the advantage of major resistance and the stability of functions over extended in vitro passages. Despite the genetic differences between the pancreatic stellate cells and skin fibroblasts,70 the use of fibroblasts from non-pancreatic origins has been helpful for understanding the crosstalk between stromal and cancer cells, as many studies have demonstrated.34,71–74 The results proved the influence of the co-culture conditions on fibroblast behavior, corresponding to an increment in IL-6 cytokine release (Fig. 1) and a change in cytoskeletal morphology (Fig. 2). In particular, we quantified a higher IL-6 concentration in the supernatant of HFF1 in co-culture with the HPDE-KRAS cells compared to the co-culture with healthy ductal cells (HPDE-wt). These outcomes are in line with the data reported by other studies in the literature showing that the expression of the oncogenic KRAS correlates with the signaling pathways involved in the inflammatory cascade and in the activation of stromal cells in pancreatic cancer.6,75–77 Moreover, the IL-6 cytokines play an important role in pancreatic tumor progression and are produced in abundance by components of stroma, including PSCs and fibroblasts.6,11,78Fig. 1 shows an increment in IL-6 produced by HFF1 in co-culture with HPDE-KRAS, with a significant increase at 48 h. These data were also confirmed by the morphological analyses performed on the HFF1 seeded in mono- and co-culture with the HPDE-KRAS on the TW_PET inserts and stained after 24 h, 48 h and 72 h (Fig. 2). The human fibroblasts exhibited a more spindle-shaped conformation at 72 h in co-culture.
To replicate the composition of the adenocarcinoma microenvironment, the stromal cells were embedded in a type I collagen gel that represents the main constituent of the PDAC stroma.79 We tested the viability of HFF1 loaded within the gel and we demonstrated that the type I collagen hydrogel exhibits good biological properties in terms of cells viability and growth (Fig. 3), supporting other studies showing the enhanced adhesion and proliferation of human fibroblasts embedded within the 3D collagen gels.80,81 We also observed the capability of stromal cells in remodeling the gel resulting in the formation of tridimensional and compact cellularized hydrogel structures (Fig. 3d).
The collagen gel was then used to incorporate the PSCs seeded within the designed multilayer microfluidic device (Fig. 4). In particular, the bottom layer was fabricated to insert the hydrogel within the central microchamber, which is delimited by two rows of micropillars (100 μm diameter and 250 μm height) aiming at both gel confinement and nutrient passage (Fig. 4b). The use of a collagen matrix to encapsulate the PSCs seeded in dedicated microchannels was also illustrated in the experimental work of Lee and collaborators.33 However, in our pancreatic cancer chip model, we employed, for the first time, a biomimetic nanofibrous membrane to control the HPDE-KRAS cells interaction with PSCs (Fig. 4c).
In addition, we investigated the fluidic properties of the bottom layer and found that the optimal distance between the micropillars was 75 μm, although larger gaps are used in literature.20,44 We have shown that a liquid medium is able to completely diffuse from the lateral microchannel into the central chamber in 30 minutes, thus ensuring the transport of nutrients to the cells in a short time (Fig. 5).
The microfluidic device designed in this work guarantees good cell survival and it allows easy monitoring of the cell response within the chip, key features for a clinically relevant experimental in vitro model of healthy and pathological tissues. The response of PSCs and HPDE-KRAS cells seeded inside the chip was analysed in terms of cell viability, proliferative capability and cell morphology. In particular, the PSCs were able to colonize the chamber by growing throughout the experiment (Fig. 6). However, the tests revealed few dead cells in the initial phase of seeding that could be associated with the embedding procedure in the gel. Thus, the thermal shock given by the temperature of the collagen solution (4 °C) could be the cause of the cell apoptosis during the first hours after seeding.82 Nonetheless, the stellate cells seemed to recover from the stress and continue to proliferate, showing a high degree of cell spreading inside the collagen network (Fig. 6b and c). The confocal microscopy analysis allowed the observation of the flat morphology of the PSCs, indicating their viability83 and the typical stellate shape suggesting their quiescent state in monoculture.6,84 Focusing on the cell behavior of HPDE-KRAS seeded in the top layer (Fig. 7), we assessed that the PDAC cells remained viable and active as confirmed by the confocal images showing the typical acinar organization of the ductal cells in vivo85 (Fig. 7c). These results underline the importance of a biomimetic substrate like the electrospun PCL/gel nanofibrous matrix in promoting the epithelial cell viability and functionality, as widely discussed in literature.86–90 The co-culture conditions implemented in our model have allowed us to preliminarily evaluate the reciprocal influence that the HPDE-KRAS cells and the PSCs have on each other. Specifically, we observed from the confocal images the morphological changes in stellate cells that appear in a spindle shape, indicating their activation as highlighted in the study of Lee and coworkers.33
This aspect has been further analyzed by culturing the HFF1 embedded in the type I collagen gel with the HPDE-KRAS supernatant to correlate the fibroblast behavior in the PDAC-on-chip device with that observed on transwell inserts. We found that HFF1 in a 3D collagen matrix changed from a relatively short and flat shape to a myofibroblast-like shape with stress fiber formation and increased expression of α-SMA (Fig. 9). In addition, a higher level of IL-6 cytokines released by HFF1 seeded in the PDAC-on-chip was measured. This result contributes to the validation of the microfluidic system since the fibroblasts cultured inside the bottom layer experienced the same stimuli as compared to the HFF1 seeded on TW inserts.
The drug sensitivity of HPDE-KRAS cells cultured in the microfluidic platform was characterized through exposure to different chemotherapeutic agents. We used bortezomib and gemcitabine, which are proteosome and DNA synthesis inhibitors, respectively.91 We preliminarily assessed the validity of the PDAC-on-chip system, which can be used to test the efficacy of drugs on pancreatic cancer cells. Our chip allows the administration of single or multiple drugs both in contact with epithelial, as well as stellate cells, and continuous monitoring of the effects of the treatment on both cell populations can be easily performed thanks to the presence of the electrospun membrane that confined cells in a specific chip compartment.
Finally, in the current tumor-stroma interplay on-a-chip scenario,74 the device developed in this work accurately recapitulates the key aspects of the relationship between the tumor and its microenvironment through a scalable and high-throughput approach. Moreover, the use of a nanofibrous and biomimetic membrane to compartmentalize the microfluidic device and thus separate the cancer component from the stromal tissue allows the study of the effect of the inflammation stimuli on the stromal cells in a controlled and specific way.
Conclusions
A multilayer PDAC-on-chip was designed and developed to model in vitro the tumor-stroma crosstalk. Pancreatic stellate cells (PSCs) embedded in a 3D type I collagen matrix and ductal cells expressing the oncogene KRAS (HPDE-KRAS) seeded on a biomimetic nanofibrous membrane were co-cultured inside the microchannels of the device. The relationship between stromal and ductal cells was evaluated both in the nanofibrous membrane alone and in the chip. Increased levels of cytokines, changes in morphology and higher expression of α-SMA were observed when human fibroblasts were cultured with HPDE-KRAS cells or HPDE-KRAS supernatant. Our work thus underlines the biological relevance that the PDAC-stroma interactions have in determining the tumor microenvironment remodeling during the pancreatic cancer insurgence.In conclusion, this study provides an in vitro model that allows the better study of the tumor-stroma reciprocal influence and the performance of drug screening towards the identification of effective personalized and high-throughput therapeutical strategies for individual patients. Furthermore, the developed device will allow additional studies to evaluate the influence of stromal cells seeded in the microfluidic device on drug diffusion.
Author contributions
VS: data curation, formal analysis, writing – original draft, investigation. SLM: conceptualization, investigation, funding acquisition, writing – review & editing. MC: conceptualization, investigation, funding acquisition, writing – review & editing. GS: investigation. AB: investigation. GC: funding acquisition, writing – review & editing. CTT: conceptualization, formal analysis, writing – original draft, investigation, methodology, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. V. Cauda and Prof. F. Bussolino for their support in cell culturing. This work was partially performed thanks to the financial support of Deflect project (Piedmont Region).References
- A. Khomiak, M. Brunner and M. Kordes, et al., Recent Discoveries of Diagnostic, Prognostic and Predictive Biomarkers for Pancreatic Cancer, Cancers, 2020, 12, 3234 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2020, CA Cancer J. Clin., 2020, 70, 7–30 CrossRef PubMed.
- M. Orth, P. Metzger and S. Gerum, et al., Pancreatic ductal adenocarcinoma: Biological hallmarks, current status, and future perspectives of combined modality treatment approaches, Radiat. Oncol., 2019, 14, 1–20 CrossRef PubMed.
- L. R. Brunet, T. Hagemann and G. Andrew, et al., Have lessons from past failures brought us closer to the success of immunotherapy in metastatic pancreatic cancer?, OncoImmunology, 2016, 5(4), e1112942 CrossRef.
- M. A. Heinrich, A. M. R. H. Mostafa and J. P. Morton, et al., Translating complexity and heterogeneity of pancreatic tumor: 3D in vitro to in vivo models, Adv. Drug Delivery Rev., 2021, 174, 265–293 CrossRef CAS PubMed.
- M. V. Apte, J. S. Wilson and A. Lugea, et al., A starring role for stellate cells in the pancreatic cancer microenvironment, Gastroenterology, 2013, 144, 1210–1219 CrossRef PubMed.
- M. F. B. Nielsen, M. B. Mortensen and S. Detlefsen, Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells, World J. Gastroenterol., 2016, 22, 2678–2700 CrossRef CAS PubMed.
- J. Watt and H. M. Kocher, The desmoplastic stroma of pancreatic cancer is a barrier to immune cell infiltration, OncoImmunology, 2013, 2(12), e26788 CrossRef.
- M. Weniger, K. C. Honselmann and A. S. Liss, The extracellular matrix and pancreatic cancer: A complex relationship, Cancers, 2018, 10 DOI:10.3390/cancers10090316 , epub ahead of print.
- S. Liot, J. Balas and A. Aubert, et al., Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins, Front. Immunol., 2021, 12, 709 Search PubMed.
- Y. Fu, S. Liu and S. Zeng, et al., The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma, Mol. Cancer, 2018, 17(1), 62 CrossRef PubMed.
- M. Hidalgo, S. Cascinu and J. Kleeff, et al., Addressing the challenges of pancreatic cancer: Future directions for improving outcomes, Pancreatology, 2015, 15, 8–18 CrossRef.
- R. R. Bynigeri, A. Jakkampudi and R. Jangala, et al., Pancreatic stellate cell: Pandora's box for pancreatic disease biology, World J. Gastroenterol., 2017, 23, 382 CrossRef CAS PubMed.
- R. H. Hruban, N. V. Adsay and J. Albores-Saavedra, et al., Pathology of genetically engineered mouse models of pancreatic exocrine cancer: Consensus report and recommendations, Cancer Res., 2006, 66, 95–106 CrossRef CAS PubMed.
- S. T. Chari, Detecting Early Pancreatic Cancer: Problems and Prospects, Semin. Oncol., 2007, 34, 284–294 CrossRef CAS PubMed.
- S. J. Cui, T. Y. Tang and X. W. Zou, et al., Role of imaging biomarkers for prognostic prediction in patients with pancreatic ductal adenocarcinoma, Clin. Radiol., 2020, 75(6), 478.e1–478.e11 CrossRef PubMed.
- D. D. Cave, R. Rizzo and B. Sainz, et al., The revolutionary roads to study cell–cell interactions in 3d in vitro pancreatic cancer models, Cancers, 2021, 13, 1–19 Search PubMed.
- D. Osuna de la Peña, S. M. D. Trabulo and E. Collin, et al., Bioengineered 3D models of human pancreatic cancer recapitulate in vivo tumour biology, Nat. Commun., 2021, 12, 1–15 CrossRef.
- M. R. Haque, T. H. Rempert and T. A. Al-Hilal, et al., Organ-Chip Models: Opportunities for Precision Medicine in Pancreatic Cancer, Cancers, 2021, 13, 4487 CrossRef CAS.
- H. Mollica, Y. J. Teo and A. S. M. Tan, et al., A 3D pancreatic tumor model to study T cell infiltration, Biomater. Sci., 2021, 9, 7420–7431 RSC.
- B. Schuster, M. Junkin and S. S. Kashaf, et al., Automated microfluidic platform for dynamic and combinatorial drug screening of tumor organoids, Nat. Commun., 2020, 11, 1–12 CrossRef.
- D. H. T. Nguyen, E. Lee and S. Alimperti, et al., A biomimetic pancreatic cancer on-chip reveals endothelial ablation via ALK7 signaling, Sci. Adv., 2019, 5, 1–10 Search PubMed.
- H. Xu, X. Lyu and M. Yi, et al., Organoid technology and applications in cancer research, J. Hematol. Oncol., 2018, 11, 116 CrossRef PubMed.
- S. F. Boj, C. I. Hwang and L. A. Baker, et al., Organoid models of human and mouse ductal pancreatic cancer, Cell, 2015, 160, 324–338 CrossRef CAS PubMed.
- M. Beer, N. Kuppalu and M. Stefanini, et al., A novel microfluidic 3D platform for culturing pancreatic ductal adenocarcinoma cells: Comparison with in vitro cultures and in vivo xenografts, Sci. Rep., 2017, 7, 1–12 CrossRef CAS.
- B. Kramer, L. De Haan and M. Vermeer, et al., Interstitial flow recapitulates gemcitabine chemoresistance in a 3D microfluidic pancreatic ductal adenocarcinoma model by induction of multidrug resistance proteins, Int. J. Mol. Sci., 2019, 20(18), 4647 CrossRef CAS PubMed.
- M. Swayden, P. Soubeyran and J. Iovanna, Upcoming Revolutionary Paths in Preclinical Modeling of Pancreatic Adenocarcinoma, Front. Oncol., 2020, 9, 1443 CrossRef PubMed.
- E. Tomás-Bort, M. Kieler and S. Sharma, et al., 3D approaches to model the tumor microenvironment of pancreatic cancer, Theranostics, 2020, 10, 5074–5089 CrossRef.
- L. A. Low, C. Mummery and B. R. Berridge, et al., Organs-on-chips: into the next decade, Nat. Rev. Drug Discovery, 2021, 20, 345–361 CrossRef CAS.
- S. N. Bhatia and D. E. Ingber, Microfluidic organs-on-chips, Nat. Biotechnol., 2014, 32, 760–772 CrossRef CAS.
- D. Gioeli, C. J. Snow and M. B. Simmers, et al., Development of a multicellular pancreatic tumor microenvironment system using patient-derived tumor cells, Lab Chip, 2019, 19, 1193–1204 RSC.
- M. J. Bradney, S. M. Venis and Y. Yang, et al., A Biomimetic Tumor Model of Heterogeneous Invasion in Pancreatic Ductal Adenocarcinoma, Small, 2020, 1–10 Search PubMed , 1905500.
- J. H. Lee, S. K. Kim and I. A. Khawar, et al., Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance, J. Exp. Clin. Cancer Res., 2018, 37, 1–12 CrossRef.
- B. F. L. Lai, R. X. Z. Lu and Y. Hu, et al., Recapitulating Pancreatic Tumor Microenvironment through Synergistic Use of Patient Organoids and Organ–on–a–Chip Vasculature, Adv. Funct. Mater., 2020, 2000545 CrossRef CAS.
- G. Giuntoli, G. Muzio and C. Actis, et al., In vitro Characterization of a Hernia Mesh Featuring a Nanostructured Coating, Front. Bioeng. Biotechnol., 2021, 8, 589223 CrossRef.
- M. Licciardello, G. Ciardelli and C. Tonda-Turo, Biocompatible Electrospun Polycaprolactone-Polyaniline Scaffold Treated with Atmospheric Plasma to Improve Hydrophilicity, Bioengineering, 2021, 8, 1–18 CrossRef PubMed.
- M. Licciardello, C. Tonda-Turo and A. Gallina, et al., Fabrication of extracellular matrix-like membranes for loading piezoelectric nanoparticles, J. Phys. Mater., 2020, 3, 034004 CrossRef CAS.
- C. Tonda-Turo, E. Cipriani and S. Gnavi, et al., Crosslinked gelatin nanofibres: Preparation, characterisation and in vitro studies using glial-like cells, Mater. Sci. Eng., C, 2013, 33, 2723–2735 CrossRef CAS.
- S. Gautam, A. K. Dinda and N. C. Mishra, Fabrication and characterization of PCL/gelatin composite nanofibrous scaffold for tissue engineering applications by electrospinning method, Mater. Sci. Eng., C, 2013, 33, 1228–1235 CrossRef CAS PubMed.
- P. Denis, J. Dulnik and P. Sajkiewicz, Electrospinning and Structure of Bicomponent Polycaprolactone/Gelatin Nanofibers Obtained Using Alternative Solvent System, Int. J. Polym. Mater. Polym. Biomater., 2014, 64, 354–364 CrossRef.
- N. S. Binulal, A. Natarajan and D. Menon, et al., PCL–gelatin composite nanofibers electrospun using diluted acetic acid–ethyl acetate solvent system for stem cell-based bone tissue engineering, J. Biomater. Sci., Polym. Ed., 2014, 25, 325–340 CrossRef CAS.
- O. Gil-Castell, J. D. Badia and E. Strömberg, et al., Effect of the dissolution time into an acid hydrolytic solvent to tailor electrospun nanofibrous polycaprolactone scaffolds, Eur. Polym. J., 2017, 87, 174–187 CrossRef CAS.
- O. Mahony, O. Tsigkou and C. Ionescu, et al., Hybrid Materials: Silica-Gelatin Hybrids with Tailorable Degradation and Mechanical Properties for Tissue Regeneration, Adv. Funct. Mater., 2010, 20, 3808–3808 CrossRef.
- J. H. Lee, S. K. Kim and I. A. Khawar, et al., Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance, J. Exp. Clin. Cancer Res., 2018, 37, 1–12 CrossRef.
- S. Y. Jeong, J. H. Lee and Y. Shin, et al., Co-culture of tumor spheroids and fibroblasts in a collagen matrix-incorporated microfluidic chip mimics reciprocal activation in solid tumor microenvironment, PLoS One, 2016, 11, e0159013 CrossRef PubMed.
- M. Fujiwara, K. Kanayama and Y. S. Hirokawa, et al., ASF-4-1 fibroblast-rich culture increases chemoresistance and mTOR expression of pancreatic cancer BxPC-3 cells at the invasive front in vitro, and promotes tumor growth and invasion in vivo, Oncol. Lett., 2016, 11, 2773 CrossRef CAS.
- P. Wang and Z. Zhu, Prognostic and Clinicopathological Significance of E-Cadherin in Pancreatic Cancer Patients: A Meta-Analysis, Front. Oncol., 2021, 11, 938 CAS.
- T. Golan, D. Atias and I. Barshack, et al., Ascites-derived pancreatic ductal adenocarcinoma primary cell cultures as a platform for personalised medicine, Br. J. Cancer, 2014, 110, 2269–2276 CrossRef CAS.
- C. Liang, S. Shi and Q. Meng, et al., Complex roles of the stroma in the intrinsic resistance to gemcitabine in pancreatic cancer: where we are and where we are going, Exp. Mol. Med., 2017, 49, e406–e406 CrossRef PubMed.
- P. Procacci, C. Moscheni and P. Sartori, et al., Tumor–Stroma Cross-Talk in Human Pancreatic Ductal Adenocarcinoma: A Focus on the Effect of the Extracellular Matrix on Tumor Cell Phenotype and Invasive Potential, Cells, 2018, 7, 158 CrossRef CAS PubMed.
- A. Matsuzawa, M. Cortesi and M. Zanoni, et al., Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight, Int. J. Mol. Sci., 2021, 23, 254 CrossRef.
- D. Thomas and P. Radhakrishnan, Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis, Mol. Cancer, 2019, 18, 14 CrossRef PubMed.
- M. Hadden, A. Mittal and J. Samra, et al., Mechanically stressed cancer microenvironment: Role in pancreatic cancer progression, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1874, 188418 CrossRef CAS PubMed.
- K. Gardian, S. Janczewska and W. L. Olszewski, et al., Analysis of Pancreatic Cancer Microenvironment: Role of Macrophage Infiltrates and Growth Factors Expression, J. Cancer, 2012, 3, 285 CrossRef PubMed.
- B. Farrow, D. Albo and D. H. Berger, The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer, J. Surg. Res., 2008, 149, 319–328 CrossRef PubMed.
- W. J. Ho, E. M. Jaffee and L. Zheng, The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities, Nat. Rev. Clin. Oncol., 2020, 17, 527–540 CrossRef PubMed.
- N. Sperb, M. Tsesmelis and T. Wirth, Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma, Int. J. Mol. Sci., 2020, 21, 1–23 Search PubMed.
- G. Lazzari, V. Nicolas and M. Matsusaki, et al., Multicellular spheroid based on a triple co-culture: A novel 3D model to mimic pancreatic tumor complexity, Acta Biomater., 2018, 78, 296–307 CrossRef CAS PubMed.
- V. Brancato, V. Comunanza and G. Imparato, et al., Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer, Acta Biomater., 2017, 49, 152–166 CrossRef CAS PubMed.
- P. Gupta, P. A. Pérez-Mancera and H. Kocher, et al., A Novel Scaffold-Based Hybrid Multicellular Model for Pancreatic Ductal Adenocarcinoma—Toward a Better Mimicry of the in vivo Tumor Microenvironment, Front. Bioeng. Biotechnol., 2020, 8, 290 CrossRef.
- K. J. Norberg, X. Liu and C. Fernández Moro, et al., A novel pancreatic tumour and stellate cell 3D co-culture spheroid model, BMC Cancer, 2020, 20, 475 CrossRef CAS.
- Y. Yu, G. Yang and H. Huang, et al., Preclinical models of pancreatic ductal adenocarcinoma: challenges and opportunities in the era of precision medicine, J. Exp. Clin. Cancer Res., 2021, 40, 1–13 CrossRef.
- B. Gündel, X. Liu and M. Löhr, et al., Pancreatic Ductal Adenocarcinoma: Preclinical in vitro and ex vivo Models, Front. Cell Dev. Biol., 2021, 9, 1–12 CrossRef PubMed.
- B. Uzunparmak and I. H. Sahin, Pancreatic cancer microenvironment: a current dilemma, Clin. Transl. Med., 2019, 8, 1–8 CrossRef.
- K. Li, T. Liu and J. Chen, et al., Survivin in breast cancer-derived exosomes activates fibroblasts by up-regulating SOD1, whose feedback promotes cancer proliferation and metastasis, J. Biol. Chem., 2020, 295, 13737–13752 CrossRef CAS PubMed.
- X. Guo, H. Oshima and T. Kitmura, et al., Stromal fibroblasts activated by tumor cells promote angiogenesis in mouse gastric cancer, J. Biol. Chem., 2008, 283, 19864–19871 CrossRef CAS.
- C. Cheng, K.-K. Yan and W. Hwang, et al., Construction and Analysis of an Integrated Regulatory Network Derived from High-Throughput Sequencing Data, PLoS Comput. Biol., 2011, 7, e1002190 CrossRef CAS PubMed.
- J. Cai, H. Tang and L. Xu, et al., Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness, Carcinogenesis, 2012, 33, 20–29 CrossRef CAS.
- M. B. Omary, A. Lugea and A. W. Lowe, et al., The pancreatic stellate cell: A star on the rise in pancreatic diseases, J. Clin. Invest., 2007, 117, 50–59 CrossRef CAS.
- M. Buchholz, H. Kestler, K. Holzmann, V. Ellenrieder, W. Schneiderhan, M. Siech, G. Adler, M. G. Bachem and T. M. Gress, Transcriptome analysis of human hepatic and pancreatic stellate cells: organ-specific variations of a common transcriptional phenotype, J. Mol. Med., 2005, 83, 795–805 CrossRef CAS.
- K. Ohuchida, K. Mizumoto and M. Murakami, et al., Radiation to Stromal Fibroblasts Increases Invasiveness of Pancreatic Cancer Cells through Tumor-Stromal Interactions, Cancer Res., 2004, 64, 3215–3222 CrossRef CAS.
- H. Damhofer, J. P. Medema and V. L. Veenstra, et al., Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma mRNA-Seq, Mol. Oncol., 2013, 7(6), 1031–1042 CrossRef CAS PubMed.
- M. Majety, L. P. Pradel, M. Gies, et al., Fibroblasts Influence Survival and Therapeutic Response in a 3D Co-Culture Model, PLoS One, 2015106e0127948 Search PubMed.
- M. V. Monteiro, L. P. Ferreira and M. Rocha, et al., Journal Pre-proof Advances in bioengineering pancreatic tumor-stroma physiomimetic Biomodels, Biomaterials, 2022, 287, 121653 CrossRef CAS.
- Z. Ji, F. C. Mei and J. Xie, et al., Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells, J. Biol. Chem., 2007, 282, 14048–14055 CrossRef CAS.
- T. A. Brentnall, L. A. Lai and J. Coleman, et al., Arousal of Cancer-Associated Stroma: Overexpression of Palladin Activates Fibroblasts to Promote Tumor Invasion, PLoS One, 2012, 7(1), e30219 CrossRef CAS.
- H. Zhou, Y. Qin and S. Ji, et al., SOX9 activity is induced by oncogenic Kras to affect MDC1 and MCMs expression in pancreatic cancer, Oncogene, 2017, 37, 912–923 CrossRef.
- A. R. Pandiri, Overview of exocrine pancreatic pathobiology, Toxicol. Pathol., 2014, 42, 207–216 CrossRef CAS PubMed.
- M. A. Shields, S. Dangi-Garimella and S. B. Krantz, et al., Pancreatic Cancer Cells Respond to Type I Collagen by Inducing Snail Expression to Promote Membrane Type 1 Matrix Metalloproteinase-dependent Collagen Invasion, J. Biol. Chem., 2011, 286, 10495 CrossRef CAS PubMed.
- K. Bott, Z. Upton and K. Schrobback, et al., The effect of matrix characteristics on fibroblast proliferation in 3D gels, Biomaterials, 2010, 31, 8454–8464 CrossRef CAS.
- F. Tabatabaei, K. Moharamzadeh and L. Tayebi, Fibroblast encapsulation in gelatin methacryloyl (GelMA) versus collagen hydrogel as substrates for oral mucosa tissue engineering, J. Oral Biol. Craniofac. Res., 2020, 10, 573–577 CrossRef PubMed.
- J. M. Baena, G. Jiménez and E. López-Ruiz, et al., Volume-by-volume bioprinting of chondrocytes-alginate bioinks in high temperature thermoplastic scaffolds for cartilage regeneration, Exp. Biol. Med., 2019, 244, 13–21 CrossRef CAS PubMed.
- H. J. Hwang, M. S. Oh and D. W. Lee, et al., Multiplex quantitative analysis of stroma-mediated cancer cell invasion, matrix remodeling, and drug response in a 3D co-culture model of pancreatic tumor spheroids and stellate cells, J. Exp. Clin. Cancer Res., 2019, 38(1), 258 CrossRef PubMed.
- J. Li, B. Chen and G. F. Fellows, et al., Activation of Pancreatic Stellate Cells Is Beneficial for Exocrine but Not Endocrine Cell Differentiation in the Developing Human Pancreas, Front. Cell Dev. Biol., 2021, 9, 1769 Search PubMed.
- C. Paoli and A. Carrer, Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation, Cancers, 2020, 12, 1–22 CrossRef PubMed.
- S. I. Cantara, D. A. Soscia and S. J. Sequeira, et al., Selective functionalization of nanofiber scaffolds to regulate salivary gland epithelial cell proliferation and polarity, Biomaterials, 2012, 33, 8372–8382 CrossRef CAS.
- S. Buscemi, G. Damiano and S. Fazzotta, et al., Electrospun Polyhydroxyethyl-Aspartamide–Polylactic Acid Scaffold for Biliary Duct Repair: A Preliminary In Vivo Evaluation, Transplant Proc., 2017, 49, 711–715 CrossRef CAS.
- M. T. Nelson, A. Short and S. L. Cole, et al., Preferential, enhanced breast cancer cell migration on biomimetic electrospun nanofiber ‘cell highways’, BMC Cancer, 2014, 14, 1–16 CrossRef PubMed.
- M. F. Leong, K. S. Chian and P. S. Mhaisalkar, et al., Effect of electrospun poly(D,L-lactide) fibrous scaffold with nanoporous surface on attachment of porcine esophageal epithelial cells and protein adsorption, J. Biomed. Mater. Res., Part A, 2009, 89A, 1040–1048 CrossRef CAS PubMed.
- T. P. Burton, A. Corcoran and A. Callanan, The effect of electrospun polycaprolactone scaffold morphology on human kidney epithelial cells, Biomed. Mater., 2017, 13, 015006 CrossRef PubMed.
- J. H. Mendle, J. Kelly and S. Voci, et al., Bortezomib and gemcitabine in relapsed or refractory Hodgkin's lymphoma, Ann. Oncol., 2008, 19, 1759 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00881e |
| ‡ Present address: Department of Electrical Engineering and Information Technology, Institute of Sensor and Actuator Systems, TU Wien, Wien, Austria. |
| This journal is © The Royal Society of Chemistry 2023 |