
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn isotope dilution mass spectrometry assay to track Norovirus-like particles in vaccine process intermediates by quantifying capsid protein VP1†
Jacquelyn R.
Jhingree
 *,
Julie
Boisvert
and
Geneviève
Mercier
*,
Julie
Boisvert
and
Geneviève
Mercier
Medicago Inc., 2552 Boulevard du Parc-Technologique, Québec, QC G1P 4S6, Canada. E-mail: jackyjhingree@gmail.com; jhingreej@medicago.com
First published on 15th May 2023
Abstract
The coronavirus disease (COVID-19) pandemic shows the rapid pace at which vaccine development can occur which highlights the need for more fast and efficient analytical methodologies to track and characterize candidate vaccines during manufacturing and purification processes. The candidate vaccine in this work comprises plant-derived Norovirus-like particles (NVLPs) which are structures that mimic the virus but lack any infectious genetic material. Presented here is a liquid chromatography-tandem mass spectrometry (LC-MS/MS) methodology for the quantification of viral protein VP1, the main component of the NVLPs in this study. It combines isotope dilution mass spectrometry (IDMS) with multiple reaction monitoring (MRM) to quantify targeted peptides in process intermediates. Multiple MRM transitions (precursor/product ion pairs) for VP1 peptides were tested with varying MS source conditions and collision energies. Final parameter selection for quantification includes three peptides with two MRM transitions each offering maximum detection sensitivity under optimized MS conditions. For quantification, a known concentration of the isotopically labeled version of the peptides to be quantified was added into working standard solutions to serve as an internal standard (IS); calibration curves were generated for concentration of native peptide vs. the peak area ratio of native-to-isotope labeled peptide. VP1 peptides in samples were quantified with labeled versions of the peptides added at the same level as that of the standards. Peptides were quantified with limit of detection (LOD) as low as 1.0 fmol μL−1 and limit of quantitation (LOQ) as low as 2.5 fmol μL−1. NVLP preparations spiked with known quantities of either native peptides or drug substance (DS) comprising assembled NVLPs produced recoveries indicative of minimal matrix effects. Overall, we report a fast, specific, selective, and sensitive LC-MS/MS strategy to track NVLPs through the purification steps of the DS of a Norovirus candidate vaccine. To the best of our knowledge, this is the first application of an IDMS method to track virus-like particles (VLPs) produced in plants as well as measurements performed with VP1, a Norovirus capsid protein.
Introduction
Noroviruses (previously known as Norwalk or Norwalk-like viruses) are non-enveloped, single-stranded RNA viruses1 known to cause a disease initially described in 1929 as the ‘winter vomiting disease’.2 It is the most common cause of viral gastroenteritis in humans with about 685 million cases worldwide, 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year, and one in five cases being acute. Vaccination against infectious viruses is the most effective method in disease control, prevention, and eradication in the masses and, to combat potential threats of epidemics and pandemics. To date, there are no licensed Norovirus vaccines available to target infections in humans, but several candidates are in development with at least four based on virus proteins.3
000 deaths per year, and one in five cases being acute. Vaccination against infectious viruses is the most effective method in disease control, prevention, and eradication in the masses and, to combat potential threats of epidemics and pandemics. To date, there are no licensed Norovirus vaccines available to target infections in humans, but several candidates are in development with at least four based on virus proteins.3
Norovirus candidate vaccine formulations in this work4 comprise protein-based virus-like particles (VLPs) that resemble and mimic the virus in that their surface antigens elicit a long-lasting immune response, but their structure lacks infectious genetic material (Fig. 1). The main capsid protein of the infectious virus is viral protein 1 (VP1).5 Norovirus-Like particles (NVLPs) are formed when VP1 is expressed in a plant-based platform.6 In this work, the NVLPs undergo a series of purification steps from the crude extract upstream to a highly purified form downstream, the drug substance (DS). To support process development, it is necessary to accurately and reproducibly quantify and track the amount of VP1 present at different stages of the purification process. Challenges in developing a quantification assay for various process intermediates can arise due to matrix effects in each sample. Moreover, in early step intermediates, the assay must be capable of detecting low amounts of the protein of interest in the presence of highly concentrated host cell proteins.
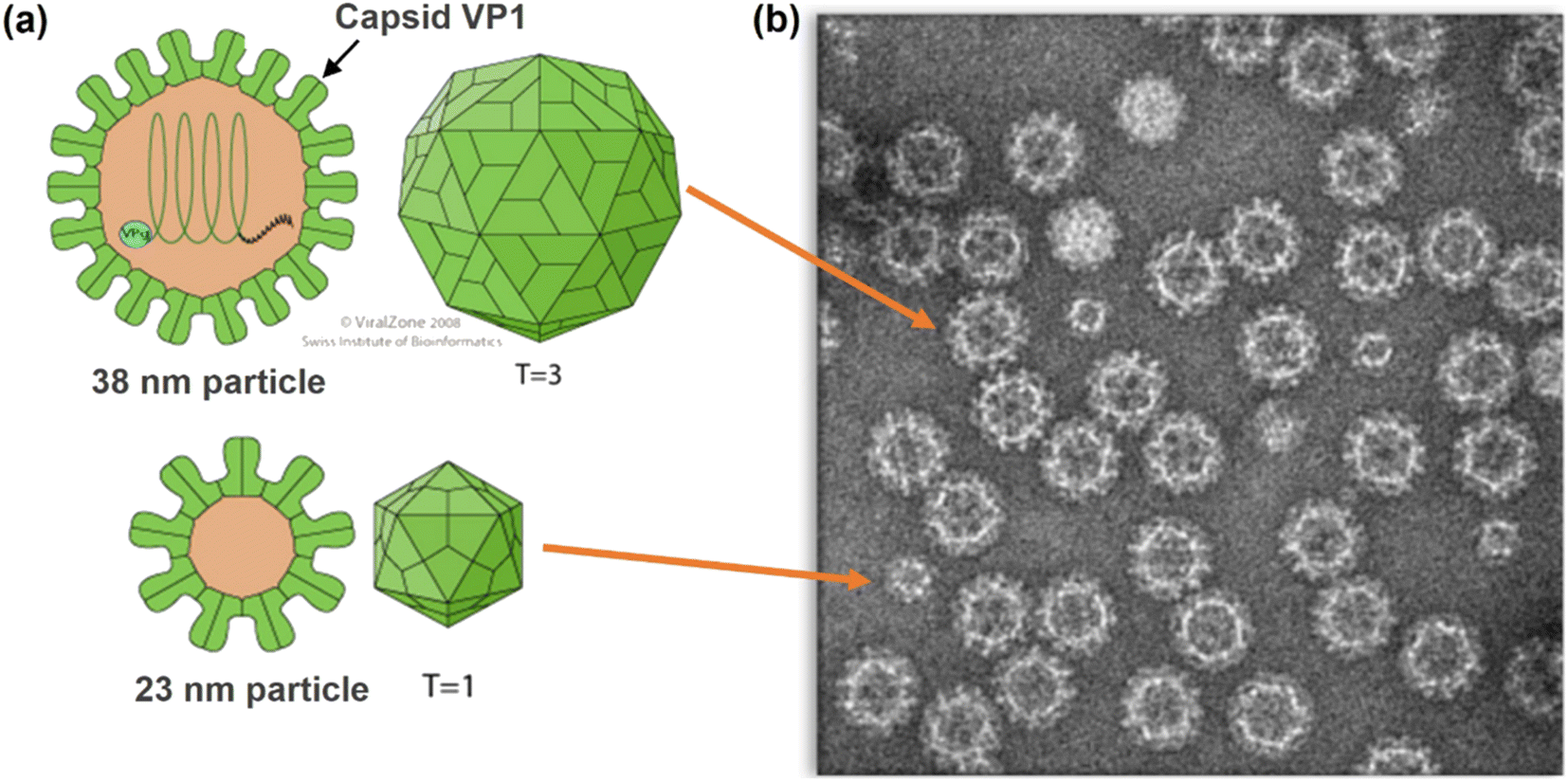 | ||
| Fig. 1 (a) Structures of Norovirus particles showing VP1 Capsid Protein. (b) Transmission Electron Microscopy (TEM) image of Norovirus-like particles (NVLPs) used in this study (copyright Medicago). | ||
Established methods for quantification of proteins in protein-based vaccine preparations are the Single Radial Immunodiffusion (SRID) assay (approved by the World Health Organization (WHO) and regulatory bodies for potency determination) and the hemagglutination assay. These methods were developed for Influenza vaccine candidates and adopted worldwide since 1978.7 The SRID assay has low sensitivity (detection limit is approximately 3–5 μg mL−1) and is time consuming (can take up 3 days to perform).8 Further, it is based upon the interaction between the Influenza virus antigen, hemagglutinin (HA), and a specific antibody, in an agarose gel, which produces a zone of precipitation whose size is directly proportional to the amount of HA; measurements can be affected by the presence of aggregates or matrix components that may interfere with HA diffusion in the gel.9,10 In addition, specific HA antigen references and their specific monoclonal antibodies need to be regularly updated.11 This update is required because the influenza genome can change via antigenic drifts and shifts resulting in mutations in the amino acid sequence of surface antigens including HA which further results in new strains or subtypes of the virus.12,13
The hemagglutination assay initially used for the determination of viral activity is based upon agglutination (clumping) of red blood cells (RBCs) by the HA protein. HA quantification, at the end of agglutination, is based on the RBC concentration where there is one influenza virus particle for each blood cell.14 It is fast compared to the SRID assay but also has limitations and these include the need for fresh red blood cells for results to be reproducible, an external standard is required for each assay as each red blood cell has a different origin and, the influenza strain can influence the agglutination reaction subsequently affecting the particle to red blood cell ratio and measurement accuracy.15 These methods can be adapted to protein quantification in other vaccine candidates but generally have the same limitations. These limitations may affect the speedy characterization of candidate vaccines and the subsequent availability of vaccines, not ideal in epidemic and pandemic situations, and therefore the WHO has recommended to researchers and pharmaceutical companies the need for the development of alternative fast and reproducible methods.16 In addition, these methods prove challenging to adapt to the VLP vaccine candidate in this study. Emerging and more recent methods for viral protein antigen, viral particle and VLP quantification include the enzyme linked immunosorbent assay (ELISA),17 assays using surface plasmon resonance (SPR),18 high performance liquid chromatography (HPLC) with Ultraviolet-visible (UV-Vis) absorption and fluorescence detection19 and, quantitative real time polymerase chain reaction (qPCR).20
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) is an accurate, fast, and sensitive method for peptide and protein quantification and is used in the method reported herein to quantify VP1 in NVLP preparations.21 The approach here is based on a strategy developed by the US Centers for Disease Control and Prevention (CDC) for the quantification of HA and neuraminidase (protein antigens) in influenza vaccines and virus preparations.22–24 It combines isotope dilution mass spectrometry (IDMS) with a multiple reaction monitoring (MRM) approach to quantify targeted proteins in vaccine preparations subjected to digestion prior to LC-MS/MS analysis.25,26 Quantification is based on the detection of peptides that are unique to the targeted protein and, assuming complete digestion, present in a stoichiometric ratio with the protein from which it originates. Herein, we have successfully adapted and extended this approached to quantify and track VP1 in NVLP preparations through different stages of DS purification; an approach that ultimately quantifies proteins in candidate vaccines and therapeutics using IDMS in general.
Experimental
NVLP vaccine preparations
All DS preparations were manufactured at Medicago Inc. (Quebec City, Canada). VP1 was tracked in different intermediates from the DS purification process. In this paper, each process step is labeled with an ascending number as it progresses through purification.Selection and synthesis of light (native) and heavy (isotopically labeled peptides)
Peptides (AQUA Ultimate Grade) were synthesized by Thermo Fisher Scientific (Burlington, Ontario, Canada). Certificates of Analysis were provided and showed peptide concentrations were within ±5–10% of the expected concentrations. There are numerous publications on the selection criteria for peptides used in quantification with MRM assays but in short, peptides chosen were those least likely to be modified in the plant cell and during sample preparation. Modifications would only serve to complicate the assay as it would produce multiple ionic forms; all of which would have to be considered for accurate quantification of the protein (VP1 in this case) from which they originate. These criteria include that they do not have oxidizable amino acids (e.g., methionine and tryptophan) or possess cysteine residues as this would add extra steps to sample preparation, i.e., sample reduction to break disulphide bonds followed by alkylation to prevent the cleaved disulphide bonds from reforming. A Peptide Synthesis and Proteotypic Peptide Analyzing Tool (Thermo Fisher Scientific, California, USA) was used to predict the ease of peptide synthesis, purification, and the compatibility of candidate peptides with MRM quantification in this work.Preparation of working standards for calibration
Peptide solutions were provided in liquid form at 5 pmol μL−1. Intermediate solutions at lower concentrations were then prepared by combining the selected synthetic light (unlabeled) and heavy (isotopically labeled) peptides respectively. Seven calibration standards were prepared from the intermediate concentration solution with a concentration range of 2.5–400 fmol μL−1 for light peptides and a limit of detection (LOD) standard was prepared at 1.0 fmol μL−1. The internal standard (IS) or heavy peptides, used to compensate for any variation in MS/MS signal, was added to each standard solution to a final concentration of 50 fmol μL−1.Sample preparation and digestion
From the total protein concentration determined by a Bradford Assay or Optical Density measurements at 280 nm, VLP samples were diluted in a 50 mM Histidine, 150 mM NaCl pH 6.7 buffer, to obtain 1 μg in 10 μL of total digestion volume. 10 μL of 0.2% Rapigest (Waters Corporation, Milford, MA, USA) in 100 mM NH4HCO3 was added to 10 μL of diluted sample in an Eppendorf tube (if the sample volume is greater than 10 μL, the same volume of Rapigest was added.) This mixture was vortexed, centrifuged and incubated for 5 minutes at 100 °C with a heating block. It was then allowed to cool to room temperature (about 5 minutes) then 10 μL of the internal standard (IS) (heavy peptide solution at 500 fmol μL−1) was added. Trypsin was added to obtain a trypsin-to-protein ratio of 1:1. Once again, the mixture was vortexed, centrifuged and incubated for 16–20 hours at 37 °C. 5 μL of 10% TFA was then added, the mixture vortexed and incubated for 30 minutes at 37 °C. The resulting peptide solution was desalted with a C18 TopTip Cartridge (Glygen Corporation, Columbia, Maryland, USA) then evaporated to dryness. The sample was then reconstituted in 100 μL of 2% acetonitrile, 0.1% formic acid, vortexed, centrifuged then put in an HPLC vial containing a polypropylene insert. This was placed in the autosampler (5 °C) for LC-MS/MS analysis or can be stored (for up to 5 days) at 2–8 °C until injection.
Spiked recovery
Matrix effects were assessed by performing spike and recovery experiments. A known amount of the light peptides or DS comprising NVLPs was spiked into process intermediates (in-process samples) prior to sample preparation and the amount of VP1 recovered was determined with the method.LC-MS/MS instruments parameters
LC-MS/MS analysis of digested solutions comprising targeted peptides was performed on a Vanquish HPLC coupled to a TSQ Altis Triple Quadrupole Mass Spectrometer (Thermo Scientific, Waltham, MA, USA). A HALO peptide ES-C18 2.7 μm fused cored analytical column (2.1 × 100 mm) along with a HALO peptide ES-C18 2.7 μm fused cored pre-column (2.1 × 5 mm) was used for peptide separation. The injector temperature was held at 5 °C while the column temperature was 30 °C. Mobile phase A was aqueous with 0.1% formic acid while mobile phase B (organic) was acetonitrile with 0.1% formic acid. A needle wash of 50% methanol was used. The flowrate was 0.3 mL min−1 with an injection volume of 10 μL. A gradient elution was done with initial mobile phase comprising 98% A and 2% B. Mobile phase B was increased to 30% from 0–6 min then to 80% from 6–6.5 min, held at 80% from 6.5–7 min then decreased to 2% from 7–7.5 min. It was held at 2% for 2.5 min. Post chromatographic separation, the effluent was introduced into the source region of the mass spectrometer where peptide charging occurred via electrospray ionization then transmitted for MRM. The peptides of interest (precursor ions) are m/z selected in the first quadrupole, fragmented in the second and product ions are mass analyzed in the third. Selected precursor/product ion pairs (MRM transitions) are monitored for quantification.Results and discussion
The goal of this method is to reproducibly track the quantity of NVLPs present in process intermediates of the drug substance to aid in process development thereby helping to optimize purification process steps. The MRM method herein quantifies specific peptides derived from VP1 from which the NVLP is assembled. VP1 is quantified by assuming that there is a stoichiometric ratio of unique peptides to protein (one peptide to one protein molecule). To ensure this stoichiometry holds it is necessary for VP1 to be fully digested during sample preparation. Digestion parameters evaluated and optimized were reduction/alkylation prior to trypsin digestion, addition of Rapigest (a surfactant which helps to solubilize a protein making it more susceptible to digestion without affecting enzyme activity), incubation time and trypsin/protein ratio for digestion. The optimized parameters are detailed in the methods section; reduction and alkylation altered the chromatographic profile, so this was not done. Further, to ensure that the NVLP samples are fully digested, quantification was done with three different peptides from different regions of VP1 including a conserved region.VP1 quantification is based on monitoring precursor/product ion pairs (MRM transitions), from LC-MS/MS analyses of the digested NVLP samples. Two MRM transitions for three VP1 peptides (internally identified as VP1-03, VP1-04 and VP1-06) were selected for monitoring after several peptides were tested using a range of collision energies for precursor ion fragmentation; MS source parameters were optimized for sensitivity. The molecular weight, the optimized collision energy for each peptide transition, as well as the two transitions of each VP1 peptide selected for quantification are shown in Table 1. Peptide sequences are not shown for confidentiality reasons. A total ion chromatogram of the three peptides shows them well resolved (Fig. 2) although even if co-elution occurred accurate quantification is possible since each MRM transition is unique for each peptide.
| Target peptide | Monoisotopic MW (Da)a |
MRM transition 1 | MRM transition 2 | ||
|---|---|---|---|---|---|
| Precursor/product (m/z) | Collision voltage (V) | Precursor/product (m/z) | Collision voltage (V) | ||
| a All masses are monoisotopic. | |||||
| VP1-03-light | 1326.64 | 664.3/792.4 | 19 | 664.3/893.4 | 20 |
| VP1-03-heavy | 1334.66 | 668.3/800.4 | 19 | 668.3/901.4 | 20 |
| VP1-04-light | 904.44 | 453.2/545.3 | 16 | 453.2/659.3 | 15 |
| VP1-04-heavy | 914.45 | 458.2/555.3 | 16 | 458.2/669.3 | 15 |
| VP1-06-light | 842.49 | 422.3/486.3 | 19 | 422.3/599.4 | 16 |
| VP1-06-heavy | 850.49 | 426.3/494.3 | 19 | 426.3/607.4 | 16 |
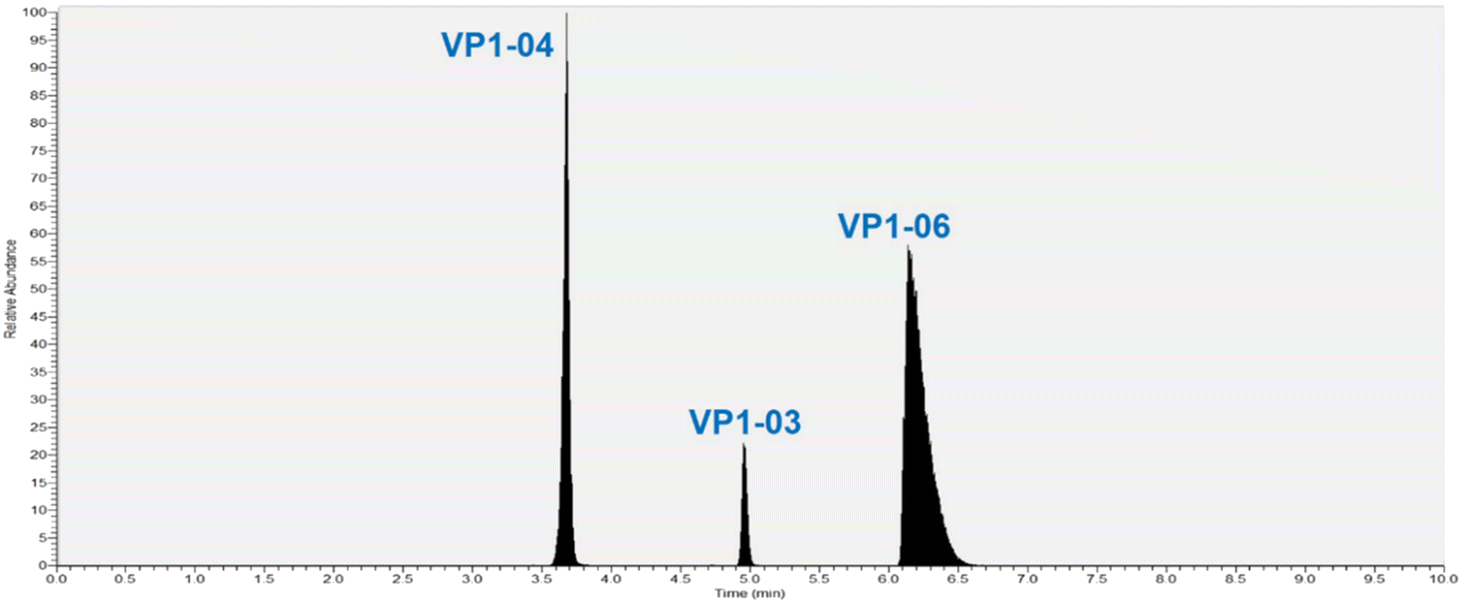 | ||
| Fig. 2 Total ion chromatogram for three peptides from protein VP1 used to track NVLPs in samples from different steps of purification. | ||
To quantify specific VP1 peptides a known amount of isotopically labeled versions (heavy peptide/internal standard) are added to digested samples. The VP1 peptide peak area-to-heavy peptide (IS) ratio is used to determine the VP1 peptide concentration from calibration curves. Calibration curves were generated from the LC-MS/MS data by plotting the peak area ratio of the light peptide-to-heavy peptide vs. the concentration of the light peptide (standard) solution [fmol μL−1]; the light peptide is the synthetic version of the native VP1 peptides of interest used in the quantitation. The limit of quantitation (LOQ) of the assay is 2.5 fmol μL−1 and the limit of detection (LOD) is 1.0 fmol μL−1. The peak area ratio varies linearly with concentration of unlabeled peptide within the calibration standard range and the coefficient of determination (R2) obtained is usually 0.995 or higher. The acceptable difference between the theoretical and experimental concentrations for each calibration standard is ≤15% except for the lowest concentration (2.5 fmol μL−1) where it is ≤20%. Note, that the guidance followed for setting these criteria, for chromatographic assays, is in accordance with regulatory authority US Food and Drug Administration (FDA) which recommends that “non-zero calibrators should be ±15% of nominal (theoretical) concentrations, except at LLOQ where the calibrator should be ±20% of the nominal concentrations in each validation run”.27 Our measurements fit well within these criteria for all peptides and therefore the broadness of VP1-06 peak in the total ion chromatogram (Fig. 2) does not affect our measurements.
Exemplar calibration curves for the two transitions used to quantify VP1-06 are shown in Fig. 3 while the corresponding data are shown in the supplementary information (Table S1†). Two injections of seven peptide standard preparations were performed with concentrations ranging from 2.5–400 fmol μL−1 for the light peptide. The difference between expected and calculated concentrations of each standard is within 11.5% and less for standards two to seven and within 12.2% for standard one. The linearity is good with coefficients of determination (R2) of 0.9997 for both VP1-06 peptide transitions and 0.9995 and higher for all peptide transitions in this study. This surpasses the criterion set – R2 > 0.990 – according to regulatory guidance. The VP1 peptides' concentration in each sample was determined using the peak area ratio of native peptide-to-heavy peptide, as samples comprise heavy peptides (IS) at the same level (concentration) as that of calibration standards.
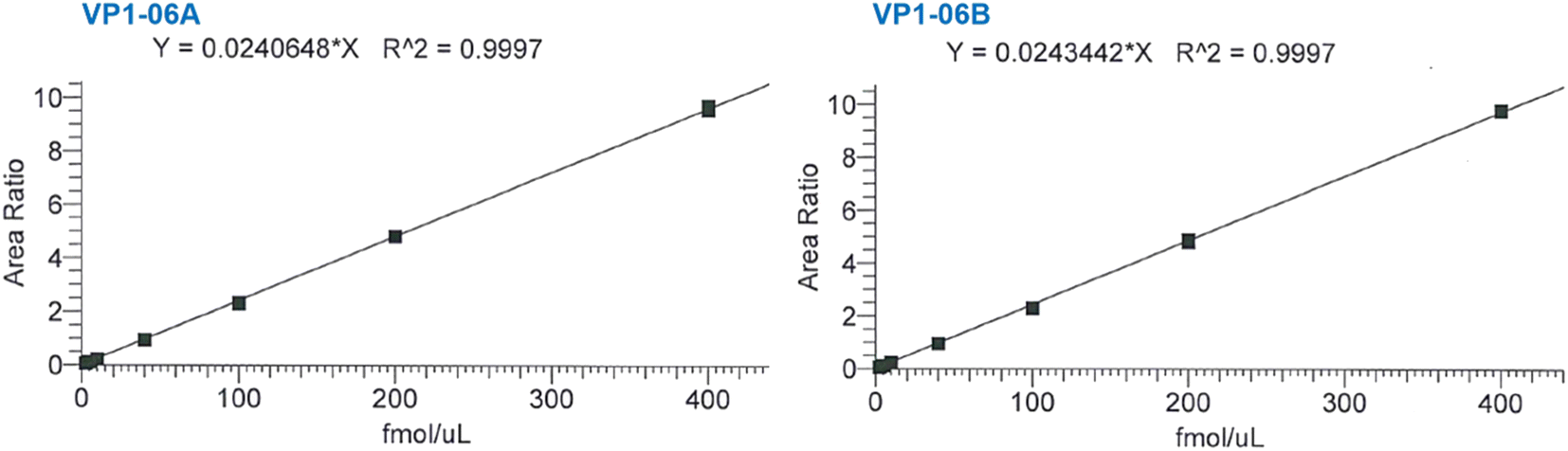 | ||
| Fig. 3 Representative calibration curves for two MRM transitions of peptide VP1-06 used in the quantification of VP1 in NVLP preparations. See Table S1† for corresponding data. | ||
The concentration of VP1 determined from measurements with two transitions (A and B) of three peptides (VP1-03, VP1-04, VP1-06) for samples from eight process steps of NVLP DS manufacturing are shown in Table 2. Concentrations were determined in fmol μL−1 and converted to μg mL−1 using the molecular weight of VP1 (for internal purposes). Three replicate measurements, including sample preparation, were done for each sample. The relative standard deviation (RSD) determined for the three replicate measurements of all six transitions, for the three peptides, ranges from 9.4% to 10.0% for the eight process steps. This indicates low variation for replicate measurements and good assay reproducibility. It is seen that the measurements for peptide VP1-06 show a small decrease in the measured concentrations of replicate measurements compared with peptides VP1-03 and VP1-04 (Table 2). However, the reliability of the measurements is set by criteria given by regulatory guidance, as previously stated, and our measurements fit these criteria well (the precision criteria is ±15% RSD, except ±20% at LLOQ, within runs and between runs).27 The good agreement between the three peptides also puts high confidence in the complete digestion of the VP1 protein and therefore in the accuracy of the determined VP1 concentration. Further, there is an increase in protein concentration with each purification step (from 1 to 8), as expected. Additionally, measurements were performed on a second triple quadrupole system for comparison. The results show that VP1 quantification is comparable between instruments with an average measurement difference of 0.2 to 10.7% for different purification steps. RSDs were less than 15% for three replicate measurements at each purification step (Table S2†). This indicates that this assay could be transferred to a different system. Assay intermediate precision was also evaluated with measurements performed by two different analysts on three days over a period of five weeks using two LC-MS systems. Measurements were in good agreement across the three days with RSDs less than 15% (Table S3†). This demonstrates good intermediate precision for VP1 quantification with this assay.
| Process step | Replicate | VP1 concentration (fmol μL−1) | Relative standard deviation (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| VP1-03A | VP1-03B | VP1-04A | VP1-04B | VP1-06A | VP1-06B | Average | |||
| 1 | 1 | 10.5 | 10.7 | 10.1 | 10.1 | 9.6 | 9.8 | 10.4 | 9.9 |
| 2 | 10.3 | 10.7 | 9.9 | 10.2 | 9.4 | 9.7 | |||
| 3 | 10.1 | 10.0 | 9.3 | 9.9 | 9.0 | 9.1 | |||
| 2 | 1 | 10.8 | 11.0 | 10.9 | 10.7 | 9.8 | 10.0 | 11.2 | 9.5 |
| 2 | 11.5 | 11.1 | 11.3 | 11.0 | 10.0 | 9.9 | |||
| 3 | 11.0 | 11.3 | 11.0 | 10.7 | 9.7 | 9.9 | |||
| 3 | 1 | 10.9 | 10.8 | 10.4 | 10.8 | 9.7 | 9.6 | 11.0 | 9.8 |
| 2 | 10.7 | 10.5 | 10.4 | 10.6 | 9.6 | 9.5 | |||
| 3 | 11.1 | 11.7 | 11.1 | 11.2 | 10.0 | 10.1 | |||
| 4 | 1 | 11.2 | 11.7 | 11.4 | 11.8 | 10.3 | 10.0 | 11.9 | 10.0 |
| 2 | 11.4 | 11.7 | 11.6 | 12.2 | 10.6 | 10.4 | |||
| 3 | 11.9 | 11.9 | 11.7 | 11.8 | 10.6 | 10.8 | |||
| 5 | 1 | 55.4 | 56.7 | 54.5 | 56.7 | 49.4 | 48.6 | 57.1 | 9.9 |
| 2 | 57.0 | 57.7 | 57.2 | 57.9 | 50.9 | 49.9 | |||
| 3 | 56.2 | 55.4 | 57.0 | 58.0 | 50.6 | 50.1 | |||
| 6 | 1 | 98.9 | 99.6 | 96.9 | 99.4 | 88.2 | 85.4 | 100.1 | 10.0 |
| 2 | 102.3 | 108.2 | 100.2 | 103.0 | 90.1 | 89.3 | |||
| 3 | 98.7 | 96.9 | 94.1 | 97.6 | 85.8 | 85.6 | |||
| 7 | 1 | 123.3 | 126.0 | 118.3 | 119.9 | 107.7 | 106.3 | 120.6 | 9.7 |
| 2 | 117.5 | 119.4 | 113.2 | 117.8 | 104.7 | 102.2 | |||
| 3 | 121.7 | 122.4 | 118.6 | 121.3 | 107.3 | 105.9 | |||
| 8 | 1 | 134.7 | 136.1 | 132.4 | 134.0 | 119.1 | 118.4 | 135.9 | 9.4 |
| 2 | 135.3 | 134.2 | 132.6 | 132.5 | 119.8 | 117.2 | |||
| 3 | 140.1 | 134.8 | 136.1 | 138.4 | 122.2 | 119.2 | |||
We recognised that it is possible that sample matrix can affect measurements, particularly in samples from early process steps, or variations may occur depending on the composition of the formulation buffer, so spike recovery experiments were done to evaluate the extent of these effects. Spiking was done in two ways – with native unlabeled peptides and with the DS comprising highly purified assembled VLPs (the active ingredient that is subsequently formulated with excipients to produce the final product). Spiking with the DS was done for two sample lots that underwent purification. The amounts recovered from spiking in both peptides and DS in samples for different process steps are shown in Table 3. Spiking with native peptides was done for eleven process steps while in the case of the DS, measurements were done with samples available. Recoveries for samples spiked with native peptides range from 94–105% for eleven steps in the NVLP purification process indicating that the LC-MS/MS detection of VP1 peptides is not impaired by matrix effects from in-process or DS samples. Recoveries for in-process samples for Lot A spiked with the DS range from 49–107%. The recovery at step 9 was significantly lower than the other measurements however, measurements done on a second Lot (B) with more process steps analyzed produced a significant improvement in recovery of 113%. In this instance the lower recovery is likely due to sample loss occurring during clean-up. The method was adjusted to minimize this loss thereby improving the recovery significantly. Recoveries ranged from 68–113% for Lot B where all measurements are within an acceptable range of 72–113%, except for process step 8 being lower (68%). This lower recovery was considered in making decisions on the process in this step and highlights the importance of this method in the decision-making process of DS purification. In developing this assay, the acceptable recovery range (70–130%) is set by regulatory guidance27,28 but the range obtained in this work is also comparable to published work on protein quantification in different matrices using IDMS.29–31 Overall, these recovery values indicate minimal matrix effects from various samples in the DS purification steps. These results also support complete digestion of NVLP particles.
Conclusions
It is clear to researchers in the biopharmaceutical industry that the development of faster and more reproducible methods for protein quantification in protein-based candidate vaccines is highly advantageous. This would provide alternative methods without the limitations in established ones and, to keep pace with vaccine development and the characterization of vaccine candidates in situations, like a pandemic, when speedy results are required. Thus, here we report a fast, sensitive, specific, and reproducible LC-MS/MS approach used to quantify the NVLP protein, VP1. It combines IDMS with MRM to quantify targeted peptides produced from digestion of VP1 for samples from the purification steps of a Norovirus vaccine candidate DS. The results from this method support the complete digestion of VP1 so the quantified peptide, present in a stochiometric ratio with VP1, allows quantification of VP1 representative of NVLP quantities. This permits accurate and reproducible tracking of NVLPs through purification steps. This is vital for decision making on the optimization of NVLP yields and purity in process development steps of vaccine DS and subsequently the final drug product (DP) development.Author contributions
G. M. conceptualized experiments; J. B. did experiments and analyzed data; J. J. drafted original manuscript; G. M., J. J. and J. B. reviewed and revised manuscript; all authors contributed to the final form of the manuscript.Conflicts of interest
The authors declare no known conflicts of interest.References
- C. Ruis, L. Thorne and J. Breuer, Norovirus, Springer, Switzerland, 2019 Search PubMed.
- J. Zahorsky, Arch. Pediatr., 1929, 46, 391–395 Search PubMed.
- Global Burden of Norovirus and Prospects for Vaccine Development, https://web.archive.org/web/20180711021911/https://www.cdc.gov/norovirus/downloads/global-burden-report.pdf, accessed March 2023 Search PubMed.
- Our Vaccine Candidates, https://medicago.com/en/our-science/our-vaccine-candidates, accessed March 2023 Search PubMed.
- M. E. Hardy, FEMS Microbiol. Lett., 2005, 253, 1–8 CrossRef CAS PubMed.
- N. Landry, S. Pillet, D. Favre, J. Poulin, S. Trépanier, B. Yassine-Diab and B. J. Ward, Clin. Immunol., 2014, 154, 164–177 CrossRef CAS PubMed.
- J. M. Wood and J. P. Weir, Influenza Other Respir. Viruses, 2018, 12, 195–201 CrossRef PubMed.
- G. C. Schild, J. M. Wood and R. W. Newman, Bull. World Health Organ., 1975, 52, 223–231 CAS.
- K. Apostolov and B. Fishman, Nature, 1967, 215, 1287–1288 CrossRef CAS PubMed.
- R. Johannsen, H. Moser, J. Hinz, H. J. Friesen and H. Gruschkau, J. Biol. Stand., 1983, 11, 341–352 CrossRef CAS PubMed.
- P. D. Mino, Vaccines, 2015, 3, 90–104 CrossRef PubMed.
- How Flu Viruses Can Change: “Drift” and “Shift”, https://www.cdc.gov/flu/about/viruses/change.htm, accessed March 2023 Search PubMed.
- Types of Influenza Viruses, https://www.cdc.gov/flu/about/viruses/types.htm, accessed March 2023 Search PubMed.
- G. K. Hirst, J. Exp. Med., 1942, 75, 49–64 CrossRef CAS PubMed.
- A. P. Manceur and A. A. Kamen, Vaccine, 2015, 33, 5913–5919 CrossRef CAS PubMed.
- Global Pandemic Influenza Action Plan to Increase Vaccine Supply, https://www.who.int/publications/i/item/WHO-CDS-EPR-GIP-2006-1, accessed March 2023 Search PubMed.
- S. Chun, C. Li, G. Van Domselaar, J. Wang, A. Farnswort, X. Cui, H. Rode, T. D. Cyr, R. He and X. Li, Vaccine, 2008, 26, 6068–6076 CrossRef CAS PubMed.
- C. E. Nilsson, S. Abbas, M. Bennemo, A. Larsson, M. D. Hämäläinen and A. Frostell-Karlsson, Vaccine, 2010, 28, 759–766 CrossRef.
- B. Lorbetskie, J. Wang, C. Gravel, C. Allen, M. Walsh, A. Rinfret, X. Li and M. Girard M, Vaccine, 2011, 29, 3377–3389F CrossRef CAS PubMed.
- I. M. Mackay, K. E. Arden and A. Nitsche, Nucleic Acids Res., 2002, 30, 1292–1305 CrossRef CAS PubMed.
- C. E. Eyers and S. J. Gaskell, New Developments in Mass Spectrometry - Quantitative Proteomics, Royal Society of Chemistry, Cambridge (UK), 2014 Search PubMed.
- T. L. Williams, L. Luna, Z. Guo, N. J. Cox, J. L. Pirkle, R. O. Donis and J. R. Barr, Vaccine, 2008, 26, 2510–2520 CrossRef CAS PubMed.
- T. L. Williams, J. L. Pirkle and J. R. Barr, Vaccine, 2012, 30, 2475–2482 CrossRef CAS.
- W. I. Santana, T. L. Williams, E. K. Winne, J. L. Pirkle and J. R. Barr, Anal. Chem., 2014, 86, 4088–4095 CrossRef CAS PubMed.
- V. Lange, P. Picotti, B. Domon and R. Aebersold, Mol. Syst. Biol., 2008, 4, 1–14 CrossRef PubMed.
- K. G. Heumann, Int. J. Mass Spectrom. Ion Processes, 1992, 118, 575–592 CrossRef.
- Bioanalytical Method Validation, Guidance for Industry, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry, accessed April 2023 Search PubMed.
- Q2B Validation of Analytical Procedures: Methodology, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q2b-validation-analytical-procedures-methodology, accessed April 2023 Search PubMed.
- D. Barnidge, M. K. Goodmanson, G. G Klee and D. C. Muddiman, J. Proteome Res., 2004, 3, 644–652 CrossRef CAS PubMed.
- R. Liu, Y. Lv, X. Hou, L. Yang and Z. Mester, Anal. Chem., 2012, 84, 2769–2775 CrossRef CAS PubMed.
- J. P. C. Coverdale, C. F. Harrington and N. Solovyev, Crit. Rev. Anal. Chem., 2023 DOI:10.1080/10408347.2022.2162811.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay00411b |
| This journal is © The Royal Society of Chemistry 2023 |