
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmbient mass spectrometry and near-infrared spectroscopy – a direct comparison of methods for the quantification of sucralose in e-liquids†
Tobias
Schlappack‡
 a,
Christoph
Kappacher‡
a,
Michela
Demetz
a,
Thomas
Jakschitz
b,
Günther K.
Bonn
ab,
Christian W.
Huck
a and
Matthias
Rainer
*a
a,
Christoph
Kappacher‡
a,
Michela
Demetz
a,
Thomas
Jakschitz
b,
Günther K.
Bonn
ab,
Christian W.
Huck
a and
Matthias
Rainer
*a
aInstitute of Analytical Chemistry and Radiochemistry, Leopold-Franzens-University Innsbruck, Innrain 80/82, A-6020 Innsbruck, Austria. E-mail: m.rainer@uibk.ac.at; Tel: +43-512-507-57307
bAustrian Drug Screening Institute-ADSI, Innrain 66a, A-6020 Innsbruck, Austria
First published on 9th May 2023
Abstract
E-liquids have become increasingly popular in society in recent years. A wide variety of flavors and nicotine strengths make it possible for every user to get a product according to their wishes. Many of these e-liquids are marketed with countless different flavors, which are often characterized by a strong and sweet smell. Sweeteners, such as sucralose, are therefore commonly added as sugar substitutes. However, recent studies have shown the potential formation of highly toxic chlorinated compounds. This can be explained by the high temperatures (above 120 °C) within the heating coils and the used basic composition of these liquids. Nevertheless, the legal situation is composed of proposals without clear restrictions, only recommendations for tobacco products. For this reason, a high level of interest lies within the establishment of fast, reliable and cost-effective methods for the detection of sucralose in e-liquids. In this study, a number of 100 commercially available e-liquids was screened for sucralose in order to identify the suitability of ambient mass spectrometry and near-infrared spectroscopy for this application. A highly sensitive high-performance liquid chromatography coupled to a tandem mass spectrometer method was used as reference method. Furthermore, the advantages and limitations of the two mentioned methods are highlighted in order to provide a reliable quantification of sucralose. The results clearly revile the necessity for product quality due to the absence of declaration on many of the used products. Further on, it could be shown, that both methods are suitable for the quantification of sucralose in e-liquids, with beneficial economic and ecological aspects, over classical analytical tools including high-performance liquid chromatography. Clear correlations between the reference and novel developed methods are displayed. In summary, these methods enable an important contribution to ensure consumer protection and elimination of confuse package labelling.
Introduction
Electronic cigarettes (e-cigarettes) were firstly introduced in the U.S. in 2007. Since then, they have gained increasing popularity within the community, especially the young adults generation.1 The replacement of tobacco cigarettes with electronic cigarettes by most consumers is being driven by the potential of less harmful physical effects. This could be proven by Bergen and Dunworth in 2013, showing that e-cigarette aerosols contain 95% less harmful chemical compounds.2 The electronic cigarettes are designed to vaporize the so-called e-liquids (“e-juice”), which is then inhaled by consumers. The evaporation is provided by a vaporization unit, which basically consists of a heated metal coil. Most of the e-liquids are based on a mixture of glycerol and propylene glycol, within different quantitative compositions. Amounts of sweeteners, aromatic compounds and nicotine are added in order to create different varieties and strengths.Synthetic high-intensity sweeteners including sucralose, cyclamates, saccharin, aspartame and acesulfame potassium are added beside bio-derived high-intensity sweeteners such as stevioside, glycyrrhizin, sugar alcohols and natural sugars in order to create intense flavors.3 In this context, sucralose gained specific attention. This chemical compound (IUPAC name: 1,6-dichloro-1,6-dideoxy-β-D-fructofuranosyl-4-chloro-4-deoxy-α-D-galactopyranoside) is known to be 600 times sweeter than table sugar (sucrose) and due to the high consumption in other products (e.g. chewing gums, mints, hard candy) it could readily be detected within the coastal region of North America.4 During the vaporization in e-cigarettes, high temperatures (above 120 °C) are reached which further leads to the formation of toxic chlorinated compounds including chloropropanols, polychlorinated naphthalenes (PCN) and dioxins.5,6 In 2021, Moser et al. were able to identify and quantify the formation of chloroacetaldehyde, 1-chloro-2-propanol, 2-chloro-1-propanol, 1,3-dichloro-2-propanol, 2,3-dichloro-1-propanol and 3-chloropropane-1,2-diol. In addition, they could demonstrate the toxicology of those compounds with reference to regulatory actions.7
In the European region, article 20 of the Tobacco Products Directive (2014/40/EU) lays down the rules for e-cigarettes sold as consumer products in the EU. This written document includes the safety and quality requirements, as well as, packaging and labelling rules. Furthermore, it is responsible for the monitoring and reporting of developments related to e-cigarettes and refill containers. Sucralose is commonly used as sweetener in various food products since the approval in the European region in 2004 and is declared as E955. The addition in e-liquids is not prohibited, despite the knowledge that toxic compounds are formed in connection with used solvents and applied heat (from around 120 °C). Many manufacturers voluntarily forego the addition of this substance however, it is still commonly added, especially in import-products. Nonetheless, it should be included within the packaging or labelling. In addition, pure flavors (aromatic compounds) are not allowed to contain any sweeteners, which is declared in the European regulation of flavorings ((EC) No. 1334/2008).
Therefore, a high level of interest lies in analytical methods which enable reliable, fast and cost-efficient analysis of refill containers for e-cigarettes, or generally e-liquids. One of the major objectives of this work was therefore to compare two state of the art analytical methods on its performance to detect sucralose in commercially available e-liquids.
First to mention, ambient mass spectrometry (MS) which fundamentally enables ionization at atmospheric pressure.8–10 In addition to the well-known ionization techniques such as electrospray ionization (ESI), newer methods such as direct analysis in real time (DART) or atmospheric solid analysis probe (ASAP) are already integrated in commercial instruments. Important features of these ambient ionization methods are higher sample throughputs and less to no solvent consumption, since solids can often be directly measured.11–13 In addition, spectra of primarily single positively or negatively charged ions are usually generated from the analytes with low fragmentation. All these features make this new subcategory of mass spectrometry a direct counterpart to NIR spectroscopy, which is already well established in industry.14
In contrast, near infrared spectroscopy (NIR spectroscopy) has presented itself as versatile and reliable tool for qualitative and quantitative tool for various analytical tasks.15–17 Emerging from the agricultural science in the early days, applications have increased drastically ranging from petrochemical, polymer, pharmaceutical, cosmetic and food industries.11,18,19 Due to their simple handling and the absence of need for sample preparation, this method has led to a gradual substitution of time-consuming conservative analytical techniques including gas chromatography (GC), high performance liquid chromatography (HPLC), nuclear magnetic resonance spectroscopy (NMR) and MS.20,21 The combination with various algorithms for multivariate data analysis, ranging from classic algorithms including partial least squares regression (PLSR) and principal component analysis (PCA) to highly complex machine learning tools (e.g. artificial neural networks, artificial intelligence) it offers reliable and quick quantification and identification of various compounds.
The aim of this study was to employ NIR and ambient MS for the quantification of sucralose in e-liquids in order to identify the existing advantages and limitations. For this purpose, 100 e-liquids were purchased and LC-MS/MS reference measurements were performed prior to the screening methods. Additionally, artificial e-liquids with varying sucralose content were produced and quantified with each method to prove the concept. This study should further serve to provide reliable and easy to handle methods to implement quality standards for the evaluation of regulations.
Materials and methods
Chemicals and solvents
Sucralose (≥98.0% HPLC grade) and propylene glycol (ACS grade) were purchased from Sigma-Aldrich (Merck KGaA, St. Louis, USA). Deuterated sucralose, which was used as internal standard was ordered from Toronto Research Chemicals (Toronto Research Chemicals, Toronto, Canada). Dry glycerine (p. a., min 99.5%), methanol (for LC-MS, min. 99.95%) and acetonitrile (for LC-MS, min. 99.95%) from Chemsolute® were bought from Th. Geyer (Th. Geyer GmbH & Co. KG, Renningen, Germany). Milli-Q water was freshly taken from a Merck Millipore Milli-Q™ Reference Ultrapure water purification system with a deionized water as feed water source.Samples
100 commercial e-liquid samples were purchased from a local distributor. Samples were ordered with the aim of analysing thirty samples with declared sucralose content, thirty samples without sucralose and 40 samples with unknown sucralose content. The sample amount for each sample set was defined with the purpose of creating significant models in the multivariate data analysis used in the spectroscopic approach.Matrix-matched calibration
To further account for matrix effects, a matrix-matched calibration set was prepared. For this purpose, the base of e-liquids consisting of 50% (v/v) propylene glycol and 50% (v/v) glycerine was used to dissolve solid sucralose within a concentration range between 0.025% and 0.700% (w/v). Due to high viscosity of the base, the solving process was carried out at 50 °C for 4 hours on an Eppendorf ThermoMixer C at 500 rpm. The readily prepared matrix-matched calibration set was used unchanged for NIR measurements and further diluted using sucralose-d6 as internal standard (IS) for MS measurements, identically to the sample preparation for the commercial e-liquids.HPLC-MS/MS
A highly selective and sensitive HPLC-MS/MS method was chosen for the reference measurements, which was based on a method from literature and only slightly modified.22 For the chromatographic method, a Waters Premier system (Waters Corporation, Milford, USA) with a Waters XBridge BEH HILIC 150 × 3 mm and a particle size of 2.5 μm was used.Tertiary gradient separation was performed using aqueous 10 mM ammonium formate buffer (pH = 3.5) (A), methanol (B), and acetonitrile (C). The flow rate was set to 300 μL min−1 with an injection volume of 1 μL. The gradient was performed as follows: 0 min (5% A, 10% B, 85% C), 8.5 min (16% A, 10% B, 74% C), 8.55 min (5% A, 10% B, 85% C), and 16 min (5% A, 10% B, and 85% C). The autosampler temperature was set at 5.0 °C to prevent evaporation of volatile compounds in the samples, while the column temperature was set at 40.0 °C. Detection was performed with a Waters Xevo triple quadrupole using nitrogen as the sheath, desolvation, and auxiliary gas and argon as the collision gas. To ensure maximum sensitivity and specificity, multiple reaction monitoring (MRM) mode was developed in negative mode using Waters' IntelliStart software. The transitions of the precursor ions of sucralose and sucralose-d6 (IS) with the corresponding fragment ions and their respective cone voltages and collision energies are shown in Table 1. Each sample and calibration point was measured in triplicate. Matrix-matched calibration measurements were adjusted by linear fitting. Data acquisition was performed using Waters MassLynx, and data analysis was performed using Waters TargetLynx.
| Analyte | Precursor ion [M − H]− | Fragment ion [M − H]− | Cone voltage/V | Collision energy/eV |
|---|---|---|---|---|
| Sucralose | 397.05 | 361.02 | 40.00 | 10.00 |
| Sucralose-d6 | 401.02 | 365.05 | 40.00 | 12.00 |
Ambient mass spectrometry
Ambient mass spectrometric measurements were conducted with an expression CMS-L from Advion (Advion Interchim scientific, Montluçon, France). An open port sampling interface was used for sample input in combination with a modified electro spray ionisation source. Nitrogen was used as sheath and auxiliary gas. Run control and sequence creation was performed with MassExpress software from Advion. The ambient mass spectrometric method could be executed in a total run time of one minute and 15 seconds. The ion source was set to default settings for ESI negative mode, while the default negative tune parameters were used. Detailed information of the ion source and tune parameters can be found in the ESI† (Table A. 1 and Table A. 2). The solvent flow of the isocratic pump used for the formation of a solvent meniscus on the open port sample interface was set to 250 μL min−1, with a mixture of 80% acetonitrile, 10% methanol and 10% Milli-Q water (all (v/v)). For sample measurements, one μL of 25-fold diluted sample was spotted on the solvent meniscus with the aid of an electronic pipette in reversed pipetting mode. Each sample and calibration point were measured in triplicate. For quantification for the masses of interest, single ion monitoring mode (SIM) was performed with m/z values of 395.0 for sucralose and 401.1 for the internal standard sucralose-d6. The span range was set to 0.3 m/z for both target ions. For automated data analysis, QuantExpress from Advion was used. Matrix-matched calibration measurements were fitted using a quadratic fit, as it showed the best coefficient of determination. Regression parameters and plot can be found in the ESI.† Smoothing was performed with a radius of seven and two performed iterations while noise removal was enabled. Peak detection was executed with the valley to valley setting.FT-NIR measurement
NIR measurements were performed on a Büchi NIRFlex N-500 (Büchi Labortechnik AG, Switzerland, Flawil) using a liquids attachment enabling detection in transmission mode. Spectral information was gained between 4000 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 (2500 to 1000 nm) and a spectral resolution of 4 cm−1 provided by a tungsten-halogen lamp and a 12 VDC HeNe laser (633 nm) for wavelength reference. The wavelength selection and detection are based on a polarization interferometer using TeO2 wedges and a single element InGaAs detector. Undiluted e-liquids were pipetted into high precision QX quartz cells (Hellma GmbH & Co. KG, Germany, Müllheim) with a layer thickness of 1 mm in order to avoid total absorption of certain wavelength regions. Measurements were performed in triplicate for each sample and 64 scans each, resulting in a total number of 300 for commercial e-liquids and 33 for matrix-matched calibration spectra.
000 cm−1 (2500 to 1000 nm) and a spectral resolution of 4 cm−1 provided by a tungsten-halogen lamp and a 12 VDC HeNe laser (633 nm) for wavelength reference. The wavelength selection and detection are based on a polarization interferometer using TeO2 wedges and a single element InGaAs detector. Undiluted e-liquids were pipetted into high precision QX quartz cells (Hellma GmbH & Co. KG, Germany, Müllheim) with a layer thickness of 1 mm in order to avoid total absorption of certain wavelength regions. Measurements were performed in triplicate for each sample and 64 scans each, resulting in a total number of 300 for commercial e-liquids and 33 for matrix-matched calibration spectra.
Spectral preprocessing
Spectral data processing and interpretation was carried out using a commercial software product called The Unscrambler X 10.5 (CAMO Software AS, Norway, Oslo). All gained raw spectra recorded in transmission mode were firstly transformed into absorbance by applying a common negative logarithm (log(1/R)). In order to identify the best results for a quantitative prediction model, different combinations of spectral pretreatments including standard normal variate (SNV), smoothing, first- and second-order derivatives and multiplicative scattering correction (MSC) were performed. Partial least squares regression (PLSR) models were generated to evaluate performed processing. In addition to spectral processing, wavenumbers with high impact on the PLSR-model were identified and selected using the resulting loadings plot and the function “uncertainty test”. The assessment was made on the basis of calculated values for the root mean square error of prediction (RMSEP), slope, offset and the coefficient of determination (COD) in full-cross validation. The following combination of spectral processing led to the most satisfying results: log(1/R), SNV and a first derivative (derivative order: 1; polynomial order: 2; smoothing points: 5).Multivariate data analysis
Chemometrics were carried out on two different issues. Firstly, the gained preprocessed spectra of all commercial e-liquids underwent a classification process based on linear discriminant analysis (LDA) in order to distinguish between liquids that contain and do not contain sucralose. LDA was chosen due to its successful application in a wide range of issues within the analysis of food and food adulteration.23 It is considered as supervised classification algorithm. For this purpose, linear boundary decisions are created, maximizing the ratio of between-class to within-class dispersion. In summary, LDA focuses on the dissimilarity between spectra and samples defined in classes.24,25 For this purpose, the sample was split into different subsample groups, due to the very low concentration of specific samples. The subsample groups consist of samples with reference concentrations of >0.05%, >0.1% and >0.15% sucralose and the whole sample set. To ensure proper validation, the sample set was further on divided into a calibration and validation (test) set by applying a ratio of 70 to 30% within each subsample group (selected by The Unscrambler X 10.5 in special intervals).In a second step, samples containing sucralose were used to create a quantification model based on partial least squares regression (PLSR). PLSR was chosen due to its proven robustness for the generation of quantitative models for spectral methods.18,26–28 Spectral pretreatments remained the same to the already mentioned LDA models. Optimization of PLSR parameters were created in full-cross validation in order to identify the best possible result.
Additionally, the sample set was identically divided into four subsample groups based on the gained reference concentration. For the calculation of the final results, the sub sample sets were divided into a calibration and validation set (as described in qualitative analysis) based on random sample selection.
For the purpose of comparability to the ambient MS methods, the matrix matched calibration set was used to create a separate PLSR model. The same spectral pretreatments were applied for this application and the results (Fig. 3) represent the predicted validation values in full-cross validation.
Results and discussion
HPLC-MS/MS
Already at the beginning of the study it was observed that several e-liquids, which according to the declaration of the distributor should contain sucralose or should be free of sucralose, did not meet the relevant requirements. Therefore, the development and establishment of a fast and simple method for quantifying sucralose in e-liquids is of utmost importance to ensure consumer protection. For this purpose, three different analysis techniques were employed, including HPLC hyphenated to MS/MS, near-infrared spectroscopy and a novel ambient MS technology. Fig. 1 shows the sucralose concentrations of the e-liquid samples measured by HPLC-MS/MS. Most of the analysed e-liquids (57%) did not contain any sucralose. The remaining e-liquids revealed sucralose concentrations between 0.1 to 0.5% and even higher. With a coefficient of determination of 0.998, the calibration curve shows sufficient linearity for the quantification of sucralose and the RSD values in the range of 0.30–3.75% ensure reliable precision of the method. Furthermore, matrix matched quality control samples in the low, medium and high concentration range (0.025, 0.200 and 0.500% (w/v)) showed bias values no greater than 4.3%, resulting in an accurate quantification of sucralose. Therefore, the HPLC-MS/MS reference method can be considered to be sufficiently correct and precise to quantify the sucralose content in e-liquids at high accuracy.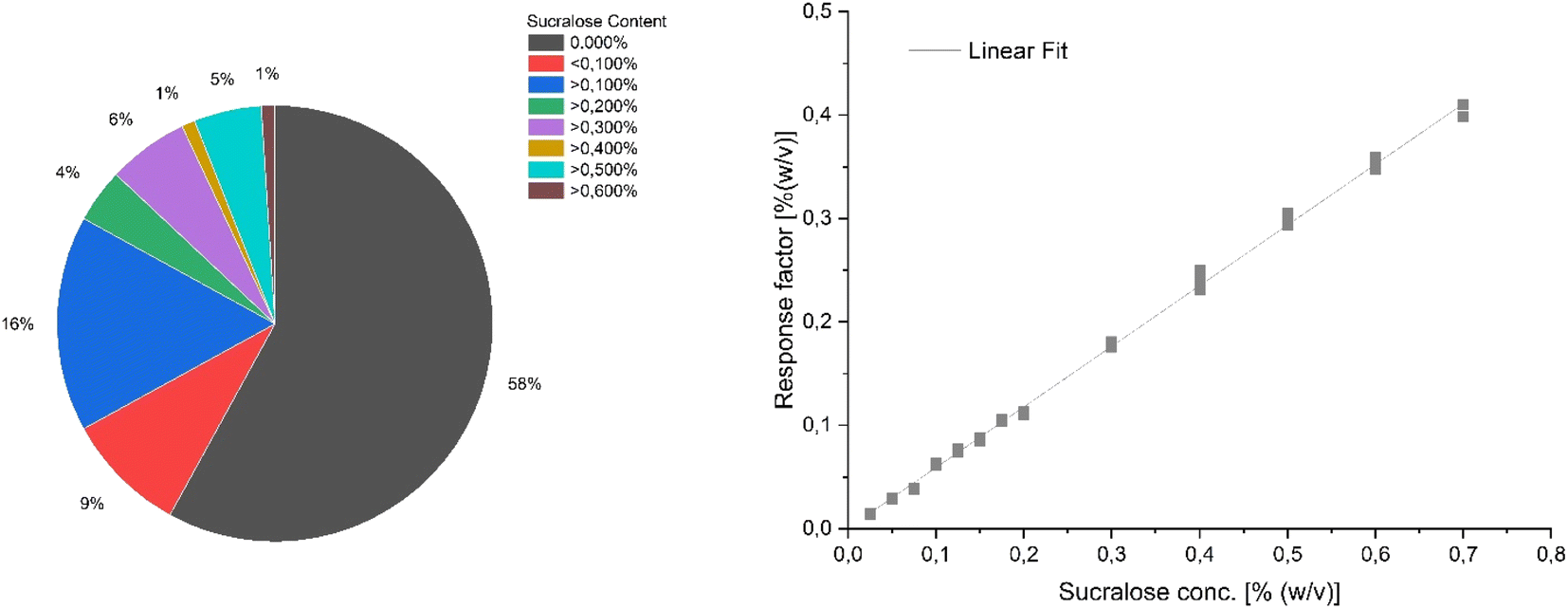 | ||
| Fig. 1 Sucralose content of e-liquid samples derived from HPLC-MS/MS measurements (left) which were quantified using a linear fitted matrix-matched calibration (right). | ||
Ambient mass spectrometry
To minimize the above-mentioned consumer declaration problems, the same e-liquids measured by the HPLC-MS/MS method were also examined by ambient MS. In addition to a reduction in the measurement time by a factor of 13.4 and a significantly lower solvent consumption by a factor of 16.0, the measurements also show reliable quantitative results. Fig. 2 reveals the correlation of the quantitative results obtained with the data obtained from the HPLC-MS/MS and ambient MS method. The linear trend line superimposed on the data shows a very good agreement of the data points with a coefficient of determination of 0.992. Thus, the accuracy of the ambient MS method presented here can be confirmed and evaluated as reliable. With RSD values between 0.45% and 6.37%, the precision can be considered as satisfactory. It should also be noted that in ambient MS random changes in the measurement conditions, such as those caused by fluctuations in the room atmosphere, can contribute significantly to measurement deviations. However, preliminary tests have also shown that the use of an internal standard is essential, as this is the only way that changes in the ionization of the analyte can be reliably considered. Finally, using a matrix matched calibration is an additional strategy to include the remaining matrix effects in the ionization. | ||
| Fig. 2 Correlation of the acquired HPLC-MS/MS reference data with the results obtained with the newly developed ambient MS method. | ||
Near-infrared spectroscopy
 | ||
| Fig. 3 Plot of the average spectra, including minimum and maximum absorbance and spectral SD. | ||
| Full sample set | >0.05% sucralose | >0.10% sucralose | >0.15% sucralose | |
|---|---|---|---|---|
| a Number of samples within the calibration set. b Number of samples within the validation set. c Accuracy for the calculated LDA. d Accuracy for the predicted independent validation set. | ||||
| n(Cal)a | 70 | 68 | 63 | 58 |
| n(Val)b | 30 | 29 | 28 | 25 |
| Acc.(Cal)c | 84.04% | 86.34% | 87.37% | 87.36% |
| Acc.(Val)d | 70.00% | 74.71% | 82.72% | 86.67% |
In Fig. 3 the deviations of all conducted spectra are illustrated. Whereas the average spectrum represents the mean of all 300 conducted spectra including the corresponding standard deviation (SD), displayed in shaded grey. The maximum and minimum absorbance are represented in red, respectively blue. Especially high deviations are depicted in the region of 5155 cm−1, representing O–H stretching and deformation vibration. This is mostly reasoned by the strongly varying composition of the e-liquids base, consisting mainly of propylene glycol and glycerol. However, high variations can also be detected in the regions around 4775 cm−1 (O–H deformation, stretching, and C–O stretching) and from 4000 to 4070 cm−1 (C–H stretching, C–C stretching). This region is of special interest for the analysis of sugars and carbohydrates, therefore an overlap of spectral information leads to a significant drop in prediction performance. Vibrations with high prediction performance could be identified by spectral interpretation of the matrix matched calibration with varying sucralose content. Therefore, the region of 4070 to 4000 cm−1 showed visibly the highest correlation to sucralose. As a result, it can be concluded that variations within these regions complicate the qualitative and quantitative analysis, especially for low concentration levels of sucralose. The results of prediction performance for the different subsets of the independent validation set and the comparably very accurate calibration using the matrix-matched calibration set underline these findings.
| Full sample set | >0.05% sucralose | >0.10% sucralose | >0.15% sucralose | |
|---|---|---|---|---|
| a Standard deviation of the predicted recovery. b Predicted recovery of the lowest results. c Predicted recovery of the highest results. d Number of detected outliers. e Number of factors/principal components used for PLSR. f Coefficient of determination for the calibration. g Coefficient of determination for the validation. h Root mean square error of calibration. i Root mean square error of validation. | ||||
| Mean recovery | 91.87% | 91.88% | 95.75% | 99.01% |
| SD recoverya | 33.51% | 33.49% | 27.30% | 36.75% |
| Lowest recoveryb | 46.77% | 50.79% | 59.75% | 54.60% |
| Highest recoveryc | 169.34% | 162.73% | 157.73% | 155.65% |
| Outlier (n)d | 1 | 1 | 0 | 1 |
| Factors usede | 7 | 7 | 7 | 7 |
| COD (Cal)f | 0.7194 | 0.7173 | 0.7101 | 0.8324 |
| COD (Val)g | 0.6587 | 0.5933 | 0.5509 | 0.7231 |
| RMSECh | 0.09352 | 0.09194 | 0.0895 | 0.0640 |
| RMSEVi | 0.1042 | 0.1116 | 0.1129 | 0.0839 |
 | ||
| Fig. 4 Box-plot of predicted recovery results. | ||
Matrix matched calibration
In order to investigate the potential influence of the existing matrix differences of commercially available e-liquids, a matrix-matched calibration set (0.025 to 0.20% sucralose) was prepared and measured identically as before. Fig. 5 presents the resulting calibration line after full cross-validation. The designed PLSR model with the data of the matrix-matched sucralose e-liquids resulted in a very accurate prediction model. This is proven by a coefficient of determination for calibration and validation of 0.9999 and 0.9879. Moreover, this is supported by mean square errors for calibration and validation of 0.00299 and 0.00719% of sucralose. These results support the assumption of high matrix effects within the commercial e-liquids, leading to worse predictions, especially within the low concentration samples. | ||
| Fig. 5 External calibration of the matrix-matched calibration set. | ||
Conclusions
The aim of this study was to develop fast and cost-effective alternative methods for the quantification of sucralose in e-liquids and to compare their advantages and limitations, in particular with regard to consumer protection. The results clearly reveal the absence of clear declarations by manufacturers, and therefore proves its necessity. Ambient mass spectrometry offers sensitive detection of sucralose, however, the usage of a deuterated internal standard and elaborate sample preparation due to high viscosity of the samples is required. Also, initial problems with the quantitative application of the sample onto the meniscus of the open-port sampling interface must be addressed, which were only manageable with an electric pipette in reverse pipetting mode. Nonetheless, the results represent a high correlation for the quantification of sucralose in comparison to the HPLC-MS/MS reference method, including the matrix-matched calibration. In contrast, near-infrared spectroscopy excels by the complete absence of sample preparation and, moreover, by fast and effortless measurements. The comparably worse results for quantification of commercial e-liquids, illustrated by the high deviation in prediction performance represents its downside. This is caused by the strongly varying composition of e-liquids with strong matrix effects leading to impaired quantification results. Quantification of low amounts of sucralose in commercial liquids is therefore hardly feasible, leading to more semiquantitative results. Nonetheless, the quantification results using the matrix-matched calibration set, clearly demonstrates the ability of low amount detection below 0.2% sucralose. Finally, two newly developed methods for the quantification of sucralose in e-liquids are presented, both of which enable less time-consuming and inexpensive analysis of sucralose in e-liquids in respect to classic HPLC analysis. Additionally, a lower consumption of consumables and organic solvents should be mentioned. These can further help to ensure consumer safety induced by the absence of sucralose-declaration on most commercial e-liquids.Author contributions
Conceptualization, T. S., C. K., M. R.; data curation, T. S. and C. K.; formal analysis, T. S., C. K., M. D.; investigation, T. S., C. K., M. D.; methodology, T. S., C. K., M. D.; project administration, M. R.; supervision, M. R.; visualization, T. S. and C. K.; writing – original draft, T. S. and C. K.; writing – review & editing, M. R., T. J., C. W. H. and G. K. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.References
- L. I. Laestadius, M. M. Wahl, P. Pokhrel and Y. I. Cho, From Apple to Werewolf: A content analysis of marketing for e-liquids on Instagram, Addict. Behav., 2019, 91, 119–127 CrossRef PubMed.
- P. Bergen and J. Dunworth, Electronic Cigarettes: what the Experts Say, 2013 Search PubMed.
- S. Miao, E. S. Beach, T. J. Sommer, J. B. Zimmerman and S.-E. Jordt, High-Intensity Sweeteners in Alternative Tobacco Products, Nicotine Tob. Res., 2016, 18, 2169–2173 CrossRef CAS PubMed.
- R. N. Mead, J. B. Morgan, G. B. Avery, R. J. Kieber, A. M. Kirk, S. A. Skrabal and J. D. Willey, Occurrence of the artificial sweetener sucralose in coastal and marine waters of the United States, Mar. Chem., 2009, 116, 13–17 CrossRef CAS.
- A. Eisenreich, R. Gürtler and B. Schäfer, Heating of food containing sucralose might result in the generation of potentially toxic chlorinated compounds, Food Chem., 2020, 321, 126700 CrossRef CAS PubMed.
- S. Andres, K. E. Appel and A. Lampen, Toxicology, occurrence and risk characterisation of the chloropropanols in food: 2-monochloro-1,3-propanediol, 1,3-dichloro-2-propanol and 2,3-dichloro-1-propanol, Food Chem. Toxicol., 2013, 58, 467–478 CrossRef CAS.
- D. Moser, P. Leitner, P. A. Filipek, S. Hussain, M. Rainer, T. Jakschitz, B. M. Rode and G. K. Bonn, Quantification and cytotoxicity of degradation products (chloropropanols) in sucralose containing e-liquids with propylene glycol and glycerol as base, Toxicol. Appl. Pharmacol., 2021, 430, 115727 CrossRef CAS PubMed.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Detection Technologies. Ambient mass spectrometry, Science, 2006, 311, 1566–1570 CrossRef CAS.
- R. M. Alberici, R. C. Simas, G. B. Sanvido, W. Romão, P. M. Lalli, M. Benassi, I. B. S. Cunha and M. N. Eberlin, Ambient mass spectrometry: bringing MS into the “real world”, Anal. Bioanal. Chem., 2010, 398, 265–294 CrossRef CAS.
- H. Lu, H. Zhang, K. Chingin, J. Xiong, X. Fang and H. Chen, Ambient mass spectrometry for food science and industry, TrAC, Trends Anal. Chem., 2018, 107, 99–115 CrossRef CAS.
- K. Losso, K. B. Bec, S. Mayr, J. Grabska, S. Stuppner, M. Jones, T. Jakschitz, M. Rainer, G. K. Bonn and C. W. Huck, Rapid discrimination of Curcuma longa and Curcuma xanthorrhiza using Direct Analysis in Real Time Mass Spectrometry and Near Infrared Spectroscopy, Spectrochim. Acta, Part A, 2022, 265, 120347 CrossRef CAS.
- S. Huber, K. Losso, G. K. Bonn and M. Rainer, Rapid quantification of cannabidiol from oils by direct analysis in real time mass spectrometry, Anal. Methods, 2022, 14, 3875–3880 RSC.
- L.-P. Li, B.-S. Feng, J.-W. Yang, C.-L. Chang, Y. Bai and H.-W. Liu, Applications of ambient mass spectrometry in high-throughput screening, Analyst, 2013, 138, 3097–3103 RSC.
- T. Schlappack, C. Kappacher, M. Rainer, C. W. Huck and G. K. Bonn, New sensitive ambient mass spectrometric method combined with chemometric modelling for the analysis of Equisetum palustre L. contaminations in the traditional herb Equiseti herba, J. Appl. Res. Med. Aromat. Plants, 2022, 30, 100396 CAS.
- C. Pasquini, Near infrared spectroscopy: A mature analytical technique with new perspectives – A review, Anal. Chim. Acta, 2018, 1026, 8–36 CrossRef CAS.
- H. Lin and Y. Ying, Theory and application of near infrared spectroscopy in assessment of fruit quality: a review, Sens. Instrum. Food Qual. Saf., 2009, 3, 130–141 CrossRef.
- C. de Bleye, P.-F. Chavez, J. Mantanus, R. Marini, P. Hubert, E. Rozet and E. Ziemons, Critical review of near-infrared spectroscopic methods validations in pharmaceutical applications, J. Pharm. Biomed. Anal., 2012, 69, 125–132 CrossRef CAS.
- C. Kappacher, M. Neurauter, M. Rainer, G. K. Bonn and C. W. Huck, Innovative Combination of Dispersive Solid Phase Extraction Followed by NIR-Detection and Multivariate Data Analysis for Prediction of Total Polyphenolic Content, Molecules, 2021, 26, 4807 CrossRef CAS.
- C. Kappacher, B. Trübenbacher, K. Losso, M. Rainer, G. K. Bonn and C. W. Huck, Portable vs. Benchtop NIR-Sensor Technology for Classification and Quality Evaluation of Black Truffle, Molecules, 2022, 27, 589 CrossRef CAS PubMed.
- H. W. Siesler, S. Kawata, H. M. Heise and Y. Ozaki, Near-Infrared Spectroscopy. Principles, Instruments, Applications, Wiley-VCH, Weinheim, 1st edn, 2008 Search PubMed.
- M. Ferrari, L. Mottola and V. Quaresima, Principles, techniques, and limitations of near infrared spectroscopy, Can. J. Appl. Physiol., 2004, 29, 463–487 CrossRef.
- J. Henschel, A. Schriewer, H. Hayen and J. Wen, Analysis of Artificial Sweeteners by HILIC-MS Method, July/ 2016, https://www.gimitec.com/file/HILICON_LCGC_Artifical_Sweeteners.pdf, accessed 25 April 2023 Search PubMed.
- L. A. Berrueta, R. M. Alonso-Salces and K. Héberger, Supervised pattern recognition in food analysis, J. Chromatogr. A, 2007, 1158, 196–214 CrossRef CAS.
- A. J. Izenman, Modern Multivariate Statistical Techniques, Springer, New York, NY, 2013 Search PubMed.
- A. J. Izenman, in Modern Multivariate Statistical Techniques, ed. A. J. Izenman, Springer, New York, NY, 2013, pp. 237–280 Search PubMed.
- J. Andrade-Garda, Basic Chemometric Techniques in Atomic Spectroscopy, Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- J.-H. Cheng and D.-W. Sun, Partial Least Squares Regression (PLSR) Applied to NIR and HSI Spectral Data Modeling to Predict Chemical Properties of Fish Muscle, Food Eng. Rev., 2017, 9, 36–49 CrossRef CAS.
- S. Mayr, K. B. Beć, J. Grabska, E. Schneckenreiter and C. W. Huck, Near-infrared spectroscopy in quality control of Piper nigrum: a comparison of performance of benchtop and handheld spectrometers, Talanta, 2021, 223, 121809 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ay00380a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |