
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of volatile short-chain fatty acids in the gas phase using secondary electrospray ionization coupled to mass spectrometry†
Cedric
Wüthrich
a,
Zhiyuan
Fan
 a,
Guy
Vergères
b,
Fabian
Wahl
b,
Renato
Zenobi
*a and
Stamatios
Giannoukos
*a
a,
Guy
Vergères
b,
Fabian
Wahl
b,
Renato
Zenobi
*a and
Stamatios
Giannoukos
*a
aDepartment of Chemistry and Applied Biosciences, ETHZ, Zurich, Switzerland. E-mail: stamatios.giannoukos@org.chem.ethz.ch; renato.zenobi@org.chem.ethz.ch
bFood Microbial Systems Research Division, Agroscope, Bern, Switzerland
First published on 23rd December 2022
Abstract
Quantification of metabolites present within exhaled breath is a major challenge for on-line breath analysis. It is also important for gauging the analytical performance, accuracy, reproducibility, reliability, and stability of the measuring technology. Short-chain fatty acids (SCFAs) are of high interest for nutrition and health. Their quantification enables a deep mechanistic understanding of a wide range of biological processes and metabolic pathways, while their high volatility makes them an attractive target for breath analysis. This article reports, for the first time, the development and testing of a modular, dynamic vapor generator for the qualitative and quantitative analysis of volatile SCFAs in the gaseous phase using a secondary electrospray ionization (SESI) source coupled to a high-resolution mass spectrometer. Representative compounds tested included acetic acid, propionic acid, butyric acid, pentanoic acid and hexanoic acid. Gas-phase experiments were performed both in dry and humid (95% relative humidity) conditions from ppt to low ppb concentrations. The results obtained exhibited excellent linearity within the examined concentration range, low limits of detection and quantification down to the lower ppt area. Mixture effects were also investigated and are presented.
1. Introduction
Short-chain fatty acids (SCFAs) are carboxylic acids containing one to six carbon atoms, that play a major role in human metabolism and have many biological functions (e.g. affecting colonic health, function, and structure, affecting colonic and intracellular pH, regulating cell proliferation and blood flow).1,2 They are a product of bacterial food fermentation in the colon and associated with gut health.1,2 Their total concentration in the human colon depends on diet and ranges from 70–140 mmol kg−1 in the proximal and 20–70 mmol kg−1 in the distal colon.2,3 SCFAs have been considered useful biomarkers for the diagnosis and mechanistic understanding of a wide selection of metabolic diseases (e.g. diversion colitis, ulcerative colitis, radiation proctitis, pouchitis, antibiotic-associated diarrhea, as well as colon cancer).4–7Recent advances in chemical sensing tools have facilitated the analysis, screening and decoding of volatile metabolites that are produced by complex and interacting biochemical processes that evolve continuously within the human body. Existing standard technologies for the qualitative investigation and quantification of SCFAs in fecal samples,8 plasma,9 or serum10 include gas chromatography (GC) or liquid chromatography (LC) coupled with mass spectrometry (MS). These methods are well-established; however, they often require invasive sampling, are not on-line, and include demanding sample preparation steps.
On the contrary, exhaled breath analysis is inherently non-invasive, requires minimum or no sample preparation, and provides on-line measurements in real-time.11 Due to these advantages, the research on breath metabolomics has gained momentum in recent years. The presence of SCFAs in human breath has been confirmed,12 but quantification studies are limited, mainly due to the lack of standardization and limited repeatability.11 A recent study13 reported the measurement of carboxylic acids in human breath up to C14 using atmospheric pressure ionization mass spectrometry (API-MS) and estimated the breath concentrations of C3–C6 fatty acids (in a semi-quantitative way) to be on the 100 ppt level which agrees with values reported in the human blood. Lee et al.14 optimized the operational parameters of secondary electrospray ionization (SESI) coupled to a mass spectrometer to measure volatile SCFAs in the headspace gas of gut microbial cultures. Recently, Meurs et al.15 calibrated proton transfer reaction mass spectrometry (PTR-MS) with gas standards of SCFA in the concentration range from 4 to 100 ppb and benchmarked the performance of the PTR-MS against a GC-MS obtaining satisfactory linearity and repeatability. Based on their measurements, the C2–C4 SCFAs exist in the low ppb range in the human breath.
In this article, the development, testing and integration of a dynamic vapor generator with an on-line SESI-Orbitrap mass spectrometer system was demonstrated. Test compounds included volatile C2 to C6 SCFAs. The described vapor generator is a modular system that operates at various levels of relative humidity (RH) and generates gaseous standards of either a single compound or mixtures in the concentration range from low ppt to high ppm in a controllable, repeatable and consistent way. The vapor generator achieves similar conditions to exhaled breath (output total flow per min, humidity, temperature) allowing quantitative investigations of breath metabolites. This paper reports and compares the analytical characteristics (i.e., linear dynamic range, limit of detection and limit of quantification) and performance of this on-line SESI-high resolution mass spectrometry system for the examined SCFA at 0% and 95% RH levels in the positive ion mode.
2. Experimental part
This work focuses on the qualitative detection and quantification of volatile SCFAs using an on-line SESI-high resolution mass spectrometry system. The SCFAs selected for this study and their relevant physical properties (e.g., vapor pressure, Henry's solubility constant, etc.) are presented in Table 1.| Compound | Formula | CAS number | Molecular weight (g mol−1) | Vapour pressure (kPa) at 20 °C | H cp (mol (m3 Pa)) at 37 °C |
|---|---|---|---|---|---|
| Acetic acid | C2H4O2 | 64-19-7 | 60.052 | 1.55 | 17.90 |
| Propionic acid | C3H6O2 | 79-09-4 | 74.079 | 0.32 | 23.30 |
| Butyric acid | C4H8O2 | 107-92-6 | 88.106 | 0.11 | 18.08 |
| Pentanoic acid | C5H10O2 | 109-52-4 | 102.133 | 0.02 | 9.06 |
| Hexanoic acid | C6H12O2 | 142-62-1 | 116.160 | 0.018 | 5.78 |
2.1 Reagents
Certified analytical standards of propanoic acid (purity ≥ 99.8%), butanoic acid (purity ≥ 99.5%), pentanoic acid (purity ≥ 99.8%), and hexanoic acid (purity ≥ 99%) were purchased from Supelco. Acetic acid (purity ≥ 99.99%) was bought from Sigma Aldrich. All reagents were provided in liquid form and stored in the fridge at 4 °C until their use. The electrospray solution was prepared from Optima LC-MS grade water purchased from Fisher Scientific, and formic acid (99.5% purity) was obtained from VWR Chemicals.2.2 Experimental setup
Experiments were carried out using a built-in-house vapor generator described previously.16,17 The operating principle of the vapor generator is based on the controlled evaporation of volatile or semi-volatile organic compounds and their diffusion into a carrier gas stream (Fig. 1).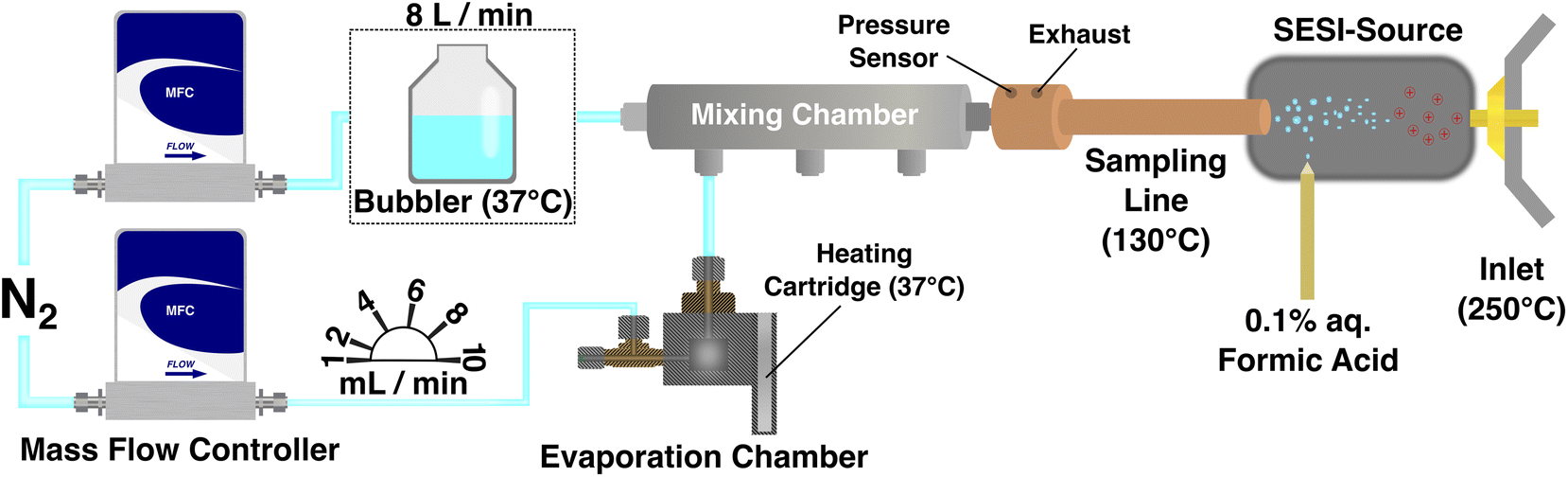 | ||
| Fig. 1 Schematic representation of the vapor generator. One mass flow controller (MFC) (the upper one in the scheme) steers a large dilution flow of 8 Liters per minute, which could be additionally humidified by a bubbler. The lower mass flow controller sets small flows from 1 to 10 mL min−1, which subsequently pass through the heated evaporation chamber. This stream takes up the gaseous molecules within the chamber and is further diluted in the mixing chamber. The gas stream then enters the ion source for ionization and is afterward analyzed within the mass spectrometer. | ||
The main components of the vapor generator are (a) a mixing chamber, (b) three individual temperature-controlled evaporation chambers, in which liquid analytes are introduced through a side injection port closed by a septum, (c) four mass flow controllers, (d) an automation platform controlled by a LabVIEW software. The vapor generator is an automated modular system allowing the controllable production of single or multi-component gas standards in either a periodic or a dynamic way. When measurements require humid conditions, a bubbler (indicated by the dashed line at the upper part of Fig. 1) kept at 37 °C can be used in conjunction with the vapor generator to produce a stable humid carrier gas flow. A schematic diagram of the modular vapor generator including only one evaporation chamber is shown in Fig. 1. For the work described in this paper, two different relative humidity levels were investigated (0% and 95%).
The mixing chamber and evaporation chambers were manufactured using grade 303 stainless steel. The mixing chamber is a cylindrically shaped chamber with a length of 80 mm, an inner diameter of 20 mm, and an outer diameter of 25 mm. The input and output ports at the ends are two 6.35 mm Swagelok stainless steel fitting unions. On the peripheral side of the mixing chamber, three 3.18 mm Swagelok fittings are positioned which could be individually connected with a separate evaporation chamber depending on measurement requirements (single or multiple analyte measurements). As such, the mixing chamber of the current version of the vapor generator has connections for three evaporation chambers. The evaporation chambers are cylindrical chambers with an inner diameter of 12 mm, an inner length of 20 mm whereas the outer diameter of the chamber is 30 mm. On the top side of the chamber, there is a 3.18 Swagelok fitting union, and on the peripheral side a Swagelok union tee (1 port connects with a mass flow controller and the other port is sealed with a thermoresistant septum (Sigma Aldrich) and is used for the introduction of the liquid analyte in the evaporation chamber), and on the opposite side, there is an input for a heating cartridge purchased from RS components International. The heater is controlled by a temperature controller with a type K sensor. The temperature range can be set from 0 to 150 °C with 0.1 °C increments and is monitored continuously by a thermocouple. During measurements, all evaporation chambers were kept at 37 °C to simulate human body temperature and the liquid analytes under investigation were loaded into the chamber using a Hamilton syringe. The carrier gas flows passing through all chambers were controlled by mass flow controllers (type GE50A) purchased by MKS Instruments UK Ltd., United Kingdom. The controller connected to the mixing chamber had a flow range between 0.5 and 10 L min−1 and the ones connected to the evaporation chambers had a flow range between 0.1 and 10 mL min−1. The controllers were driven by an automation platform operating through a custom-made software on LabView, which enabled their simultaneous control and the sequence generation of time steps and gas flows for each individual controller. All controllers were calibrated for N2 gas and their input pressure was kept stable at 1.5 bar constantly. The relative humidity was measured at two points (at both ends of the mixing chamber) using two separate sensors at the ends. The first one was an ALMEMO 2590-2A/-4AS (Ahlborn Mess-und Regelungstechnik GmbH, Germany) and the second was a digital humidity and temperature sensor (model SHT45) purchased from Sensirion, Switzerland.
On-line analysis of the produced gas standards was done using a commercial SESI source (Fossil Ion Tech, Spain) coupled to a Q-Exactive Plus Orbitrap (Thermo Fischer, Germany) mass spectrometer. A flow meter (EXHALION, Fossil Ion Tech, Spain) connected at the front-end part of the sample transfer line was used to measure the sample flow rate and volume. All experiments were done at 8 L min−1 corresponding to the average human exhaled breath flow. Gas samples were introduced into the SESI source using a custom-made adaptor attached to the sampling line of the SESI source. The sampling line was constantly heated at 130 °C and the ionization chamber was kept at 90 °C. The electrospray solution was a 0.1% aqueous formic acid solution passing through a nano-electrospray capillary (inner diameter of 20 μm and an outer diameter of 365 μm, Fossil Ion Tech, Spain) with an overpressure of 0.8 bar. In the ionization chamber, the charged electrospray droplets were interacting with the gaseous sample for charge transfer prior to introduction into the mass spectrometer for analysis. Sheath and auxiliary gas values were set at 15 psi and 2 a.u. respectively. The electrospray solution voltage was +3.5 kV and the inlet capillary was heated to 250 °C. The automatic gain control (AGC) target was adjusted to 106 and the maximum injection time was set to 500 ms. Scans were recorded with a m/z range of 50–500 and with a mass resolution of 140![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
2.3 Sample preparation
Gaseous standards of individual compounds were prepared using the built-in-house vapor generator described in Section 2.2. To achieve the generation of gaseous standards at low concentrations, target analytes in the liquid phase and at specific concentrations were introduced into the evaporation chambers. Liquid stock solutions of the tested SCFAs were prepared individually in LC-MS grade water at the following concentrations (mol L−1): C2 (0.0035, 0.0347, 0.3409), C3 (0.0003, 0.0027, 0.0266), C4 (0.0002, 0.0022, 0.0216), C5 (0.0002, 0.0018, 0.0182), and C6 (0.0016, 0.0156). For each concentration level, 20 μL of the liquid stock solution were introduced into a purged chamber. Subsequently, four measurements were conducted using different flow programs. A decreasing, increasing, random 1 and random 2 N2 flow program passing through the evaporation chambers were tested as shown in Fig. 2. Between each measurement, the chamber was doped with 5 μL of the corresponding stock solution. The flow of the N2 gas passing through the mixing chamber was kept at 8 L min−1 constantly to simulate the rate of human exhalation. The duration of each step was 90 seconds, whereas the first and last segment (background flow of N2 passing through the mixing chamber) lasted for 120 s. After each use, the chambers were washed multiple times and ultra-sonicated with Milli-Q water for 15 minutes and then dried in an oven at 50 °C overnight. | ||
| Fig. 2 (a) Stepwise decrease, (b) stepwise increase, (c) random 1 and (d) random 2 flow profiles passing through the evaporation chambers tested for each individual SCFA. | ||
All experiments were conducted under two conditions, dry and humid (95% RH) in the positive ion mode allowing a comparison of the analytical performance of instrumentation under these two different conditions. The gas phase concentration of each tested SCFA in the evaporation chambers was calculated based on Henry's law.18 The Henry's law solubility constants of the tested SCFAs were obtained by Johnson et al.19 for C2, and the values measured by Khan et al.20 for C3–C6. The Henry's solubility constants for C2, C5 and C6 were measured in the range from 5 to 35 °C and were extrapolated to 37 °C. For C3 and C4, Henry's constants were measured at 25 °C, and were extrapolated to 37 °C using the enthalpy of dissolution reported by Abraham.21 The output of the evaporation chambers was then diluted by the N2 carrier gas flow passing through the mixing chamber.
To examine mixture effects in the gas phase, individual liquid stock solutions for C2, C3 and C4 were prepared at the following concentrations: 0.035, 0.027, and 0.022 mol L−1 respectively. 20 μL of each stock solution were then introduced into three separate evaporation chambers and a fully randomized N2 flow program per chamber was applied: 6, 2, 4, 10, 1, 8 mL min−1 for C2, 1, 8, 2, 10, 4, 6 mL min−1 for C3 and 2, 1, 8, 6, 10, 4 mL min−1 for C4. The N2 flow passing through the mixing chamber was kept again at 8 L min−1 throughout the whole duration of the measurement.
2.4 Data processing and analysis
Measurements and their corresponding mass spectra were processed with a custom-written Python script (v 3.7), which used the data files in the mzML-format22 converted from the proprietary RAW-format through ProteoWizard23 as input. In the first step, scan averaging over all measurements was performed to yield a composite mass spectrum. With this mass spectrum, peak picking was performed, employing a height filter set at an intensity of 104 a.u. The widths of the individual peaks were determined at 90% of the peak height. The time trace of each peak within the individual measurement was subsequently obtained by integration within the extension of the individual peak width obtained from the composite mass spectrum of the signal.To obtain a calibration curve, the time traces of each compound were further processed. In each time trace, the scans corresponding to flow through the evaporation chamber were identified using the ruptures package,24 which helps to identify change points using the L2 regularization model. Within each pulse in the time trace corresponding to the evaporation chamber activity, the last 15 scans were averaged, thus obtaining the response to the applied concentration. Each pulse was then compared to the limit of detection (LOD) and limit of quantification (LOQ), which were calculated from the baseline scans. The criteria for a pulse being higher than the limit of detection was that the intensity of the pulse had a higher intensity than the average of the baseline plus three times the standard error of the baseline. For the limit of quantification, ten times the standard error was used. It is important to note that the concentrations obtained for these limits were calculated by applying the inverse calibration curve on the corresponding intensity values of these limits. To calculate the regression curves, the baseline subtracted averages of the different flow program measurements were taken. To achieve optimal coverage over the different concentration ranges, a linear regression was drawn through the double logarithmically transformed data.
Accuracy estimation for the individual compounds and RH condition was determined by the exclusion of one data point per technical replicate for each stock solution. A linear regression was fitted over the log-transformed remaining data and the expected value for the omitted data point was compared with the actual value. Reproducibility was assessed through the determination of the coefficients of variation calculated from the technical replicates.
3. Results and discussion
3.1 Individual short-chain fatty acids experiments
 | ||
| Fig. 3 Calibration curves obtained for (a) acetic acid, (b) propionic acid, (c) butyric acid, (d) pentanoic acid and (e) hexanoic acid in the gas phase at 0% RH. Each point of the calibration curves corresponds to the average concentration at a specific flow of the four technical replicate measurements (four different flow programs passing through the evaporation chambers). The error bars correspond to the standard error per data point. | ||
000
| Compound | Exact m/z [M + H]+ | Measured m/z [M + H]+ | Mass error (ppm) |
|---|---|---|---|
| Acetic acid | 61.0284 | 61.0285 | 1.6 |
| Propionic acid | 75.0441 | 75.0440 | −1.3 |
| Butyric acid | 89.0597 | 89.0595 | −2.3 |
| Pentanoic acid | 103.0754 | 103.0751 | −2.9 |
| Hexanoic acid | 117.0910 | 117.0907 | −2.5 |
Calibration curves were obtained over three orders of magnitude concentration for acetic, propionic, butyric, and pentanoic acid, except for hexanoic acid which showed a linear regression over two orders of magnitude. No stable signal was observed for a lower stock concentration for this compound compared to the other compounds. In terms of sensitivity (slope of the linear fit), hexanoic acid showed the steepest slope, followed by acetic acid. For the other three acids, a similar regression slope was obtained. The R2 values obtained were in the range from 0.97 to 0.99. The corresponding LODs and LOQs are reported in Table 3 with average values ranging from low to a few hundreds of ppt's respectively. The calibration curves and the corresponding linearities (R2 values) obtained for each stock solution for the individual analytes under dry conditions are shown in the ESI, Fig. S2.†
| Compound | 0% RH | 95% RH | ||||
|---|---|---|---|---|---|---|
| R 2 | LOD (ppt) | LOQ (ppt) | R 2 | LOD (ppt) | LOQ (ppt) | |
| Acetic acid | 0.9969 | 420.63 | 507.36 | 0.9386 | 157.42 | 261.25 |
| Propionic acid | 0.9789 | 5.78 | 7.29 | 0.9915 | 3.40 | 5.27 |
| Butyric acid | 0.9721 | 1.85 | 2.32 | 0.9838 | 0.71 | 1.00 |
| Pentanoic acid | 0.9702 | 2.53 | 3.16 | 0.9890 | 21.89 | 28.13 |
| Hexanoic acid | 0.9971 | 40.63 | 47.42 | 0.9802 | 53.67 | 66.81 |
 | ||
| Fig. 4 Calibration curves obtained for (a) acetic acid, (b) propionic acid, (c) butyric acid, (d) pentanoic acid and (e) hexanoic acid in the gas phase at 95% RH. Each point of the calibration curves corresponds to the average concentration at a specific flow of the four technical replicate measurements (four different flow programs passing through the evaporation chambers). The error bars correspond to the standard error per data point. | ||
In terms of analytical performance, the fits of the double logarithmically transformed data show satisfactory linearity with the R2 of the fits, possessing values from 0.9802 to 0.9915. A notable exception was the fit obtained for acetic acid, for which an R2 of 0.9386 was obtained. The lower performance relates back to the high variance and stability of the time trace over the pulse experiments. The calibration curves and the corresponding linearities (R2 values) obtained for each stock solution for the individual analytes under humid conditions are shown in the ESI, Fig. S3.†
3.2 Multi-compound detection experiments
An experiment with three evaporation chambers and acetic, propionic, as well as butyric acid, was performed to show the capability of generating a multi-component gas standard mixture. This experiment aimed to examine a fully randomized flow rate experiment and whether the obtained calibration curves corresponded to single compound standard experiments. Experiments were performed as described in Section 2.4. Gas standards of acetic, propionic, and butyric acid were prepared at 0% RH and the output of the mixing chamber was directly connected with the inlet of the SESI system for sampling and analysis. The acquired time trends for acetic, propionic, and butyric acid are shown in Fig. 5a whereas the corresponding calibration curves obtained on a linear scale under these conditions are shown in Fig. 5b. | ||
| Fig. 5 (a) Time traces of acetic acid, propanoic acid and butyric acid, (b) calibration curves of the individual fatty acids, when a mixture of all was used. The curves are given on a linear scale. | ||
Compared to the individual calibration curves reported in Fig. S2,† the slopes are different, but the magnitudes are on the same level. The sensitivity behavior of the SCFAs (Fig. 5b) compared to each other is the same in the mixture as in a single pure compound calibration (butyric acid > propionic acid > acetic acid). The individual slope differences likely arise from instrumental drift; therefore, a proper calibration must be performed on a daily basis.
3.3 Evaluation of the method
The performance of the SESI system was evaluated by addressing the following analytical criteria: (a) linear dynamic range within the examined concentration area, (b) LODs, and (c) LOQs in dry and humid conditions. Table 3 summarizes the obtained results. In overall, calculated LODs and LOQs are in the range of low to medium ppt concentrations. Acetic acid has the highest LOD and LOQ values for both conditions, whereas butyric acid exhibits the lowest. LODs and LOQs are also graphically indicated on the calibration curves in Fig. S4 and S5 (ESI†).In Table 4, the accuracy and reproducibility estimates are given for each compound and RH condition. The estimated accuracy remains for most compounds within ±15% of the expected value apart from butanoic acid, which shows a slightly higher deviation under dry and acetic acid under humid conditions. The variation between technical replicates is low with the trend of humid conditions being less precise, although almost with a negligible difference.
| Compound | 0% RH | 95% RH | ||
|---|---|---|---|---|
| Accuracy (%) | Reproducibility (%) | Accuracy (%) | Reproducibility (%) | |
| Acetic acid | 94–107 | 4.35 | 99–118 | 7.78 |
| Propanoic acid | 90–109 | 2.34 | 96–107 | 4.36 |
| Butanoic acid | 88–116 | 3.93 | 97–108 | 3.72 |
| Pentanoic acid | 97–112 | 4.50 | 95–106 | 4.49 |
| Hexanoic acid | 94–105 | 5.26 | 90–103 | 6.06 |
4. Conclusions
For the first time, the integration of a dynamic vapor generator, capable of producing gaseous standards in a precise and reproducible way, with a SESI source coupled to a high-resolution mass spectrometer for the qualitative and quantitative analysis of volatile SCFAs was demonstrated. The analytical performance of this approach was investigated by addressing essential analytical criteria such as LODs, LOQs of individual compounds, and the response in multi-component mixtures under dry and humid conditions in the full mass range m/z from 50 to 500. Under both conditions, gas concentrations down to low ppt were successfully recorded, thus exhibiting the system's potential to serve as a quantification tool for SCFAs in exhaled breath. Even though lower LODs and LOQs were calculated for humid conditions, the generation of standards close to these limits partially failed as signal instability increased. The slopes of all calibration curves in both dry and humid conditions became steeper (increased) from propionic to hexanoic acid, inversely proportional to the decreasing Henry's solubility constant. However, this was not the case for acetic acid in dry conditions. Hydrophobicity seemed to play a major role in determining the slope of the calibration curves, as fatty acids with a longer aliphatic tail showed a larger dependence on a change in concentration.The methodology reported here can be used in human studies to quantitatively evaluate how diet quality and the health status of the consumer modulate the production of SCFAs by the human organism.
Data availability
The original data used in this publication are made available in a curated data archive at ETH Zürich (https://www.research-collection. ethz. ch) under the DOI: 10.3929/ethz-b-000578573.Author contributions
SG designed and fabricated the vapor generator. CW, ZF and SG designed and performed the experiments. CW analysed the data. CW, ZF, GV, FW, RZ and SG interpreted the compiled results. SG, CW and ZF prepared the manuscript. All authors contributed to the discussion and revision of the paper.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The research leading to these results has received funding from the NutriExhalomics project (grant agreement No. 15668), the Ascher Trust, and the Jubiläumsstiftung der Von Roll Holding AG. The authors gratefully acknowledge Marco Zesiger and Christian Marro from the mechanical workshop of the Department of Chemistry and Applied Biosciences for the technical support of this work. The authors thank Jérôme Käslin for the valuable discussions and input on data processing.References
- S. Cook, Review Article: Short Chain Fatty Acids in Health and Disease, Aliment. Pharmacol. Ther., 1998, 12(6), 499–507 CrossRef CAS PubMed
.
- J. M. W. Wong, R. de Souza, C. W. C. Kendall, A. Emam and D. J. A. Jenkins, Colonic Health: Fermentation and Short Chain Fatty Acids, J. Clin. Gastroenterol., 2006, 40(3), 235–243 CrossRef CAS PubMed
- D. L. Topping and P. M. Clifton, Short-Chain Fatty Acids and Human Colonic Function: Roles of Resistant Starch and Nonstarch Polysaccharides, Physiol. Rev., 2001, 81(3), 1031–1064 CrossRef CAS PubMed
- D. Friedel and G. M. Levine, Effect of Short-Chain Fatty Acids on Colonic Function and Structure, J. Parenter. Enteral Nutr., 1992, 16(1), 1–4 CrossRef CAS
- P. G. Farup, K. Rudi and K. Hestad, Faecal Short-Chain Fatty Acids - A Diagnostic Biomarker for Irritable Bowel Syndrome?, BMC Gastroenterol., 2016, 16(1), 51 CrossRef PubMed
- M. Müller, M. A. G. Hernández, G. H. Goossens, D. Reijnders, J. J. Holst, J. W. E. Jocken, H. van Eijk, E. E. Canfora and E. E. Blaak, Circulating but not Faecal Short-Chain Fatty Acids Are Related to Insulin Sensitivity, Lipolysis and GLP-1 Concentrations in Humans, Sci. Rep., 2019, 9(1), 12515 CrossRef
- D. Smith, K. Sovová, K. Dryahina, T. Doušová, P. Dřevínek and P. Španěl, Breath Concentration of Acetic Acid Vapour is Elevated in Patients with Cystic Fibrosis, J. Breath Res., 2016, 10(2), 021002 CrossRef PubMed
- M. Primec, D. Mičetić-Turk and T. Langerholc, Analysis of Short-Chain Fatty Acids in Human Feces: A Scoping Review, Anal. Biochem., 2017, 526, 9–21 CrossRef CAS
- C. Shin, Y. Lim, H. Lim and T.-B. Ahn, Plasma Short-Chain Fatty Acids in Patients With Parkinson's Disease, Mov. Disord., 2020, 35(6), 1021–1027 CrossRef CAS PubMed
- J. Tarini and T. M. S. Wolever, The Fermentable Fibre Inulin Increases Postprandial Serum Short-Chain Fatty Acids and Reduces Free-Fatty Acids and Ghrelin in Healthy Subjects, Appl. Physiol., Nutr., Metab., 2010, 35(1), 9–16 CrossRef CAS
- T. Bruderer, T. Gaisl, M. T. Gaugg, N. Nowak, B. Streckenbach, S. Müller, A. Moeller, M. Kohler and R. Zenobi, On-Line Analysis of Exhaled Breath, Chem. Rev., 2019, 119(19), 10803–10828 CrossRef CAS PubMed
- B. de Lacy Costello, A. Amann, H. Al-Kateb, C. Flynn, W. Filipiak, T. Khalid, D. Osborne and N. M. Ratcliffe, A Review of the Volatiles from the Healthy Human Body, J. Breath Res., 2014, 8(1), 014001 CrossRef CAS PubMed
- P. Martínez-Lozano and J. Fernández de la Mora, Direct Analysis of Fatty Acid Vapors in Breath by Electrospray Ionization and Atmospheric Pressure Ionization-Mass Spectrometry, Anal. Chem., 2008, 80(21), 8210–8215 CrossRef
- J. H. J. Lee and J. Zhu, Optimizing Secondary Electrospray Ionization High-Resolution Mass Spectrometry (SESI-HRMS) for the Analysis of Volatile Fatty Acids from Gut Microbiome, Metabolites, 2020, 10(9), 351 CrossRef CAS
- J. Meurs, E. Sakkoula and S. Cristescu, Real-Time Non-Invasive Monitoring of Short-Chain Fatty Acids in Exhaled Breath, Front. Chem., 2022, 10, 853541 CrossRef CAS PubMed
- M. Statheropoulos, G. C. Pallis, K. Mikedi, S. Giannoukos, A. Agapiou, A. Pappa, A. Cole, W. Vautz and C. L. P. Thomas, Dynamic Vapor Generator That Simulates Transient Odor Emissions of Victims Entrapped in the Voids of Collapsed Buildings, Anal. Chem., 2014, 86(8), 3887–3894 CrossRef CAS PubMed
- S. Giannoukos, A. Marshall, S. Taylor and J. Smith, Molecular Communication over Gas Stream Channels using Portable Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2017, 28(11), 2371–2383 CrossRef CAS PubMed
- R. Sander, Compilation of Henry's law Constants (Version 4.0) for Water as Solvent, Atmos. Chem. Phys., 2015, 15(8), 4399–4981 CrossRef CAS
- B. J. Johnson, E. A. Betterton and D. Craig, Henry's Law Coefficients of Formic and Acetic Acids, J. Atmos. Chem., 1996, 24(2), 113–119 CrossRef CAS
- I. Khan and P. Brimblecombe, Henry's Law Constants of Low Molecular Weight (<130) Organic Acids, J. Aerosol Sci., 1992, 23, 897–900 CrossRef
- M. H. Abraham, Thermodynamics of Solution of Homologous Series of Solutes in Water, J. Chem. Soc., Faraday Trans., 1984, 80(1), 153–181 RSC
- L. Martens, M. Chambers, M. Sturm, D. Kessner, F. Levander, J. Shofstahl, W. H. Tang, A. Römpp, S. Neumann, A. D. Pizarro, L. Montecchi-Palazzi, N. Tasman, M. Coleman, F. Reisinger, P. Souda, H. Hermjakob, P. A. Binz and E. W. Deutsch, mzML—a Community Standard for Mass Spectrometry Data, Mol. Cell. Proteomics, 2011, 10(1), R000133 CrossRef PubMed
- M. C. Chambers, B. MacLean, R. Burke, D. Amodei, D. L. Ruderman, S. Neumann, L. Gatto, B. Fischer, B. Pratt, J. Egertson, K. Hoff, D. Kessner, N. Tasman, N. Shulman, B. Frewen, T. A. Baker, M. Y. Brusniak, C. Paulse, D. Creasy, L. Flashner, K. Kani, C. Moulding, S. L. Seymour, L. M. Nuwaysir, B. Lefebvre, F. Kuhlmann, J. Roark, P. Rainer, S. Detlev, T. Hemenway, A. Huhmer, J. Langridge, B. Connolly, T. Chadick, K. Holly, J. Eckels, E. W. Deutsch, R. L. Moritz, J. E. Katz, D. B. Agus, M. MacCoss, D. L. Tabb and P. Mallick, A cross-platform toolkit for mass spectrometry and proteomics, Nat. Biotechnol., 2012, 30, 918–920 CrossRef CAS PubMed
- C. Truong, L. Oudre and N. Vayatis, Selective review of offline change point detection methods, Signal Processing, 2020, 167, 107299 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01778d |
| This journal is © The Royal Society of Chemistry 2023 |