
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDichloromethane replacement: towards greener chromatography via Kirkwood–Buff integrals†
Julie
Lynch
 *a,
James
Sherwood
a,
C. Rob
McElroy
a,
Jane
Murray
b and
Seishi
Shimizu
*c
*a,
James
Sherwood
a,
C. Rob
McElroy
a,
Jane
Murray
b and
Seishi
Shimizu
*c
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, North Yorkshire YO10 5DD, UK. E-mail: jl3362@york.ac.uk
bMerck KGaA, Frankfurter Strasse 250, 64293 Darmstadt, Germany
cYork Structural Biology Laboratory, Department of Chemistry, University of York, York, YO10 5DD, UK
First published on 13th January 2023
Abstract
Dichloromethane (DCM) is a useful and advantageous solvent used in pharmaceutical development due to its low cost, miscibility with other organic solvents, high volatility, and ability to solubilize drug molecules of variable polarities and functionalities. Despite this favourable behaviour, efforts to identify safer and more sustainable alternatives to hazardous, halogenated solvents is imperative to the expansion of green chemistry. In this work, bio-derived esters tert-butyl acetate, sec-butyl acetate, ethyl isobutyrate, and methyl pivalate are experimentally identified as safe and sustainable alternatives to directly replace DCM within thin-layer chromatography (TLC) in the analysis of small, common drug molecules. To elucidate the intermolecular interactions influencing retardation factors (Rf) a statistical thermodynamic framework, which quantifies the driving molecular interactions that yield empirical TLC measurements, is presented. Within this framework, we are able to deduce Rf dependence on polar eluent concentration, in the presence of a low-polar mediating solvent, between the stationary and mobile phases. The strength of competitive analyte–eluent (and analyte–solvent interactions) are quantified through Kirkwood–Buff integrals (KBIs); resulting KBI terms at the dilute eluent limit provide a theoretical foundation for the observed suitability of alternative green solvents for the replacement of dichloromethane in TLC.
1 Introduction
Efforts to replace halogenated organic solvents with safer and environmentally benign options are a challenging yet pertinent issue. Predominantly, identifying replacements for the chlorocarbon dichloromethane (DCM), or methylene chloride is noted in numerous green solvent selection guides,1,2 reviews,3 and surveys4–7 as a difficult task due to its both useful and distinctive solvation behaviours, particularly when utilised in pharmaceutical chemistry. Adhering to the Twelve Principles of Green Chemistry8 and recommendations by the ACS Green Chemistry Institute Pharmaceutical Roundtable,9 identifying operational replacements for halogenated solvents are of immediate importance commencing at the research stage of drug development.Dichloromethane is a suspected category 2 human carcinogen10,11 and is recognised for its acute and chronic toxicity leading to negative effects on the respiratory, nervous, and reproductive systems. This has led to conditional restrictions under the European Union REACH regulation.12 Moreover, large-scale use of DCM requires industrial scrubbing systems wherein waste streams are rid of halogenated volatile organic compounds (VOCs). DCM is currently categorised as a very short-lived substance (VSLS) thus, is not regulated under the Montreal Protocol on Substances that Deplete the Ozone Layer.13 However, recent studies have demonstrated that the environmental release of DCM does, in fact, contribute to ozone depletion and is observably counteracting ozone repair directives.14,15
Despite its hazards, DCM is a valuable solvent in medicinal chemistry3,9 due to its ability so solubilize both polar and nonpolar molecules with varying functionalities, including heterocyclic compounds. It is non-flammable, inert, with high volatility and a low boiling point. These characteristics are advantageous within drug discovery, particularly for liquid chromatography where solvent use is high.16 When separating drug-like molecules, DCM is employed as a low-polarity eluent, which allows the chromatographic system to be tuned according to the analytes by use of additives with high polarity, traditionally, methanol (MeOH). The intrinsic and versatile solvation abilities of DCM, in tandem with its lower polarity and low cost, contribute to the uniqueness of this solvent making it very difficult to replace.
Advancements in identifying DCM replacements for chromatographic systems have used thin-layer chromatography (TLC) as an inexpensive, rapid, and facile chromatographic method to identify and separate analytes.4,6,7 These studies have explored a number of green solvents and solvent mixtures through their effectiveness at separating small, drug-like molecules. The most significant development to date is the commercially available 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 blend of ethyl acetate and ethanol (EtOAc–EtOH).4 This blend behaves instead as the polar component that, when mixed with heptane, works effectively at replacing the traditional DCM–MeOH binary eluent system. In this case, the hydrocarbon behaves as the non-polar component, tuning the polarity of its mixture with EtOAc–EtOH and resulting in a favourable range of retardation factors (Rf). Solvent replacement strategies require an understanding of solubility, usually provided by solubility parameters. Examples include the Hansen Solubility Parameters (HSPs)17 and the Kamlet–Taft (KT) parameters.18,19 These parameters are useful within general solvent replacement efforts, providing pertinent information describing solvent–solute interactions.20–23 However, within chromatographic applications the effects of the stationary phase on empirical results must also be considered.24
:1 blend of ethyl acetate and ethanol (EtOAc–EtOH).4 This blend behaves instead as the polar component that, when mixed with heptane, works effectively at replacing the traditional DCM–MeOH binary eluent system. In this case, the hydrocarbon behaves as the non-polar component, tuning the polarity of its mixture with EtOAc–EtOH and resulting in a favourable range of retardation factors (Rf). Solvent replacement strategies require an understanding of solubility, usually provided by solubility parameters. Examples include the Hansen Solubility Parameters (HSPs)17 and the Kamlet–Taft (KT) parameters.18,19 These parameters are useful within general solvent replacement efforts, providing pertinent information describing solvent–solute interactions.20–23 However, within chromatographic applications the effects of the stationary phase on empirical results must also be considered.24
One method that incorporates the stationary phase is the solvent strength parameter (ε°) which is commonly used when predicting solvent behaviour within adsorption chromatography applications.24 This parameter considers solvent molecular size, dipolarity, and polarisability in tandem with intrinsic properties of the stationary phase, which in the case of this work is hydrogen-bond acidic silica.
The ability to interpret chromatographic results from the perspective of specific and non-specific interactions is valuable when developing and testing alternative solvents for such applications. Herein, through a statistical thermodynamic framework, we will quantify the driving molecular interactions that determine empirical TLC measurements, aiding in the further understanding of solvation behaviours.25–29 Within this framework, we are able to deduce Rf value dependence on polar eluent concentration between the stationary and mobile phases in the presence of a low-polarity mediating solvent. The competitive strength of the analyte–eluent and analyte–solvent interactions will be quantified through the Kirkwood–Buff integrals (KBIs)25–29 of the radial distribution functions, describing the dispersion of molecules around a given analyte. The resulting KBIs at the dilute eluent limit will allow us to interpret the chromatographic data of green replacement solvents in the practical substitution of DCM. Bio-derived esters tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3), and methyl pivalate (4), were experimentally identified as safe and sustainable replacements to directly replace DCM for TLC and used herein to analyse the common bio-active molecules acetylsalicylic acid, ketoprofen, caffeine, and acetaminophen. The chromatographic data produced herein is used to calculate the KBIs for each system, quantifying all relevant intermolecular interactions and deriving links to relevant solubility predictors.
2 A statistical thermodynamic approach to solvent selection
Quantifying the specific and non-specific intermolecular interactions in chromatography, including solvents, analytes, and the stationary phase, is possible within a singular statistical thermodynamic framework. Consider a TLC system wherein the mobile phase is a binary eluent mixture consisting of low polarity and high polarity components. Conventionally, the tunability of this system has been interpreted based on the overall polarity of the eluent, which increases incrementally with the concentration of the polar component. Replacements for DCM would likewise require the ability to facilitate favourable interactions between an analyte and a polar additive.The interactions between a polar eluent component (here referred to as eluent) and analyte, in the presence of a mediating low-polar component (here referred to as solvent), can be quantified through experimental TLC data by applying statistical thermodynamics and preferential solvation theory. One way to interpret data through these frameworks is to use Kirkwood–Buff integrals (KBIs), a widespread quantification method of molecular interactions in multi-component solutions of varying complexities,30,31 encompassing both weak and strong as well as specific and non-specific molecular interactions. KBIs quantify the excess distribution of solvent (or eluent) molecules around a solute (local) compared to its absence (bulk) (Fig. 1). This provides insight into the solvent–analyte and eluent–analyte interactions that take place in solution. The KBI's capacity to unite and measure attractive and repulsive interactions is critical in quantifying these non-specific interactions.26,27,29,32
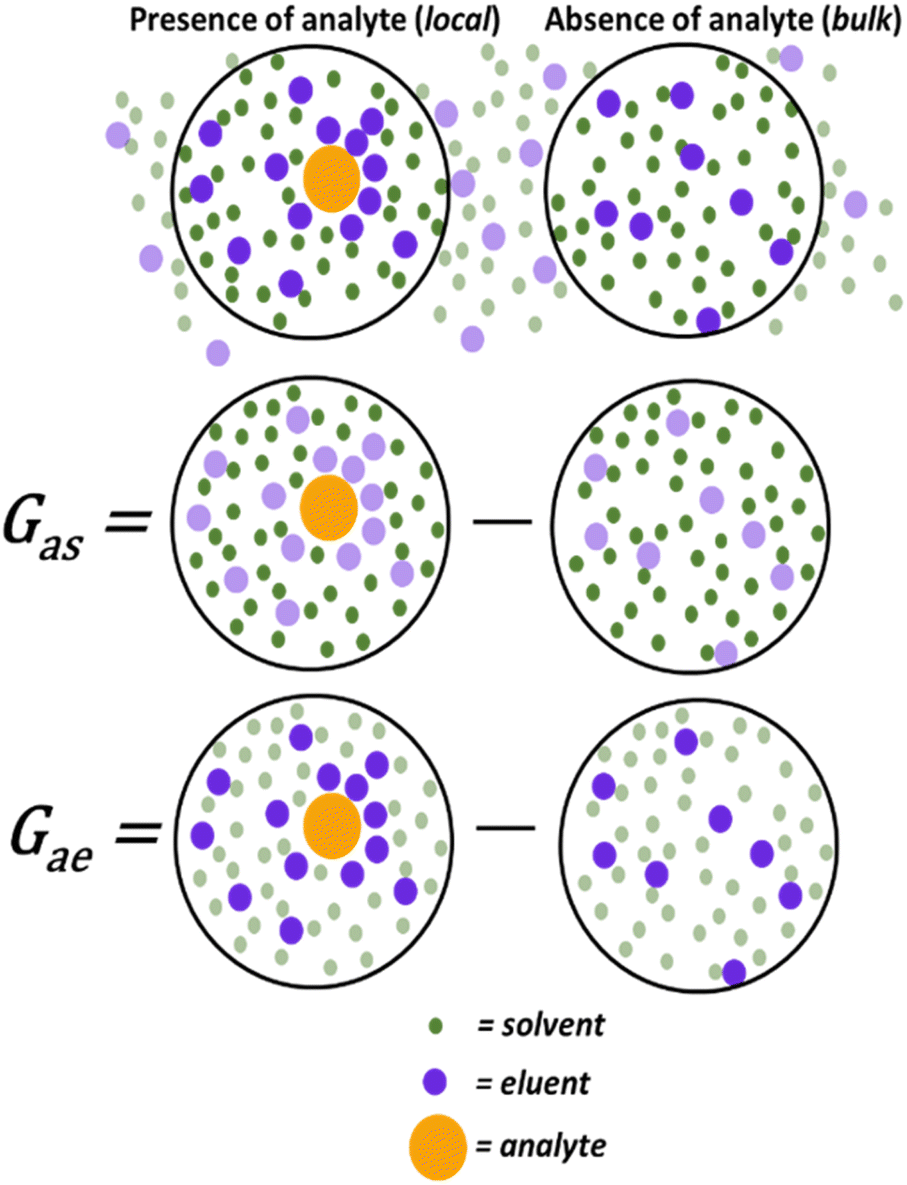 | ||
| Fig. 1 Preferential solvation represented by Kirkwood–Buff integrals (KBIs) where the distribution difference of solvent and eluent molecules in the presence (local) and absence (bulk) of an analyte molecule can be quantified. Figure adapted from ref. 29. | ||
2.1 Thin-layer chromatography and the partition coefficient
Consider an analyte (i = a), and a mobile phase containing solvent species (i = s), and up to two eluent species (i = e1 and e2) each with the concentration ci. Under instantaneous equilibrium assumption, standard chromatography theory defines analyte partitioning between a mobile and stationary phase as | (1) |
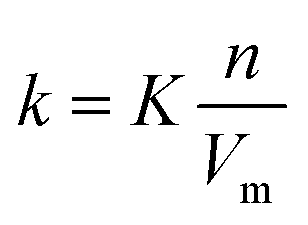 | (2) |
Within applications to thin-layer chromatography, retardation factor, Rf, is commonly used. Rf is related to the retention factor k through the well-known equation
 | (3) |
 | (4) |
| ΔGai = G(M)ai − G(S)ai | (5) |
Experimentally, we measure how the retention factor k (or Rf) changes with the eluent concentration, ce. The gradient at the dilute eluent limit gives the KBI difference ΔGae − ΔGas at this limit, via
 | (6) |
2.2 Binary eluent mixtures
We can extend our theory to binary eluent mixtures. Following the derivation in ESI Appendix A,† in the presence of dilute e1 and e2, eqn (6) can be generalized asand

 | (7) |
In Appendix A (see ESI),† we have shown that the eluent effects are additive at the dilute limit. Combining eqn (7) with eqn (A8),† the dependence of k on the combined eluent concentration is shown to be an addition of analyte–e1 and analyte–e2 interactions, as
 | (8) |
2.3 The role of the solvent
Through this derivation, it is shown that the competitive difference of analyte–eluent and analyte–solvent interactions between the stationary and mobile phases is crucial to the observed change of k (or Rf) upon the addition of eluent(s). The analyte–eluent and analyte–solvent interaction difference between the stationary and mobile phases allows us to calculate a combined term, which quantifies all present interactions (Fig. 2). In our case, the substitution of DCM with an alternative test solvent may lead to changes in ΔGae1 − ΔGas and ΔGae2 − ΔGas as alternative solvents will mediate the analyte–eluent(s) interaction differently: a favourable analyte–solvent or eluent–solvent interaction for instance would weaken an analyte–eluent interaction. Solvent replacement can be investigated by comparing how ΔGae − ΔGas, of the same sets of analyte and eluents, vary when the solvent is replaced, a mechanism that has not been captured by traditional solubility models. | ||
| Fig. 2 Determination of the analyte–eluent, analyte–solvent KBI difference between the stationary (S) and mobile phases (M) within a TLC system. Quantification between competing analyte–eluent and analyte–solvent interactions provide insight regarding the intermolecular behaviours facilitating experimental data. | ||
3 Experimental
3.1 Materials and methods
The solvents used in this project, including DCM (99%), MeOH (99.9%), EtOAc (98%), EtOH (99.8%), were obtained from Fisher Scientific (Loughborough, England). Heptane (99%), ethyl isobutyrate (99%), and methyl pivalate (99.9%) were obtained from Sigma-Aldrich (Merck). tert-Butyl acetate (99%), and sec-butyl acetate were obtained from Fluorochem (UK) and Thermo Scientific, respectively. Analytes ibuprofen (99.1%), ketoprofen (99.51%), and acetaminophen (99.96%) were obtained from APExBIO (USA), caffeine (99%) and 4-aminophenol (99%) were obtained Sigma-Aldrich (Merck), and o-acetylsalicylic acid (99%) was obtained from Alfa Aesar and used without any further purification. For Kamlet–Taft tests, dyes N,N-diethyl-4-nitroaniline (97%) and 4-nitroaniline (99%) were obtained from Fluorochem (UK) and Sigma-Aldrich, respectively. TLC plates (TLC Silica gel 60 F254, aluminium support, 20 cm × 20 cm, Merck) were cut to 2.0 cm × 8.0 cm plates and analysed via a 254 nm UV lamp (UVP UVGL-55 Handheld UV Lamp, 254/365 nm, multi-band split tube) and viewing cabinet.3.2 Application of test solvents: TLC analysis of drug molecules
Analytes were spotted consecutively on TLC plates using glass capillary tubes. Plates were run in triplicate with each test solvent (including DCM) over a range of increasing polar eluent concentrations. Polar eluents included MeOH and 3:1 EtOAc–EtOH. All TLC plates were visualised via UV lamp at 254 nm.
:1 EtOAc–EtOH (25%) as the polar eluent additive. Structurally similar ibuprofen and ketoprofen were run as one mixture and acetaminophen and its primary degradation product 4-aminophenol were run as the second mixture. TLC plates were run and visualised similarly as in Section 3.2.
3.3 Kamlet–Taft parameters
Kamlet–Taft (KT) parameters of DCM and test solvents were obtained from the literature with the exception of tert-butyl acetate and sec-butyl acetate. N,N-Diethyl-4-nitroaniline and 4-nitroaniline dyes were used to calculate the π* and β parameters, respectively. The α parameter was not experimentally determined as it was assumed to be zero for both butyl acetates. The calculation method for KT parameters can be found in the ESI.†4 Results and discussion
4.1 Solvent and analyte selection
A number of green solvents, including structural isomers tert-butyl acetate, sec-butyl acetate, ethyl isobutyrate, and methyl pivalate (Fig. 3), were previously identified as potential alternatives to traditional low-polarity solvents.23 On this basis, it was postulated that they would provide an eluent system wherein analytes could be sufficiently separated upon incremental addition of a polar additive. Their low viscosities and high volatilities were also considered to facilitate a saturated environment within the TLC vessel and encourage solvent mobility up the stationary phase. The performance of these alternative solvents was considered in terms of chromatographic separation and greenness compared to DCM. Traditional hydrocarbons were not tested due to their health hazards, environmental toxicity, lack of functionality, and UV activity (i.e., toluene) as TLC plates were to be visualized under UV light. | ||
| Fig. 3 Chemical structures, molecular weights (MW), and boiling points (BP) of the four green solvents tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3), and methyl pivalate (4). These esters are identified as suitable DCM alternatives for the TLC analysis of small, bio-active molecules. | ||
Evidence of the adequate separation of analytes was held to the commonly accepted ideal range for TLC of Rf = 0.3–0.7. The polar additives used in testing were MeOH and a 3:1 blend of EtOAc–EtOH. MeOH was used to facilitate a direct comparison to traditional DCM–MeOH systems as well as provide a single polar eluent system. The alternative 3:1 EtOAc–EtOH blend was used to provide an overall greener TLC system and to observe the molecular interactions of a binary polar eluent mixture.
Two acidic analytes, o-acetylsalicylic acid (A) and ketoprofen (B), as well as the basic analyte caffeine (C), and neutral acetaminophen (D) were chosen as analytes to represent common drug molecules with varying functionalities, including aromatic rings, ketones, carboxylic acids, and heterocycles (Fig. 4).
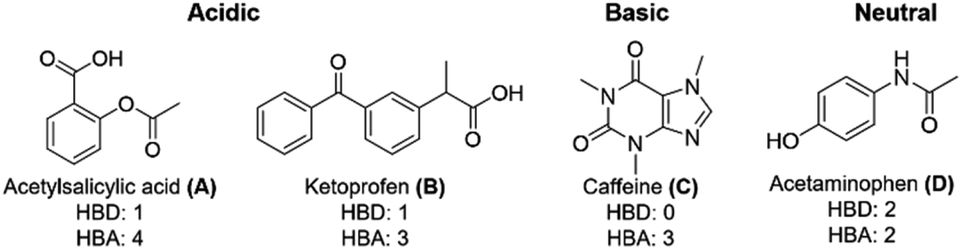 | ||
| Fig. 4 Analytes acetylsalicylic acid (A), ketoprofen (B), caffeine (C), and acetaminophen (D) chosen to represent common, small, bio-active molecules. Analytes were chosen to offer a variety of functionalities and acid/base properties. | ||
4.2 Solubility parameters of test solvents
HSPiP (ver. 5.3.06) was used to determine the HSPs of the test solvents. Kamlet–Taft parameters of test solvents were obtained from the literature, with the exception of tert-butyl acetate (1) and sec-butyl acetate (2), which were obtained experimentally for this work. The estimated solvent strength parameters of DCM and test solvents were calculated in this work (see ESI†) from the KT parameters seen in Table 1.| Parameters | DCM | Test solvents | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| a Obtained from the HSPiP software. b Obtained from ref. 19. c Obtained in this work. d Obtained from ref. 23. | |||||
| δ D (MPa1/2) | 17.0a | 15.0a | 15.0a | 15.5a | 15.1a |
| δ P (MPa1/2) | 7.3a | 3.7a | 3.7a | 4.6a | 4.0a |
| δ H (MPa1/2) | 7.1a | 6.0a | 7.6a | 5.3a | 5.1a |
| π* | 0.82b | 0.45c | 0.50c | 0.49d | 0.49d |
| α | 0.13b | 0.00c | 0.00c | 0.00d | 0.00d |
| β | 0.10b | 0.46c | 0.46c | 0.48d | 0.48d |
| ε° | 0.32c | 0.34c | 0.35c | 0.35c | 0.35c |
The test solvents possess similar HSP values, but can be considered as less polar than DCM. The higher δP value of DCM compared to the test solvents is counterintuitive to the empirical TLC results, in which DCM appears to behave as a slightly less polar solvent than all four test solvents (see Section 4.3). Similarly, the π* (dipolarity/polarizability) KT parameter too describes DCM as having a much higher polarity when compared to the esters. As discussed, there is an inherent difficulty when using traditional solubility parameters to screen for solvent replacements within chromatographic applications. Eluent behaviours can only be rationalized by considering specific and non-specific intermolecular interactions, a highly acidic, silica stationary phase, and solute specific preferential solvation. By using the solvent strength parameter, we are now able to see a rational trend between the performance of DCM and test solvents, with very close estimated ε° values of 0.32 (DCM), 0.34 (tert-butyl acetate), and 0.35 (sec-butyl acetate, ethyl isobutyrate, and methyl pivalate).
4.3 TLC results – test solvent Rf ranges
TLC was performed to observe the influence of polar eluents on Rf ranges. Analytes acetylsalicylic acid, ketoprofen, and acetaminophen ran successfully in all test solvent mixtures where MeOH was the polar eluent (0–20%), with Rf ranges averaging 0.23–0.67, 0.35–0.71, and 0.15–0.64 respectively (Fig. 5). These plots were directly compared to the traditional TLC solvent blend DCM–MeOH, where Rf values averaged from 0–0.85. The DCM–MeOH binary mixture produced a larger Rf value range for caffeine (0–0.84) in comparison to the test solvents, which produced lower Rf ranges between 0.0 and 0.37, just reaching the desired separation at 20% MeOH. It is probable that higher percentages of MeOH (>20%) would increase the Rf value range observed, however this was not tested as direct comparison to DCM–MeOH systems was decidedly limited to 0–20% MeOH. | ||
| Fig. 5 TLC analysis (n = 3) of drug molecules using test solvents tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3), and methyl pivalate (4). Test solvents are directly compared to dichloromethane (DCM) systems modified with MeOH and 3:1 EtOAc–EtOH. Drug molecules analysed: acetylsalicylic acid (A), ketoprofen (B), caffeine (C), and acetaminophen (D). The lines are a guide to the eye. | ||
All test solvents were shown to successfully replace DCM in the TLC analysis of acetylsalicylic acid, ketoprofen, and acetaminophen when a 3:1 EtOAc–EtOH binary blend was used as the polar eluent instead of MeOH. The DCM-3:1 EtOAc–EtOH blend produced average Rf values ranging from 0.01–0.75, with caffeine having the smallest range of 0–0.54. Test solvents modified with the EtOAc–EtOH mixture produced similar chromatographic behaviour to the DCM–MeOH blend, with Rf ranges averaging at 0.23–0.75, 0.35–0.83, 0.02–0.46, and 0.15–0.83 for acetylsalicylic acid, ketoprofen, caffeine, and acetaminophen respectively (Fig. 5). Caffeine displayed an improved range of Rf values when compared to test solvent–MeOH systems with increased variability in the ideal range. To further test their efficacy, a mixture of structurally similar analytes, ketoprofen and ibuprofen, were adequately separated in all test solvent systems (tert-butyl acetate, sec-butyl acetate, ethyl isobutyrate and methyl pivalate with 25% 3:1 EtOAc–EtOH). A mixture of acetaminophen and primary degradation product 4-aminophenol could also be well separated using the same solvent systems. Resulting separations were effectively comparable to the same mixtures run in DCM/10% 3:1 EtOAc–EtOH solvent blend (Fig. 6).
 | ||
| Fig. 6 Good separation of structurally similar analytes ibuprofen and ketoprofen, as well as acetaminophen and major degradation product 4-aminophenol, can be seen in test solvents 1–4 modified with 25% 3:1 EtOAc–EtOH. Results are compared to a mixture of DCM/10% 3:1 EtOAc–EtOH. | ||
Replacing DCM within chromatography is particularly challenging due to the lack of safe, green, low polarity solvents that can aid in the solvation of functionalised analytes. It is particularly difficult when separating very polar, heterocyclic molecules such as caffeine. DCM is an excellent solubilizer for such molecules and, when mixed with MeOH, is seen to synergistically solvate33,34 solutes through proposed weak hydrogen and halogen bonding networks. This type of preferential solvation can increase the polarity of the binary mixture, exceeding that of both individual neat solvents, at given mole fractions. This behaviour can account for the large Rf value range for caffeine in DCM–MeOH (0–0.85) compared to a much smaller range for the DCM-3:1 EtOAc–EtOH (0–0.54) solvent blend.
Despite the advantageous properties of DCM solvent mixtures, the test solvents used herein indeed demonstrate the suitability of functionalised green solvents as low-polarity, mediating components within chromatographic separations; this is observed predominantly when analysing acetylsalicylic acid, ketoprofen, and acetaminophen analytes that offer hydrogen-bond donating abilities to the hydrogen-bond accepting esters. The polarities of the esters are low enough to yield similar, and in some cases improved, Rf value ranges where the mobile phase is more tuneable in the ideal range (0.3–0.7), particularly when the 3:1 EtOAc–EtOH eluent is used.
In addition to the observed experimental behaviour, the esters greatly improve the greenness of the system. Each test solvent candidate can be synthesised from renewable resources via catalytic routes.23 For example, tert-butyl acetate can be synthesized from bio-isobutene.35,36 Additionally, these solvents are not restricted under REACH, and their use remediates the health and environmental dangers present with DCM, the only concern being high flammability with the expectation of methyl pivalate, which also possesses an ingestion hazard.
Green solvents tert-butyl acetate, sec-butyl acetate, ethyl isobutyrate and methyl pivalate offer safer, more sustainable alternatives to DCM in a TLC mobile phase. These readily available solvent replacements for the separation and analysis of small drug molecules also offer theoretical insights for expanding chlorinated solvent replacement. We aim to quantify the molecular interactions influencing empirical TLC measurements within the framework of statistical thermodynamics in Section 4.4.
4.4 Statistical thermodynamic interpretation of results
Here, we implement a method wherein chromatographic data can be interpreted within a singular statistical thermodynamic framework, probing the intermolecular behaviours of solvents we are looking to replace. Accessing this quantitative information begins with analysing the eluent concentration dependence ce on the retention factor, k. Recall that k can be calculated from empirically obtained Rf values and linked to statistical thermodynamics through partition coefficient K using eqn (2) and (3). The KBIs can then be calculated for solvent systems of interest.The competitive difference among the analyte–eluent and analyte–solvent KBIs between the stationary and mobile phases will allow us to directly compare the behaviours of replacement solvents through this quantitative framework. To determine the system KBIs, ln(k) values are plotted over ce (Fig. 7) for a given TLC dataset. Data sets containing an undefined ln(k) value (i.e. when Rf = 0) pose experimental difficulties as such results would lead to divergent KBIs, which is unphysical. The inherent limitation in TLC is accuracy, and as such, these data sets are fit to a simple linear regression model (ANOVA) to yield physical results and make KBI calculation possible (see ESI† for fitting parameters and linear regression data). The approach to plot the relationship between retardation factor and eluent concentration (percent by volume) linearly follows the tactic used by previous papers.4,6 The gradients of these functions, at the dilute eluent limit, yields the combined KBI term between the mobile and the stationary phases (eqn (6) and (8) for single and binary eluents, respectively). This will allow us to observe and elucidate trends between DCM and the test solvents, offering rapid insights to the molecular level from experimental data.
 | ||
| Fig. 7 Variance of ln(k) as of function of eluent concentration in each solvent system including DCM, tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3) and methyl pivalate (4). Plots A, B, C, and D represent analytes acetylsalicylic acid, ketoprofen, caffeine, and acetaminophen, respectively. | ||
Calculating the combined KBI terms supplies a theoretical background to chromatographic behaviours by quantifying the molecular interactions that can not be elucidated from chromatographic data or solubility predictors alone. In Fig. 8 we can observe the distribution of KBI terms for each mobile phase system and analyte as we exchange the solvent. Plots A, B, C, and D represent analytes acetylsalicylic acid, ketoprofen, caffeine, and paracetamol, respectively. Solvents include DCM and test solvents tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3), and methyl pivalate (4). KBIs calculated in each polar eluent are shown on either axis, the X-axis representing the KBIs calculated in MeOH and the Y-axis representing KBIs calculated in the 3:1 blend of EtOAc–EtOH.
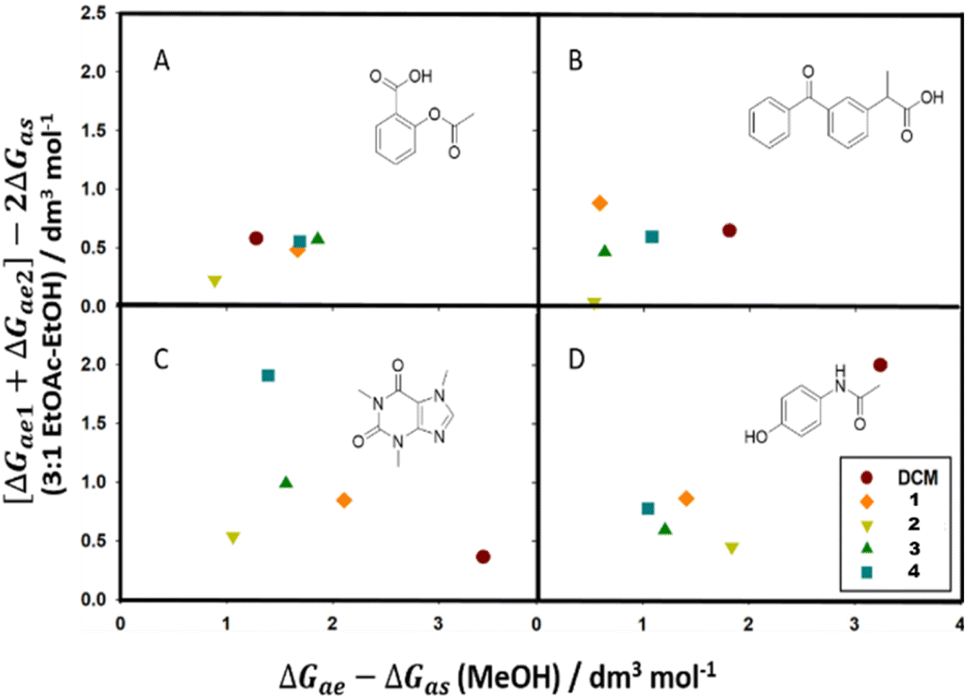 | ||
| Fig. 8 Scatter plots comparing the combined KBI difference terms of each analyte in each solvent–eluent mixture. Plots A, B, C, and D represent analytes acetylsalicylic acid, ketoprofen, caffeine, and paracetamol, respectively. Solvents: DCM and test solvents tert-butyl acetate (1), sec-butyl acetate (2), ethyl isobutyrate (3), and methyl pivalate (4). | ||
Comparing chlorinated and bio-based oxygenated solvents, and rationalising their comparable experimental behavior, is inherently difficult due to the divergent characteristics and varying intermolecular interactions that take place when solvating target molecules. The solubility parameters in Table 1 convey the high polarisability of DCM (0.82) in comparison to the generally dipolar esters which have π* values between 0.45 and 0.50. Strong hydrogen bonding interactions between oxygenated solvents and analytes containing nitrogen or hydroxy groups enhance solvation over the weaker hydrogen bonding between DCM and those same analytes. This can be observed in the high hydrogen bond accepting ability (β) in the esters compared to that of DCM. Conversely, where the esters have zero hydrogen bond acidity (∝), DCM has a small ability (0.10) through the opportunity for halogen bonding.37 Merging the effects of all specific and non-specific intermolecular interactions, including the acidic silica stationary phase, makes it difficult to use typical solvation parameters and predicators to explain empirical TLC results.
We can estimate that because DCM is more polar (using HSP and KT parameters), that the analytes should travel further up the TLC plate in 100% solvent when compared to the less polar test solvents. Experimentally, we can confirm that this is not the case as Rf values for all analytes were zero, or close to zero, when run in pure DCM and much greater than zero (with the exception of heterocyclic caffeine) when run in pure test solvents 1–4. Additionally, DCM and the test solvents act differently when the same amount of polar eluent is added. When MeOH is added incrementally to the mobile phase, the overall polarity increases far more drastically, with steeper changes to Rf values for DCM systems than in test solvent systems. This can be contributed to the previously proposed synergistic solvating behaviour of DCM–MeOH systems.33,34
We can propose elucidations of the experimental results through the quantification of the KBIs for the analyte–mobile phase systems. For example, the chromatographic data for acetylsalicylic acid in MeOH modified solvent blends (Fig. 5(A)) infers that there is a stronger competition present between the analyte–solvent and analyte–eluent interactions in DCM and sec-butyl acetate compared to tert-butyl acetate, ethyl isobutyrate and methyl pivalate test solvents; as ce → 0, there is a greater eluent concentration influence on the retention factor. This suggests even more competition between analyte–eluent and analyte–solvent interactions, where the eluent concentration influence on chromatographic data is reduced by a less mediating solvent.
These quantitative results, which include the specific analyte and stationary phase used, bridge a connection to solubility predictors. For all the analytes, sec-butyl acetate is seen to have the poorest mediating ability, yielding the smallest KBI difference overall. Compared to the other test solvents (Table 1), it has the highest hydrogen bonding ability (HSP) and polarisability (Kamlet–Taft). This can be corroborated with an increase in ΔGas. As pure solvents, ethyl isobutyrate and methyl pivalate show an increased estimated ε° and π* (KT) compared to tert-butyl acetate and a larger δP (HSP) parameter compared to both esters. As such, they should be less mediating, with a smaller ΔGae − ΔGas value, or larger ΔGas value, due to increased polarity. This behaviour can be observed through the comparatively smaller ΔGae − ΔGas values (Tables 2 and 3) for analytes C and D (modified with MeOH) and analytes B and D (modified with the EtOAc–EtOH blend). In these cases, one or both of the acetates prove to be more mediating, yielding larger ΔGae − ΔGas values, where the solvent contribution ΔGas is small. Contrarily, for analytes A and B when modified by MeOH, and for A and C when modified with the EtOAc–EtOH blend, their larger ΔGae − ΔGas difference suggest they are more mediating than the acetates. This prompts a deeper investigation into the solubility predictors.
| Analyte | DCM | Test solvents | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| A | 1.28 | 1.67 | 0.889 | 1.86 | 1.69 |
| B | 1.81 | 0.588 | 0.534 | 0.634 | 1.08 |
| C | 3.42 | 2.11 | 1.06 | 1.56 | 1.39 |
| D | 3.24 | 1.41 | 1.84 | 1.21 | 1.05 |
:1 EtOAc–EtOH
| Analyte | DCM | Test solvents | |||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| A | 0.581 | 0.485 | 0.226 | 0.571 | 0.551 |
| B | 0.652 | 0.886 | 0.044 | 0.469 | 0.601 |
| C | 0.362 | 0.845 | 0.536 | 0.986 | 1.91 |
| D | 2.00 | 0.864 | 0.451 | 0.595 | 0.778 |
It can be proposed that hydrogen bonding plays a large role in the quantification of interactions here. Hydrogen bonding is an important specific intermolecular interaction in TLC that can be captured in KBIs. Using the HSP scale (Table 1), sec-butyl acetate has the largest hydrogen bonding ability of the test solvents, and for all solvent–eluent–analyte combinations (with the exception of analyte D in MeOH), is the least mediating. Additionally, sec-butyl acetate has the largest π* value. The increased polarity and hydrogen bonding ability of this solvent suggests that the observed non-mediating ability, through a smaller competitive ΔGae − ΔGas difference, is corroborated through increased competition with the polar eluent.
Ethyl isobutyrate and methyl pivalate hydrogen bonding forces (δH) are reduced compared to both acetates. This can be observed in the larger ΔGae − ΔGas difference for analytes A and B (MeOH) and A and C (EtOAc–EtOH). In these cases, the solvent is less mediating and ΔGas is more negative. For cases where this does not apply, tert-butyl acetate is often found to be the most mediating of the test solvents.
When comparing both eluents, one difference of note is, for MeOH (as an eluent), DCM (as a solvent) appears to be more consistently mediating compared to the EtOAc–EtOH as a solvent mixture. Using caffeine as an example analyte, the ranking suggests that DCM is the least mediating solvent when modifying with EtOAc–EtOH. Caffeine's favourable interaction with DCM, which could be evidenced by its smallest estimated ε° value as well as high caffeine solubility in DCM, would compete with effects from the added polar eluent. The difference between the favourable ΔGae and ΔGas can still be small, through which DCM would seem a poor mediator in EtOAc–EtOH systems. Comparatively, when MeOH is used as the eluent, DCM is observed to be the most mediating with the largest ΔGae − ΔGas difference. As proposed, DCM–MeOH binary mixtures are seen to synergistically solvate certain probe molecules through specific bonding networks (H- and X-bonding) between them. When MeOH is added to the TLC system, DCM–analyte hydrogen bonding is present, but the potential synergistic solvation would greatly increase analyte solubility in the mobile phase, increasing the KBI difference via a steep change in Rf values at the dilute eluent limit.
A final observation to explore is the magnitudes of the competitive ΔGae − ΔGas values evaluated in each eluent system. The KBI difference is much larger in the MeOH system versus the EtOAc–EtOH system where it is are seemingly halved. A straightforward comparison of polarisability (through both HSP and Kamlet–Taft) of MeOH, EtOAc, and EtOH suggest that small amounts of MeOH would increase the polarity of the system greater and faster than additions of EtOAc–EtOH would. This rapid increase in polarity, at the dilute eluent limit, would result in an increased effect on the Rf and subsequent KBI difference through enlarged analyte–eluent interaction effects.
5 Conclusions
Despite its reported environmental and health hazards, DCM is an attractive TLC solvent due to its effective low-polarity, allowing chromatographic systems to be modified according to specific analytes through the addition of high polar eluents. Here, DCM mediates analyte–eluent interactions in thin-layer chromatography (TLC) where Rf values are dependent on the concentration of added eluent (ce). Ideal replacement solvents would behave in a similar mediating manner. Less suitable solvents would be increasingly favourable for an analyte, strengthening analyte–solvent or eluent–solvent interactions and weakening analyte–eluent interactions.Green solvents tert-butyl acetate, sec-butyl acetate, ethyl isobutyrate and methyl pivalate are safer, more sustainable alternatives to DCM in TLC mobile phases analysing common, small drug molecules. The polarities are low enough to yield comparable, and sometimes improved, Rf value ranges wherein the mobile phase is more tuneable in the ideal range.
Herein, through a statistical thermodynamic framework, we are able to interpret chromatographic results that capture the specific and non-specific interactions present in experimental measurements. Understanding these interactions is necessary when probing alternative solvents for such applications and is not always achieved through the interpretation of traditional solubility parameters.
Within this framework, we are able to deduce Rf value dependence on polar eluent concentration between the stationary and mobile phases, where the competitive strength of the analyte–eluent and analyte–solvent interactions are quantified through the Kirkwood–Buff integrals (KBIs). Our probed replacement solvents provide a suitable alternative for DCM that can be observed empirically and theoretically through a statistical thermodynamic framework where both the TLC data is comparable, as well as the calculated KBIs presented herein.
Author contributions
J. L.: conceptualization, methodology, formal analysis, investigation, writing – original draft J. S.: conceptualization, writing – review and editing, supervision. C. R. M.: conceptualization, writing – review and editing, supervision. J. M.: conceptualization, funding acquisition. S. S.: conceptualization, formal analysis, writing – review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Merck KGaA, Dr Tony Wild and the Wild Fund for the funding contributions supporting this study.References
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef.
- A. Jordan, P. Stoy and H. F. Sneddon, Chem. Rev., 2021, 121, 1582–1622 CrossRef CAS PubMed.
- J. P. Taygerly, L. M. Miller, A. Yee and E. A. Peterson, Green Chem., 2012, 14, 3020–3025 RSC.
- L. Moity, M. Durand, A. Benazzouz, C. Pierlot, V. Molinier and J.-M. Aubry, Green Chem., 2012, 14, 1132–1145 RSC.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2012, 14, 3016–3019 RSC.
- A. Sharma, E. Yu, G. Morose, D. T. Nguyen and W.-T. Chen, Separations, 2021, 8, 1–14 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- Agency for Toxic Substances and Disease Registry (ATSDR), 2000 Toxicological profile for Methylene Chloride, U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, https://www.epa.gov/sites/default/files/2014-04/documents/methylene_chloride_toxicology_profile_tp14_3v.pdf, accessed, January 14, 2022 Search PubMed.
- World Health Organization, International Labor Organization, International Chemical Safety Cards (ICSCs). ICSC 0058 – Dichloromethane, https://www.ilo.org/dyn/icsc/showcard.display?p_card_id=0058#:%7E:text=ICSC0058%2DDICHLOROMETHANE%26text=Flammableunderspecificconditions, accessed, January 14, 2022 Search PubMed.
- European Chemicals Agency Substance Info Card: Dichloromethane, https://echa.europa.eu/substance-information/-substanceinfo/100.000.763, accessed, December 7, 2021 Search PubMed.
- L. J. Carpenter, S. Reimann, J. B. Burkholder, C. Clerbaux, B. D. Hall, R. Hossaini, J. C. Laube and S. A. Yvon-Lewis, Scientific Assessment of Ozone Depletion: 2014, in Global Ozone Research and Monitoring Project – Report No. 55, World Meteorological Organization, Geneva, Switzerland, 2014, ch. 1, pp. 1.1–1.101 Search PubMed.
- R. Hossaini, M. P. Chipperfield, S. A. Montzka, A. A. Leeson, S. S. Dhomse and J. A. Pyle, Nat. Commun., 2017, 8, 15962 CrossRef CAS PubMed.
- M. An, L. M. Western, D. Say, L. Chen, T. Claxton, A. L. Ganesan, R. Hossaini, P. B. Krummel, A. J. Manning, J. Mühle, S. O’Doherty, R. G. Prinn, R. F. Weiss, D. Young, J. Hu, B. Yao and M. Rigby, Nat. Commun., 2021, 12, 7279 CrossRef CAS PubMed.
- E. A. Peterson, B. Dillon, I. Raheem, P. Richardson, D. Richter, R. Schmidt and H. F. Sneddon, Green Chem., 2014, 16, 4060–4075 RSC.
- C. M. Hansen, Hansen Solubility Parameters, CRC Press, Boca Raton, FL, 2nd edn, 2007 Search PubMed.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409 RSC.
- P. G. Jessop, Green Chem., 2011, 13, 1391 RSC.
- P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- A. A. C. Pacheco, J. Sherwood, A. Zhenova, C. R. McElroy, A. J. Hunt, H. L. Parker, T. J. Farmer, A. Constantinou, M. De Bruyn, A. C. Whitwood, W. Raverty and J. H. Clark, ChemSusChem, 2016, 9, 3503–3512 CrossRef CAS PubMed.
- F. P. Byrne, B. Forier, G. Bossaert, C. Hoebers, T. J. Farmer and A. J. Hunt, Green Chem., 2018, 20, 4003–4011 RSC.
- S. K. Poole and C. F. Poole, Chromatographia, 2001, 53, S162–S166 CrossRef CAS.
- S. Shimizu, S. Abbott, K. Adamska and A. Voelkel, Analyst, 2019, 144, 1632–1641 RSC.
- S. Shimizu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1195–1199 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, J. Phys. Chem. B, 2014, 118, 3922–3930 CrossRef CAS PubMed.
- S. Shimizu and N. Matubayasi, J. Mol. Liq., 2019, 273, 626–633 CrossRef CAS.
- S. Shimizu, Curr. Opin. Colloid Interface Sci., 2020, 48, 53–64 CrossRef CAS.
- A. Ben-Naim, J. Chem. Phys., 1977, 67, 4884–4890 CrossRef CAS.
- S. Shimizu and N. Matubayasi, Phys. Chem. Chem. Phys., 2017, 19, 23597–23605 RSC.
- S. Shimizu and N. Matubayasi, Langmuir, 2021, 37, 7380–7391 CrossRef CAS PubMed.
- S. Gupta, A. Chakraborty and P. Sen, J. Mol. Liq., 2016, 223, 274–282 CrossRef CAS.
- K. Polok, N. Subba, W. Gadomski and P. Sen, J. Mol. Liq., 2022, 345, 117013 CrossRef CAS.
- Global Bioenergies, Focus Catal., 2019, 5, 7 Search PubMed.
- X. Chen, J. Fen, Z. Fei, W. Sun, J. Tang, P. He, M. Cui and X. Qiao, J. Porous Mater., 2016, 23, 255–262 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimangi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116(4), 2478–2601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ay01266a |
| This journal is © The Royal Society of Chemistry 2023 |