
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of the volatile monoterpene composition of citrus essential oils by photoelectron spectroscopy employing continuously monitored dynamic headspace sampling†
Hassan
Ganjitabar‡
 a,
Rim
Hadidi§
b,
Gustavo A.
Garcia
b,
Laurent
Nahon
b and
Ivan
Powis
*a
a,
Rim
Hadidi§
b,
Gustavo A.
Garcia
b,
Laurent
Nahon
b and
Ivan
Powis
*a
aSchool of Chemistry, The University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: ivan.powis@nottingham.ac.uk; Tel: +44 (0)115 9513467
bSynchrotron SOLEIL, l'Orme des Merisiers, Saint Aubin BP 48, 91192 Gif sur Yvette Cedex, France
First published on 13th November 2023
Abstract
A new photoelectron spectroscopic method permitting a quantitative analysis of the volatile headspace of several essential oils is presented and discussed. In particular, we focus on the monoterpene compounds, which are known to be the dominant volatile components in many such oils. The photoelectron spectra of the monoterpene constituents may be effectively isolated by accepting for analysis only those electrons that accompany the production of m/z = 136 ions, and by using low photon energies that restrict cation fragmentation. The monoterpene isomers are then identified and quantified by regression modelling using a library of terpene standard spectra. An advantage of this approach is that pre-concentration of the volatile vapour is not required, and all steps are performed at ambient temperature, avoiding the possible deleterious effects (such as isomerisation/decomposition) that may sometimes arise in gas chromatographic (GC) procedures. As a proof-of-principle demonstration, three citrus oils (lemon, lime, bergamot) are analysed with this approach and the results are compared with reported GC composition profiles obtained for these oils. Potential advantages of the methodology that include multiplex detection and real-time, in situ analysis are identified and discussed. Alternative and faster experimental implementations concerning laboratory-based ionization and detection schemes are proposed and considered, as is the possibility of a straightforward extension towards simultaneous determination of enantiomeric excesses.
1. Introduction
Essential oils (EOs) extracted from aromatic plants find a variety of applications in pharmacology, the cosmetics and food industries1–6 and this creates requirements for their composition to be fully characterized for purposes of authentication, identifying geographical origin, method of extraction, and quality of the oils, as well as revealing possible adulteration or contamination.7–10 The major components of the EOs are volatiles, providing the often-prized scents, odours and tastes of these natural extracts. Chemically, these volatile compounds include various terpene hydrocarbons, their oxygenated derivatives, as well as aliphatic aldehydes, alcohols, and esters. Among these, monoterpene (C10H16) isomers are easily the dominant class, for example constituting 88% to 98% of the total volatile components in citrus oils.7,11Currently, the most common approaches for characterising the volatile composition of the EOs use some variant of gas chromatography (GC)6,12–14 in combination with a volatile headspace sampling method.15,16 Headspace (HS) sampling is generally preferred over direct injection of the whole EO sample matrix, as it simplifies sample preparation, and avoids the potentially deleterious loading of the column with the non-volatile residue of the oil. Nevertheless, sensitivity considerations usually require pre-concentration of the volatile headspace sample, e.g. by means of cryogenic trapping or, more frequently, by an absorption/desorption process. Solid phase microextraction (SPME) using absorption on a coated fibre has come to be widely used for this purpose.17
Non-specific eluate detection can be achieved by flame-ionization detection (FID). Equipment for more selective mass spectrometry detection (GC-MS) is widely available but, it may be noted, provides little advantage with monoterpene eluates as these are isomers and so possess the same parent mass, and similar principal fragments and fragmentation patterns. Tandem mass spectrometry detection (GC-MS/MS)18–20 and ion mobility detection (GC-IMS)21,22 offer increased selectivity and with it improved signal-to noise levels. A recent detector development that does, however, provide discrimination for isomers of the monoterpenes (and others) is the coupling of compact vacuum ultraviolet (VUV) absorption spectrometer to a column output (GC-VUV).23
Faster analyses, appropriate for quality control purposes, have used Raman24,25 and NIR26,27 vibrational spectroscopy, applying chemometric methods to provide EO “fingerprints”. Regression modelling using a library of standard spectra allows determination of relative composition of the principal components in an EO sample.
In this article we demonstrate a somewhat related approach using a sensitive, universal, and background-free photoionization-based spectroscopy technique and that could, in principle, be similarly adapted for continuous real-time monitoring of a process sample stream. Spectra of the monoterpene (C10H16 – m/z = 136) constituents from a volatile headspace sampled citrus oil are first isolated by recording mass-tagged photoelectron spectra. Regression modelling is then used to predict the EO spectrum from a library basis of standard spectra, selected to comprise the major terpenes encountered in these oils. The present results constitute a proof of principle determination of terpene composition, but photoelectron spectroscopy does offer additional prospects of rapid and precise enantioselective analysis28–31 for exploitation in future developments.
Generally, analysis of monoterpenes needs to guard against their isomerization caused by derivatization procedures or applied heating which would clearly have adverse consequences for the accuracy of measured isomer ratios. Kinetic studies32,33 have examined thermal isomerization of, for example, the pinenes at temperatures above 350° C. In the context of GC analysis, it has been speculated that with surface catalyzed assistance this may potentially pose problems in heated metal traps and transfer lines, or during thermal desorption from adsorbent sampling traps.34–38 While volatile headspace sampling eliminates the strongly heated injector required to volatilize a complete liquid EO sample, typical injector temperatures around 250 °C (ref. 35, 36 and 39) are nevertheless required for SPME desorption from traps. Aggressive column temperature ramping, used particularly with so-called fast and very fast GC,14 can extend beyond this to 300° C.40 In contrast, the sensitivity of the method to be described here is sufficient to allow the dynamically sampled headspace vapour to be streamed directly into the photoionization spectrometer in real time, never exceeding ambient temperature.
Below we present results for the relative headspace compositions of the principal monoterpene constituents of lemon, lime, and bergamot citrus oils. Specifically for this first demonstration, slow-photoelectron spectra (SPES) and ion time-of-flight (ToF) mass spectra are recorded for photon energies within a few eV of the terpene ionization thresholds (∼8 eV) using identical conditions for both the oils and the monoterpene standards. In the following sections we describe the experimental method, the theoretical model for the oil spectra, and data analysis, and then the results for the citrus oils we examined. Finally, we conclude and present a perspective for the further development and improvement of this approach.
2. Methods
2.1. Experimental
Commercial samples of citrus oils – Californian lemon oil (cold-pressed), Argentinian lemon oil (rectified), Mexican lime oil (distilled), Italian bergamot oil – were obtained from Sigma Aldrich. The terpene standards of 1R,5R-(+)-α-pinene, 1S,5S-(−)-β-pinene, 1S,6R-(+)-3-carene, 4R-(+)-limonene, 5R-(−)-α-phellandrene, and γ-terpinene were obtained from Sigma-Aldrich and used without further purification. Pure sabinene samples were not commercially available so we obtained a natural extract of sabinene of stated purity ≥75% (Sigma-Aldrich). The method for estimation of the pure Sabinene standard spectrum from this sample, whose precise composition is known from two dimensional GC measurements, has been given in a previous paper.41Measurements were made using the DELICIOUS 3 double imaging (i2PEPICO) spectrometer42 mounted in the SAPHIRS molecular beam source43 at the DESIRS undulator-based variable-polarization vacuum ultraviolet (VUV) beamline44 at Synchrotron SOLEIL. This provides velocity map imaging (VMI) of the electrons, and simultaneous time-of-flight mass determination of coincident ions. The individual electron-ion pair temporal correlation makes it possible to accept only electrons associated with specific ion masses and thus generate mass-tagged photoelectron images and, ultimately, mass-tagged photoelectron spectra. Further experimental information, including an explanation of the Slow Photoelectron scanning technique (SPES) used to obtain the high-resolution photoelectron spectra used for this current study, are provided in ESI.†
The liquid sample was contained in a glass bubbler at room temperature (regulated at 21 °C). Helium gas was passed through at 0.5 bar pressure, providing a continuous gentle agitation of the liquid sample, and the He/vapour mix flowed directly to the spectrometer where it was admitted into vacuum by expansion through a 70 μm nozzle to form a seeded molecular beam. This was passed via a double skimmer arrangement into the spectrometer chamber, where it was intersected in the DELICIOUS 3 source region by the ionizing synchrotron radiation beam.
For these experiments the very mild nozzle expansion conditions (0.5 bar backing pressure) were adopted since it was assessed by considering previous temperature dependent spectra and simulations for the photoelectron spectrum of limonene45 that there was little benefit from strong vibrational cooling in the molecular beam, which might also increase the possibility of unwanted dimer formation.
2.2. Data analysis
The EO and terpene standard scanned spectra were recorded under standardised conditions and processed identically, applying the SPES technique46,47 to optimise the trade-off between resolution and S/N in the spectra. The effective resolution of all reported spectra was thus set during this processing to be ∼20 meV.The EO spectra, SEO, were then fit using a robust linear regression model with the terpene standard spectra, S0a, as predictors:
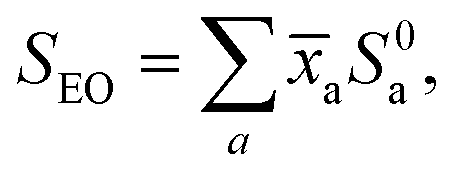 | (1) |
In practice, a constant term was added into the right side of eqn (1) to allow fitting to any residual background in the oil spectrum. Following an initial pass, the model was refined by removing those standards reported to have unphysical negative coefficients (in practice never >2% in magnitude) before being rerun. As an alternative for later comparison, the citrus oil spectra were re-analysed without applying any m/z = 136 mass filtering—effectively non-coincidence mode recordings of the SPES.
3. Results
3.1. Photoionization mass spectra
In Fig. 1 we present, as an example of the citrus oil photoionization mass spectra, that of lime oil recorded using 9.5 eV photon energy. This is above the ionization thresholds of likely constituent monoterpenes (see Table 1) but precedes the onset of significant fragmentation of the parent ions (see supplementary information for ref. 41). Monoterpenes have been reported11 to constitute 88–98% of the volatile fraction of citrus oils, and the principal peak (m/z = 136) is clearly that of unfragmented monoterpene ions. All other peaks observed have relative intensities of 2% or much less. Some of the more prominent of these are examined in the insets (a) and (b) of Fig. 1. These insets show expanded views around the bases of the m/z = 136 and m/z = 154 peaks. All these time-of-flight peak shapes are narrow, with no evidence of broadening by kinetic energy release in dissociative ionization. We thus assume that these are parent ions, indicating molecular weights of corresponding neutrals present in the oil vapour. The m/z = 154 peak we attribute to C10H18O+ ions. Examples of these monoterpenoids, including terpineols, citronellal, linalool etc., are commonly reported in the literature on essential oils. The very much weaker m/z = 152 peak might then suggest C10H16O+ ions. Plausible examples for this mass include neral, citral or limonene oxide. Perhaps surprisingly, there is no corresponding sharp peak at m/z = 204 to indicate the expected presence of sesquiterpenes (C15H24+) such as caryophyllene. Typical ionization energies of terpenoids48 and sesquiterpenes49 are reported to fall in the range 8.3–8.7 eV (but to our knowledge no information exists on the ion fragmentation energies of these compounds) so their ionization may be expected at 9.5 eV photon energy. Of course, the relative peak intensities observed depend not just upon composition but also ionization cross-sections and fragmentation thresholds, and so need not reflect accurately the vapour composition ratios.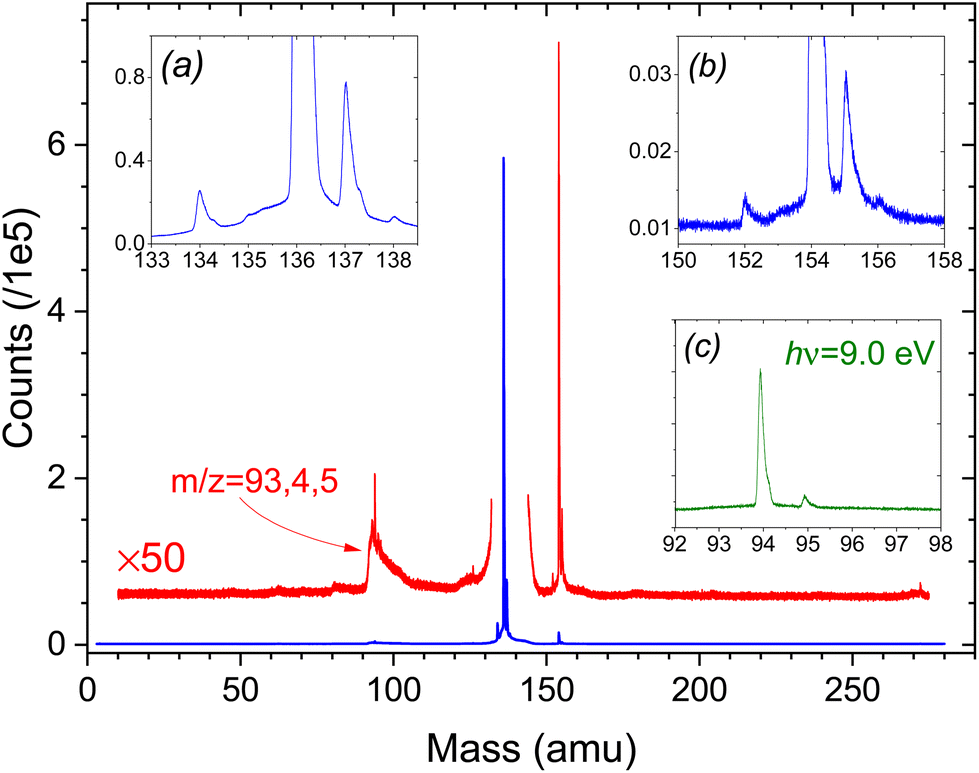 | ||
| Fig. 1 Time-of-flight ion mass spectrum of lime oil headspace recorded with 9.5 eV photon energy. The red trace is a ×50 expansion to better show the minor peaks. The insets (a) and (b) offer expanded views, with horizontal and vertical zoom, of the base regions of, respectively, the m/z = 136 and m/z = 154 peaks. Note that the m/z 137, 138 peaks (inset (a)) are 13C isotopic substituted terpenes at natural abundance. Likewise, the m/z 155 (156) peaks (inset (b)) are attributed to 13C substituted monoterpenoid compounds. Inset (c) shows a section of the m/z 94 region of the hν = 9.0 eV ToF spectrum with vertical scale commensurate to the magnified trace in the 9.5 eV ToF spectrum (see text). | ||
| Sample | Adiabatic IE (eV) | Vertical IE (eV) | Observed AE (eV) m/z = 93/94 |
|---|---|---|---|
| a Supplementary information for ref. 41. b From ref. 73. c From ref. 48. d From ref. 45. e Estimated values, this work. | |||
| α-Pinene | 8.16a | 8.98a | |
| β-Pinene | 8.45b | 8.73c | 9.34a |
| Sabinene | 8.00–8.39a | 9.38a | |
| Limonene | 8.505d | 8.53c | 9.33a |
| 3-Carene | 8.38a | 9.05a | |
| γ-Terpinene | 8.32e | 8.30c | 9.24e |
| α-Phellandrene | 7.91e | 9.31e | |
The m/z = 137, 138 peaks in inset (a) are 13C isotopologues of the principal m/z = 136 terpene peak at natural abundance and appear identically in the mass spectra of the pure terpene standards. However, the small m/z = 134 peak is only seen in the more complex oil samples, indicating that it is unrelated to the C10H16 terpenes. Isomers of cymene (C10H14) have sometimes been noted at levels up to a few % in certain lime oil samples8 and so may account for this m/z = 134 peak.
The cluster around m/z = 94 corresponds to previously observed fragmentation channels of monoterpene cations,41 with the ToF peak shapes clearly now broadened by dissociative kinetic energy release. However, we cannot rule out the possibility that the m/z = 94 cluster also contains contributions from fragmentation of the heavier volatile species.
Superficially, the ToF mass spectra of the other citrus oils studied appear very similar, dominated by an intense m/z = 136 peak, but all other observed peaks have an intensity ≪1% of the principal m/z = 136 terpene peak. Thus, the m/z = 154 peak is much weaker in bergamot oil, while the whole 140–270 amu range of the lemon oil spectra is essentially featureless. Conversely, bergamot displays a sharp, distinct m/z = 196 peak, and both bergamot and lemon ToF spectra show a rather more pronounced sharp peak than does lime oil at m/z = 271, perhaps some dimer formation. No m/z = 134 peak is observed in the lemon oil ToF spectra.
3.2. Mass-filtered slow photoelectron spectra
While it might be considered that the apparently low photoionization intensities of species with mass greater than the monoterpenes would render their presence negligible for an analysis focussed on the monoterpene composition, we nevertheless exploit the advantages of our electron-ion coincidence detection to select electrons originating solely from these terpenes. This is achieved by filtering out electrons not coincident with the sharp m/z = 136 peak noted in the mass spectra. As well as rejecting all electrons correlated with heavier mass species (and the m/z = 134 peak), this also removes contributions associated with lighter fragment ions, since we cannot be sure that these originate solely from terpene ion fragmentation.The terpene standards for our study were selected as those frequently reported as major volatile oil constituents.7,8,11 The full, non-filtered SPES of α- and β-pinenes, 3-carene, sabinene, and limonene have been published previously with accompanying simulations of their vibrational structure.41,45 (More extensive vibrational analyses have been reported for the ground state cations of limonene45 and 3-carene.50) For the present work the raw data of these published SPES have been re-analysed, applying m/z = 136 mass filtering, and with the SPES electron bandwidth increased to 100 meV to trade off resolution for better statistics. These standards have been extended with new recordings, similarly processed, of another bicyclic chiral terpene, α-phellandrene, and the achiral γ-terpinene. An overview of these mass-filtered SPES is provided in Fig. 2. These do not constitute a fully comprehensive set of all plausible terpene constituents that have been reported in the literature on citrus oils, for example camphene and thujene, but those omitted all have typical reported abundances ≪1%.8 The only exception, which we were unable to include, is β-myrcene, that has been reported with a typical abundance ∼1.5%.8,11 As will be seen, the evidence from the oil analyses here is that these omissions have little significance.
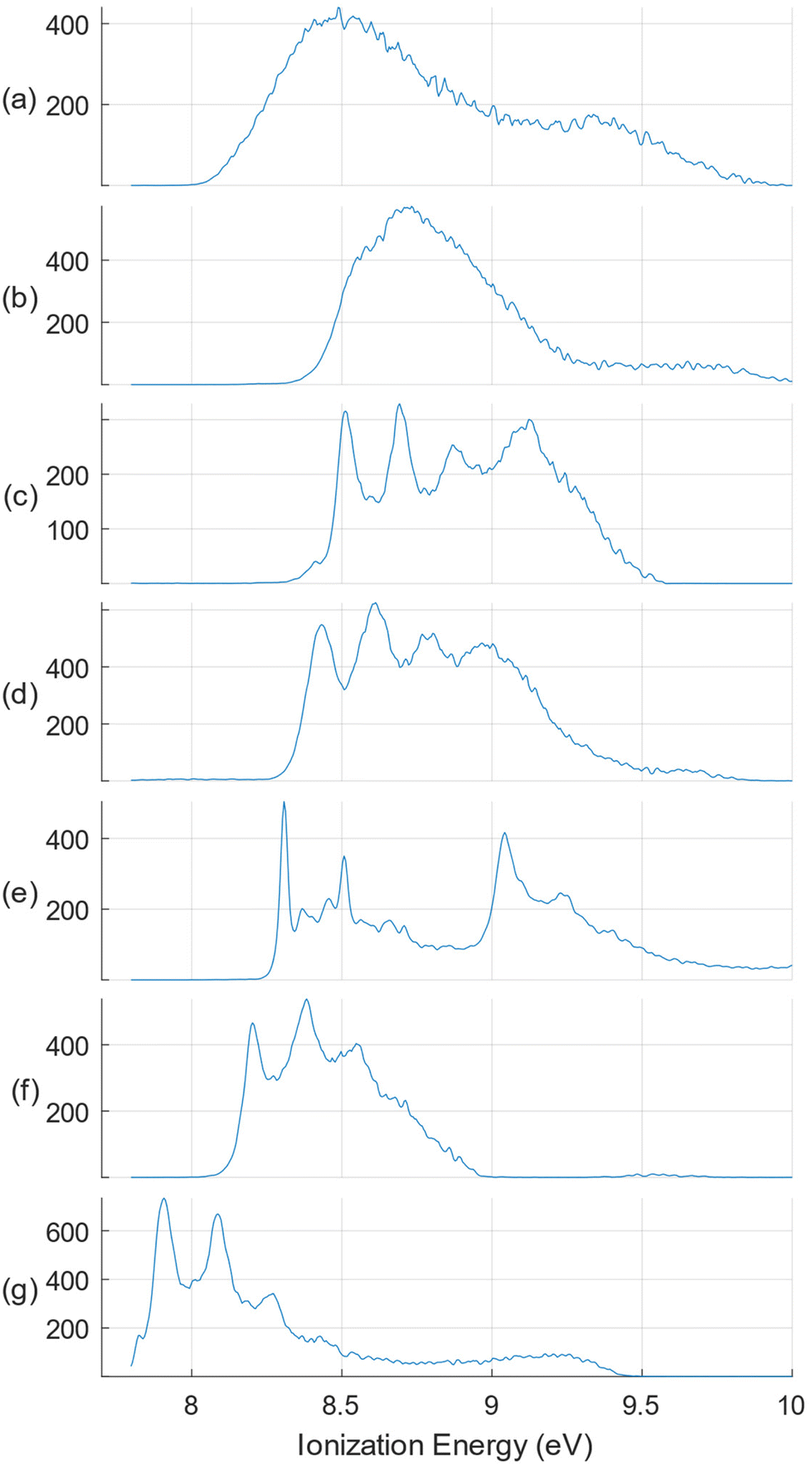 | ||
| Fig. 2 Overview of the m/z = 136 mass filtered SPES monoterpenes selected for this study. Top to bottom: (a) sabinene; (b) β-pinene; (c) limonene; (d) 3-carene; (e) γ-terpinene; (f) α-pinene; (g) α-phellandrene. The SPES are shown normalized for experimental conditions, but with relative intensities expressed in arbitrary units. | ||
In Fig. 3–5 we show the SPES recorded for the citrus oils. There are clear similarities in their appearance and referring to the terpene standards in Fig. 2 it is easy to identify the four distinct peaks of the limonene spectrum, between 8.5 and 9. 2 eV, immediately suggesting that limonene may be a major component of all three. At the same time there are less intense but nevertheless distinct features visible in the 8.0–8.5 eV regions of the three citrus spectra that hint at the presence of other molecules with distinctive structure in this lower ionization energy region, specifically α-pinene, γ-terpinene, or possibly α-phellandrene. The lemon oil spectrum is nevertheless visually distinguishable, having shallower valleys between the limonene peaks; perhaps this may result from broad underlying spectral contributions from sabinene or β-pinene. Just how well these intuitive empirical analyses fare will be discussed in the following section.
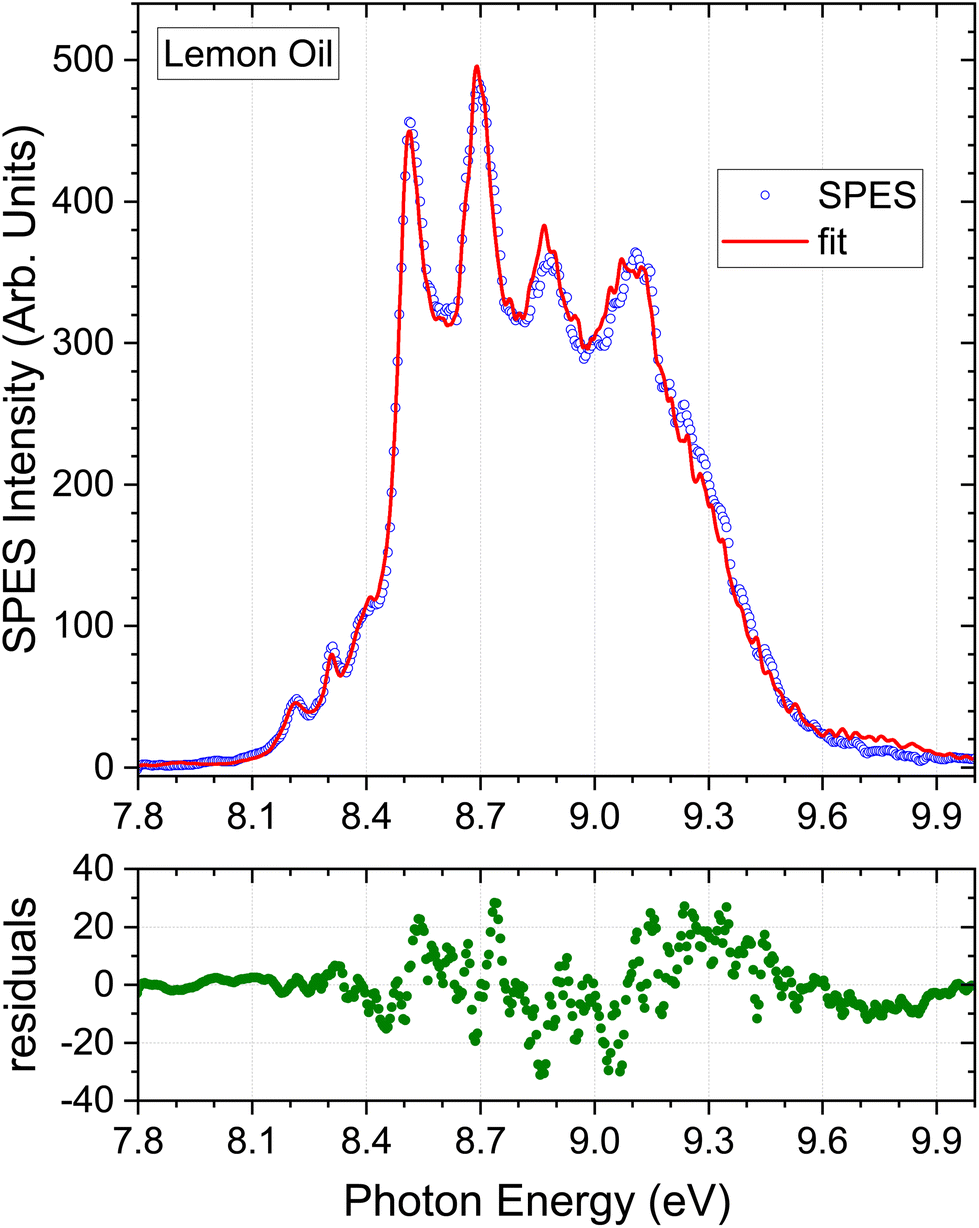 | ||
| Fig. 3 Monoterpene parent mass filtered (m/z = 136) SPES of lemon oil and the best fit obtained using the m/z = 136 mass-tagged terpene standard spectra. The SPES region above 9.3 eV was excluded from the fitting process, but the extrapolation beyond this cut-off, using the derived population coefficients, is included in the plot. The lower panel shows the residuals from the simulation fitted to the experimental lemon oil SPES. | ||
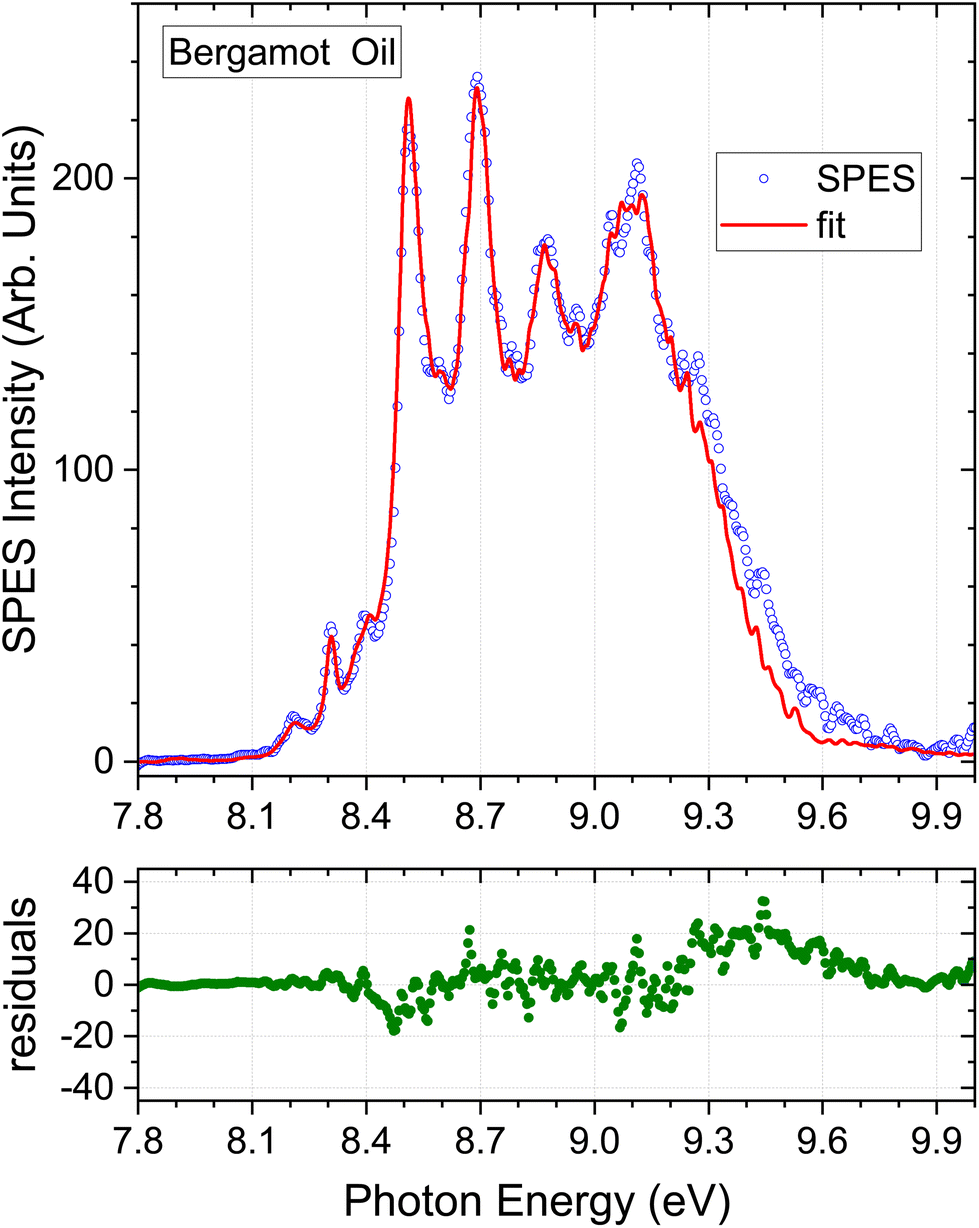 | ||
| Fig. 4 Bergamot oil results. Detail as Fig. 3. | ||
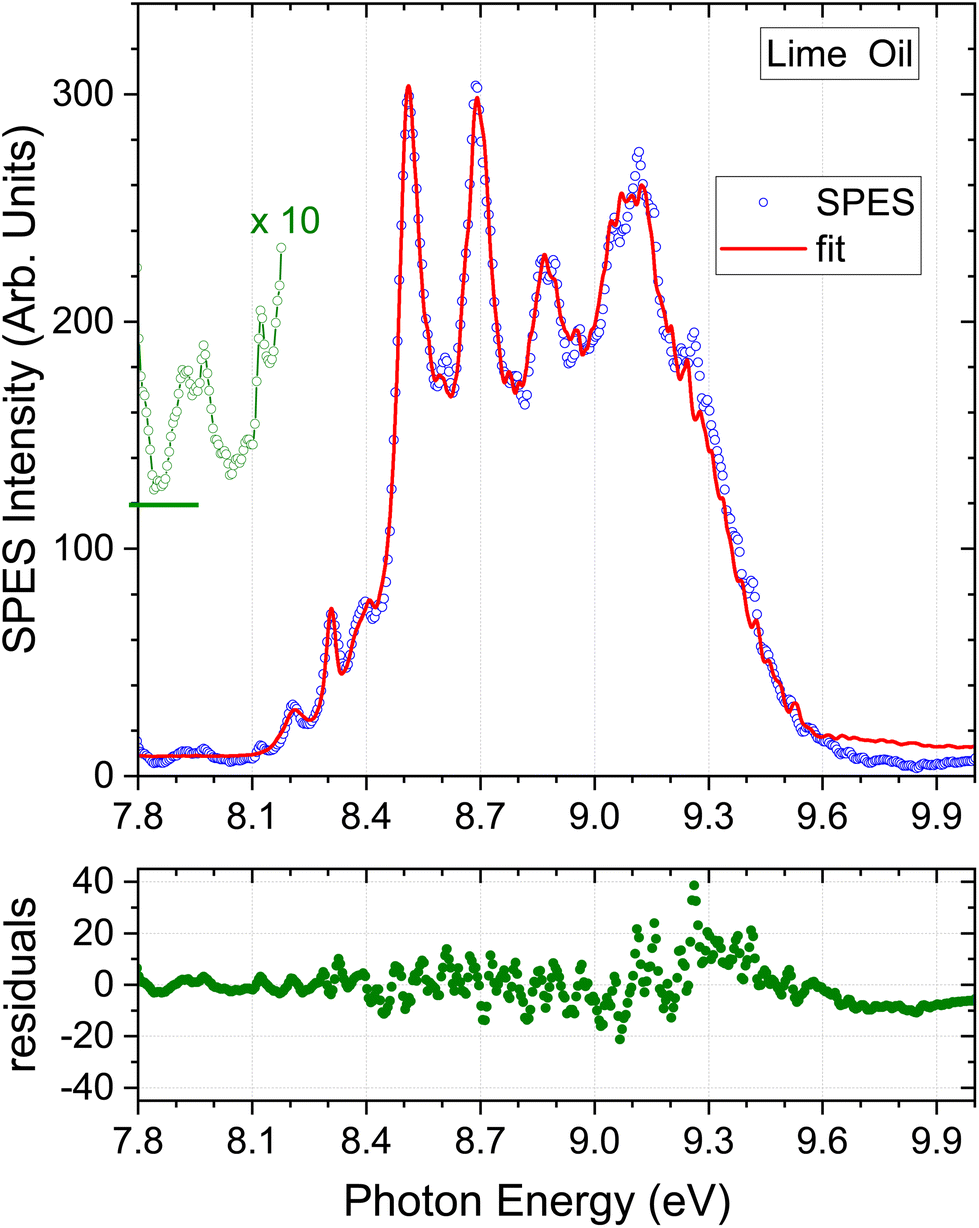 | ||
| Fig. 5 Lime oil results. A ×10 expansion of the region below 8.2 eV is added, but vertically offset for clarity. Other detail as Fig. 3. | ||
Considering the m/z = 136 filtered terpene standard SPES appearing in Fig. 2, it can be seen that the intensity diminishes rapidly above 9.1 eV, and nearly all distinguishing spectral features lie below this energy. In consequence, little additional information can be expected from fitting with the higher energy regions of these spectra. We have therefore applied an upper energy cut-off to exclude higher energies from the regression analysis, trialling a number of values between 9.0 and 9.5 eV. It was found that going beyond a 9.3 eV cut-off, for example to 9.5 eV, produced negligible changes either in the statistical quality of the achieved fit or the deduced composition. The simulated SPES obtained by regression analysis of the essential oil spectra with an applied 9.3 eV cut-off are included in Fig. 3–5, and the normalised regression coefficients are presented in Tables 2–4 accompanied by their standard error estimates.
| SPES analysisa,b | GC analyses | ||||
|---|---|---|---|---|---|
| Pressed lemon (California) | Non-mass-filtered lemon (Ca.) | Cold pressed lemon oilc | Cold pressed lemon oild | Algerian varietiese | |
| a Where necessary, the data have been renormalized to express composition as % of the total reported C10H16 monoterpene content in a sample. Entries appearing as – were not reported. b This work. c Terpene composition ranges derive for industrial cold-pressed lemon oils appearing in columns 1, 2(a–d), 3, 4, 5(a,b), 6, 7 of Table 1.20 in ref. 8. d Composition ranges derived for samples of cultivars of varied (non-Italian) geographic origin appearing in columns 2(d,e), 7(a–c), 8, 9a of Table 1.21 in ref. 8. e Ranges spanned by samples of two Algerian varieties and comparing three different extraction procedures. Data reported in ref. 11. | |||||
| α-Pinene | 5.2 ± 0.4 | 5.2 ± 0.7 | 1.9–2.4 | 1.4–2.9 | 1.1–1.9 |
| β-Pinene | 18.4 ± 0.5 | 22.2 ± 0.8 | 11.8–19.4 | 9.1–15.6 | 7.9–16.7 |
| Sabinene | 3.9 ± 0.7 | 4.8 ± 1.1 | 0.0–2.1 | 0.0–2.7 | — |
| Limonene | 67.0 ± 0.8 | 61.1 ± 1.5 | 64.3–73.6 | 65.6–79.4 | 71.0–81.2 |
| δ-3-Carene | — | 0.0–0.0 | 0.0–0.3 | 0.1–0.2 | |
| γ-Terpinene | 5.2 ± 0.5 | 6.4 ± 0.8 | 7.7–11.2 | 6.7–12.0 | 7.5–9.6 |
| α-Phellandrene | 0.2 ± 0.1 | 0.30 ± 0.16 | 0.0–0.1 | 0.0–1.7 | — |
| Myrcene | — | — | 1.4–1.8 | 0.0–2.3 | 1.4–1.7 |
| α-Thujene | — | — | 0.0–0.5 | 0.0–0.4 | — |
| Camphene | — | — | 0.0–0.1 | 0.0–0.1 | — |
| (E)-β-Ocimene | — | — | 0.0–0.1 | 0.0–0.1 | — |
| (Z)-β-Ocimene | — | — | 0.1–0.2 | 0.0–tr. | — |
| α-Terpinene | — | — | 0.1–0.2 | 0.0–0.2 | — |
| Terpinolene | — | — | 0.2–0.4 | 0.3–0.7 | — |
| Tricyclene | 0–tr. | — | — | ||
| SPES analysisa,b | GC analysesa | |||||
|---|---|---|---|---|---|---|
| m/z 136 filtered | Non-mass-filtered | Cold pressedc | Cold extractedd | Static headspace samplinge | ||
| 22° C | 40° C | |||||
| a Where necessary data have been renormalized to express composition as % of the total reported C10H16 monoterpene content in a sample. Entries appearing as – were not reported. b This work. c Ranges appearing in columns 2, 3, 5, 6, 10–14 of Table 1.17 in ref. 8. d Ranges appearing in columns 1a, 1b, 6b of Table 1.18 in ref. 8. e Data for single extract of Citrus bergamia taken from Table 4, ref. 51. Analysis was performed by static headspace sampling at different regulated temperatures. | ||||||
| α-Pinene | 3.4 ± 0.4 | 3.0 ± 0.6 | 1.3–2.9 | 1.4–3.4 | 6.26 ± 0.12 | 2.21 ± 0.05 |
| β-Pinene | 7.7 ± 0.6 | 7.6 ± 1.3 | 6.4–16.2 | 8.8–19.8 | 22.89 ± 0.36 | 12.17 ± 0.09 |
| Sabinene | 1.1 ± 0.7 | 0.7 ± 1.1 | 0.0–2.2 | 1.9–3.3 | — | — |
| Limonene | 78.7 ± 1.1 | 78.1 ± 2.3 | 60.1–79.8 | 56.5–71.5 | 54.96 ± 0.33 | 63.69 ± 0.50 |
| δ-3-Carene | — | 1.5 ± 0.7 | — | — | — | — |
| γ-Terpinene | 8.8 ± 0.6 | 8.8 ± 0.8 | 7.6–16.3 | 12.7–14.1 | 15.27 ± 0.34 | 20.95 ± 0.17 |
| α-Phellandrene | 0.30 ± 0.04 | 0.3 ± 0.1 | 0.0–0.2 | 0.0–0.1 | 0.31 ± 0.02 | 0.67 ± 0.03 |
| Myrcene | — | — | 1.5–7.5 | 1.5–2.7 | — | — |
| α-Thujene | — | — | 0.0–0.8 | — | — | — |
| Camphene | — | — | 0.0–0.1 | 0.0–0.1 | — | — |
| (E)-β-Ocimene | — | — | 0.0–4.0 | 0.0–0.1 | — | — |
| (Z)-β-Ocimene | — | — | 0.0–2.2 | — | — | — |
| α-Terpinene | — | — | 0.0–0.4 | 0.2–0.3 | 0.31 ± 0.01 | 0.31 ± 0.03 |
| Terpinolene | — | — | 0.0–0.9 | 0.5–0.6 | — | — |
| Terpene | SPES analysisa,b | GC analysesa | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Key lime | Persian lime | |||||||||
| m/z 136 filtered | Non-mass-filtered | Distilled | Pressed | Various | Distilled | Industrial | Cold pressed | |||
| a Where necessary data have been renormalized to express composition as % of the total reported C10H16 monoterpene content in a sample. Entries appearing as – were not reported. b This work. The non-mass-filtered SPES was analysed using a nonnegative constrained least squares procedure; statistical error estimates are not then available. c Data from Jantan et al. ref. 74. d Summary of data from columns 10a & 13(a,b) in Table 1.6 ref. 8. e Average reported in Table 4 ref. 75. f Average derived from data reported for three different Key lime extraction methods in ref. 11. g Summary of data from columns 7–9 of Table 1.7 ref. 8. h Values from 2, 3 of Table 1.7 ref. 8. i Values from laboratory cold-pressed extraction reported in Table 1.5 ref. 8. j Averaged value reported in Table 5 ref. 75. k Sabinene and β-pinene are commonly reported to co-elute. Where no value is reported for Sabinene it was most likely was not resolved. In such cases it may be reasonable to expect that sabinene constitutes a few % of the reported abundance for β-pinene. | ||||||||||
| α-Pinene | 4.5 ± 0.5 | 3.9 | 2.0 | 2.0–4.0 | 2.9 | 2.0 | 1.9–2.4 | 2.0–2.3 | 3.6 | 2.4 |
| β-Pinene | 5.4 ± 0.7 | 3.8 | 39.1 | 19.9–27.3 | 27.4 | 14.1 | 12.4–15.8 | 11.6–12.2 | 14.5 | 14.6 |
| Sabinenek | 0.3 ± 0.8 | 2.1 | — | 0.0–2.1 | — | — | 0.0–2.0 | 1.6–2.0 | 2.2 | — |
| Limonene | 79.1 ± 1.1 | 77.3 | 53.9 | 43.8–66.6 | 57.9 | 70.5 | 60.7–66.5 | 64.2–65.9 | 58.1 | 64.1 |
| δ-3-Carene | — | 0.0 | 0.7 | — | tr. | — | — | — | tr. | tr. |
| γ-Terpinene | 10.8 ± 0.5 | 9.1 | 1.1 | 5.2–21.1 | 9.1 | 11.8 | 15.7–16.6 | 15.9–16.2 | 18.9 | 15.3 |
| α-Phellandrene | 0 ± 0.001 | 3.7 | 0.1 | 0.0–0.7 | tr. | — | — | 0.0–0.1 | tr. | tr. |
| Myrcene | — | — | 1.4 | 0.4–3.3 | 1.5 | 1.6 | 1.5–1.8 | 1.4–1.7 | 1.4 | 1.6 |
| α-Thujene | — | — | 0.1 | 0.0–0.3 | 0.5 | — | 0.0–0.6 | 0.6–0.7 | 0.1 | 0.6 |
| Camphene | — | — | 0.2 | 0.1–0.7 | 0.1 | — | 0.0–0.1 | 0.0–0.1 | 0.1 | 0.1 |
| (E)-β-Ocimene | — | — | — | 0.0–0.5 | — | — | 0.0–0.1 | 0.0–0.1 | tr. | 0.1 |
| (Z)-β-Ocimene | — | — | — | 0.0–1.3 | — | — | — | — | tr. | 0.1 |
| α-Terpinene | — | — | 1.5 | 0.0–1.9 | 0.6 | — | 0.0–0.3 | 0.0–0.3 | 0.3 | 0.3 |
4. Discussion
A primary objective for many essential oil headspace analyses is the creation of a sample profile, which identifies the component species and characterises them as major, minor, trace (or possibly absent) constituents, but often stopping short of achieving full quantification. For complex plant-derived matrices there may be significant matrix-effects in the liquid/vapour partitioning, and precise equilibrium ratios can be expected to be temperature dependent.51 Likewise, when headspace pre-concentration procedures are adopted prior to presentation of the vapour for analysis, the adsorption and desorption temperatures will influence the vapour/adsorbate equilibria, and the choice and preparation of adsorbent material may introduce additional selectivity in the transfer stages.17,52–54 It is not then uncommon for relevant experimental conditions to be empirically optimised to improve the resolution (reduce co-elution) of the components at the expense of quantitative precision. Preparation for fully quantitative analysis can be an onerous procedure,10 and the complete calibration factors are unlikely to be directly transferable to other studies.With these caveats about reported quantitative accuracy, a natural step at this proof-of-principle stage of our photoelectron method is to make comparison with composition profiles obtained by representative GC analyses taken from the literature.
4.1. Lemon oil
Our lemon oil sample (citrus limon) was stated to be cold-pressed and of Californian origin but no more specific information on cultivar was provided. Table 2 lists the composition deduced from the regression analysis. As anticipated from visual examination the dominant component is limonene, followed by β-pinene. γ-Terpinene and α-pinene contribute around 5% each. The table also summarises various profiles of lemon oil samples obtained by GC analysis that have been reported in the literature. As already noted, fully quantitative agreement is not to be expected. Measured profiles should also, of course, show purely sample-dependent variations: the region of growth, species and cultivar, and method of extraction will be crucial factors.55 These uncertainties and variations are fully evident in the composition ranges evident in the summarised GC results.The semi-quantitative match of the GC composition profiles shown in Table 2 and the present SPES results for the seven selected terpene standards, is however, very good. A potential error may arise from the failure to include myrcene in our library of SPES standards. As seen in Table 2 GC analysis generally suggests ∼1.5% abundance for this species. Little information on ionization/fragmentation thresholds is available for myrcene, although a low resolution He I photoelectron spectrum has been reported, with a vertical ionization energy of ∼8.68 eV.48 The pertinent region of this spectrum has been replotted and can be seen in ESI, Fig. S1.† This offers some approximate indication of how the high-resolution mass-tagged SPES might look. The position and overall HOMO band envelope appear, as best can be judged, to quite closely map the β-pinene profile. We see no evidence for systematic errors in the corresponding central region of the essential oil SPES fits that could be attributed to myrcene. It may then be expected that any small missing myrcene SPES contribution may be partially disguised by a slight raise in the apparent abundance of β-pinene, but we conclude that the exclusion of this minor component from the fitting procedure does not for now result in significant overall error.
In light of the preceding comments on variability of associated instrumental factors in the GC literature, and the omission of myrcene from the fitting process, the SPES for a second lemon oil sample stated to be a rectified extract of Argentinian origin was recorded and analysed. The instrumental factors and methods are maintained constant, and only the lemon oil sample varies. The low energy region of this Argentinian lemon is expanded in Fig. 6. The overall quality of the fit obtained by the regression analysis is again good. A small systematic deviation can just be observed around 8.45 eV, possibly attributable to the missing standards in the regression model. Of greater significance, a clear discrimination between the two lemon extracts can be seen from the increase in relative intensity of the 8.3 eV structure (attributable to γ-terpinene) in the Argentinian lemon oil when a comparison is made under our identical experimental conditions.
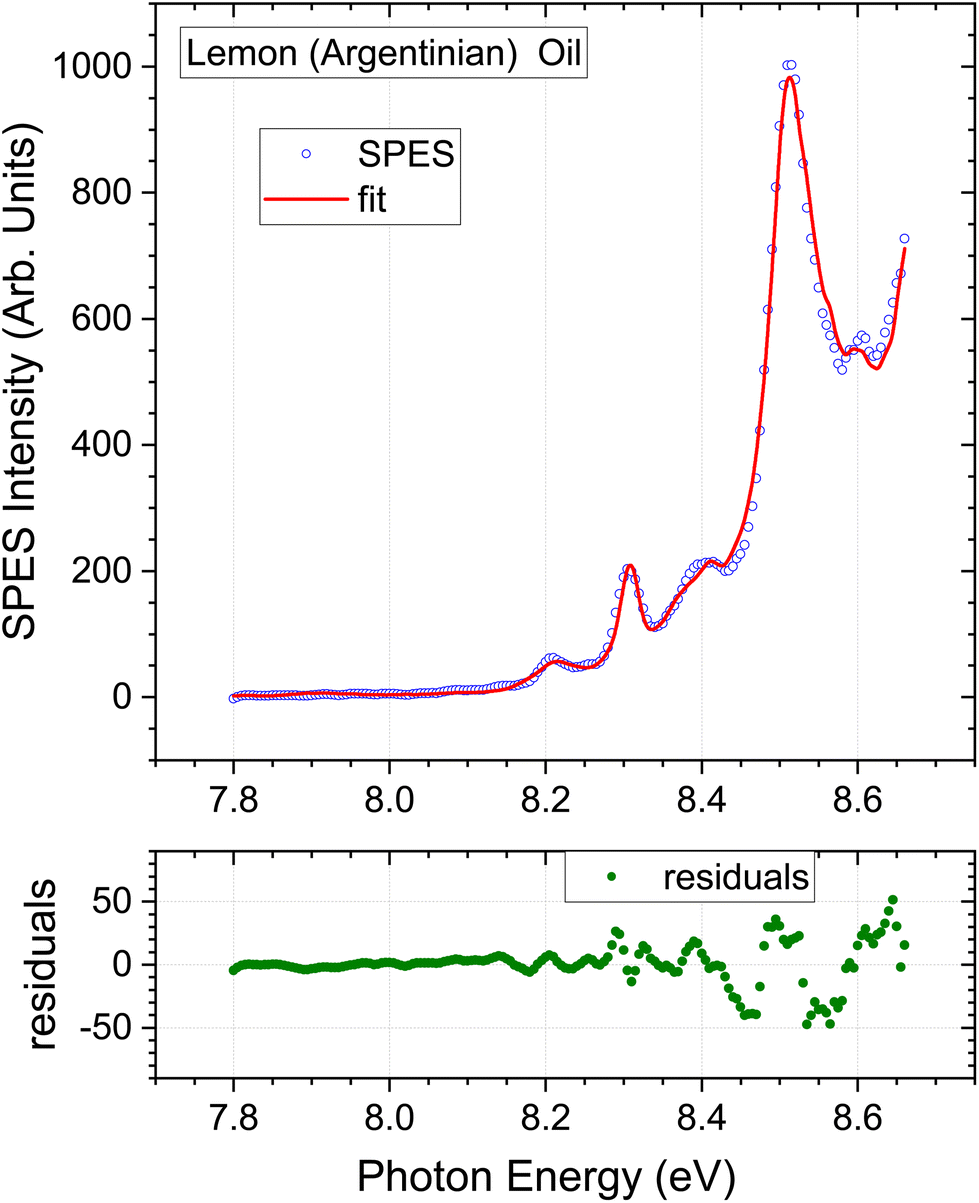 | ||
| Fig. 6 Lemon oil (distilled) of argentinian origin. Expanded view of low energy region. Other detail as Fig. 3. | ||
4.2. Bergamot oil
The composition determined by the present SPES analysis is compared with selected GC results from the literature in Table 3. Again, we caution against expecting an exact match between these methods due to the number of sample and instrumental variables, and once again these limitations are implicit in the ranges evident in the GC results alone. Of particular note, the final columns in Table 3 contain a study of a single sample that used a static headspace sampling, with direct injection via a gas syringe onto the GC column.51 A different selectivity is evident between the 22° and 40° C equilibrated sampling. No value was reported for sabinene in these studies. Difficulties in resolving sabinene and β-pinene due to their co-eluting are commonly noted, and it may be inferred that the “β-pinene” abundance here may represent the sum of β-pinene and sabinene components.With the foregoing caveats, the concentration profile obtained from the fit shown in Fig. 4 matches that of the GC studies from the literature that have been included in Table 3, with all seven of our terpene standard concentrations falling within the quoted ranges established by GC, albeit the SPES value for limonene is at the upper limit. As was the case with lemon oil, the omission of a myrcene component from the SPES regression model is, in principle, a source of error given an expected abundance of a few % indicated by Table 3. The small discrepancy between the bergamot SPES and its fit seen above 9.3 eV (Fig. 4) seems unlikely to be attributable to myrcene given that the intensity of the myrcene He I PES (Fig. S1 ESI†) intensity is already low and rapidly falling further in this energetic region. However, no other explanation can be advanced at present. None of the other oils (Fig. 3, 5 and 6) evidence the same difficulty in this region.
4.3. Lime oil
Uniquely among the samples studied here, the lime oil SPES has weak structure apparent below 8.15 eV. This region has been replotted with a ×10 vertical scale expansion in Fig. 5. Peaks appear at 8.12, 7.97, and 7.92 eV. Unfortunately, our scans do not extend below 7.8 eV as such low ionization thresholds had not been anticipated, but a further relatively strong peak looks to be emerging at or below this cut-off. The lowest ionized of our terpene standards, α-phellandrene, has structure in this region, but not closely matching these lime oil features (see e.g., Fig. S2 ESI†). There is little data concerning ionization energetics of many other terpenes. To our knowledge, the only other terpene, known to have even lower ionization thresholds is α-terpinene, for which an adiabatic ionization energy of 7.47 eV (ref. 56) and vertical energies of 7.57–7.67 eV (ref. 48 and 57) have been reported. We can thus make this a speculative attribution for these low energy features in the lime oil spectrum.Because the low energy lime SPES structure is not matched in our current library of terpene standard SPES, the regression analysis tends, in consequence, to visibly overestimate the base-line at the extremes of the spectrum (Fig. 5). This will in turn induce some minor inaccuracies in the remaining regression coefficients that determine the composition. With this caveat, the lime oil composition result is shown in Table 4. The bergamot and lime SPES spectra (Fig. 4 and 5) have a similar overall appearance, and so it is no surprise that the composition profiles are also quite similar.
An initial comparison with typical results obtained in previous GC measurements of lime, appearing alongside in Table 4, is not helped by the high variability these exhibit. Even so, the SPES value for β-pinene lies significantly below, and the limonene result slightly above, the ranges quoted in Table 4. Myrcene once again seems to be the major component excluded from our SPES regression model but, again, with our limited knowledge of the photoionization of this molecule we do not detect any significant perturbation that might obviously be attributed to this omission. Many studies also identify α-terpinene as a minor (sub 1%) component of lime oils, making our speculative visual identification of this molecule in the SPES more plausible.
The lime sample supplied for this study was stated to originate from Mexico, but its botanical origin was not specified. There are two main varieties of lime, Key lime (citrus aurantifolia) and Persian lime (citrus latifolia). “Mexican lime” is often understood to be synonymous with Key lime, but paradoxically Mexico is also the largest grower of Persian limes. The categorization of our sample is thus ambiguous.
In a comprehensive review of the published studies of citrus oil composition, Dugo et al.8 remark on the many different lime cultivars and geographical growth areas that lead to wide variation in reported composition and so may cloud detailed comparisons. Nevertheless, it is suggested that useful distinguishing characteristics of Persian limes are a lower concentration of β-pinene and higher concentration of γ-terpinene and limonene than found in Key limes. The columns headed Key and Persian lime types in Table 4 clearly exemplify such differences. While quantitatively imperfect, the SPES composition profile, in particular for these three distinguishing components, follows more closely the Persian lime examples in that table. One may therefore deduce that our provided sample extract was most likely Persian lime.
4.4. Non-mass-filtered SPES analyses
From the ToF mass spectra (e.g.Fig. 1) it is clear that in the photon energy range that has been considered (≤9.5 eV) the citrus oil photoionization yields are very strongly dominated by parent monoterpene (m/z = 136) cations. It is consequently of interest to examine the benefits achieved by applying the m/z mass filtering to explicitly reject the other minor species. Such filtering requires the spectra to have been recorded in electron-ion coincidence mode, and this imposes additional experimental complexity and constraints, a critical one being the need of continuous or very high repetition rate photon sources, another one imposing a lowering of the photon flux to limit, even with a pseudo-continuous source as synchrotron radiation, the amount of false coincidences. There would, therefore, be potential advantages in speed and cost if the mass filtering could be dispensed with. Accordingly, the regression analyses were repeated, but using the essential oil spectra with no mass-filtering, by simply disregarding the coincident ion data.Regression modelling results for the three citrus oils are included in the relevant Tables 2–4 and plots of the unfiltered SPES and best fit simulations are available in ESI (Fig. S3–S5†). One general observation from the figures is that without applying mass-filtering the SPES S/N is typically worsened above ∼8.7 eV. Not only does the mass filtering reject electrons associated with specific ToF peaks, it also rejects all of the uncorrelated electrons found in the SPES baseline, and hence helps eliminate random noise in the spectrum. This may partially account for an approximate doubling of the statistical error estimates accompanying the un-filtered regression coefficients in the tables.
The Californian lemon oil results (Table 2) maintain a very similar composition profile with or without the coincident mode mass filtering, the biggest change being a drop of 6% in the reported abundance of the major component limonene. Better yet, the reported composition of the bergamot oil (Table 3) is virtually unchanged by the removal of the m/z = 136 mass filtering.
The unfiltered lime oil SPES presents its own challenge. Fully evident visually, the unfiltered lime SPES (Fig. S5 ESI†) unexpectedly reveals intense new structure (∼25% as intense as the limonene peaks) in the low energy region around 8 eV. No corresponding features are found in spectra of any of the terpene standards or other oil samples. On closer examination of the data, this feature is associated very specifically with the m/z = 94 mass channel. Fig. 1 shows the m/z = 94 eV region of the lime oil ToF spectrum. While fragmentation is evident in the broad m/z = 93–95 cluster with 9.5 eV photoionization, the 9 eV ToF spectrum (inset (c)) consists of a single, sharp mass 94 peak. From its width we conclude that at 9 eV, and presumably down to an ionization threshold, this is an undissociated parent ion, and the coincident SPES is that of a m/z = 94 neutral species.
The first two peak positions in the m/z = 94 filtered SPES we measure at 7.96 and 8.14 eV. Close examination (see e.g. Fig. S2 ESI†) shows a poor correspondence with the low energy structure noted in the m/z = 136 mass-selected lime SPES (provisionally identified as α-terpinene) re-affirming their independence and helping corroborate that at such low ionization energies the m/z = 94 feature is not a fragmentation channel. A search for C7H10 species with known first ionization energies in this region returns trans-1,3,5 heptatriene as the only candidate, and indeed its reported ionization energy of 7.96 eV and He I PES58 convincingly match the m/z = 94 channel SPES. We are not aware of heptatriene being a reported constituent in other studies.
A consequence of such an intense unaccounted feature in the unfiltered SPES is that the quality of the achievable fit using the preselected standard monoterpenes is degraded. The composition figures appearing in Table 4 were obtained using a non-negative constrained least squares procedure. While an imperfect fit necessarily results (see Fig. S5 ESI†), a comparison of the filtered- and non-filtered composition figures in the table shows rather remarkable agreement with differences in the percentage compositions less than ±2%.
5. Conclusions and perspective
We have shown a new approach for the analysis of the volatile terpene content using high resolution photoelectron spectroscopy. By recording electrons in coincidence with their mass-selected cations, the electron spectra can be mass-tagged, allowing the ionization yield to be selectively filtered to represent, in this case, just the monoterpene (m/z = 136) components. Using dynamic headspace sampling to feed the volatiles directly to the photoionization spectrometer, slow-photoelectron spectra (SPES) of four essential oil samples—lemon (×2), bergamot, and lime—were recorded and the volatile monoterpene content of each determined by regression modelling with a small library of seven pure terpene standard spectra. Examination of the standards’ SPES shows that five have very strongly structured spectra between 7.8 and 8.5 eV, sufficiently distinct to allow a qualitative visual identification in the essential oil spectra; a quantitative regression analysis is shown to identify these down to the sub 1% abundance level, with typical statistical error bars at most in the % range. With fragmentation channels of the terpene cations slowly opening from ∼9 eV photon energy, it has furthermore been established that little to no additional information would be provided by extending the SPES measurement and analysis to energies beyond 9.3 eV.The terpene concentration profiles determined in this proof-of-principle demonstration match well previously measured composition profiles obtained by GC, with the caveat that these themselves have a wide scatter in the literature that can be attributed to: (i) differences between cultivars of a given species; (ii) natural variability between individual samples of a cultivar, differences due to geographic origin, growth history, ripeness; (iii) sample extraction methods and matrix pre-processing; (iv) choice of sample injection procedures, injection temperature and column temperature programming, co-elution (resolution of components), detector efficiency and response functions, methods used to prepare reports from the chromatograms; (v) (in the case of headspace sampling) selectivity exhibited by pre-concentration stages due to of adsorbent material, its pre-treatment, adsorption and desorption temperatures. As has been noted by other authors, the enormous variability in instrumental factors (iii) – (v) may impede cross-comparisons for the more interesting variations arising from (i) and (ii). The present results do successfully demonstrate a clear discrimination between the two distinct lemon oil samples investigated.
The headspace sample handling described in this study offers an easy approach to standardisation of many of these instrumental variables. The dynamic headspace sampling requires no sample preparation steps beyond the extraction process. No pre-concentration process is required as the photoionization efficiency and sensitivity permit the vapour stream to be fed directly and continuously to the spectrometer without compromising sensitivity, and ambient temperature is maintained throughout, avoiding any risks of temperature induced degradation.
It is not contended that the technique as described here represents a viable alternative to existing analytical approaches, despite the simplicity and advantages of this headspace sampling arrangement, not least because of the restricted availability of synchrotron beamtime, and relatively slow SPES scan rates. Rather, our purpose was to prove and demonstrate the potential of photoelectron spectroscopy to filter out and then distinguish the monoterpene components of such analytes. This approach to isomeric/isobaric discrimination in complex media has been well established in other contexts59 including combustion,60 pyrolysis,61 and catalytic62 studies and including also atmospheric chemistry.63
Looking beyond this, photoelectron spectroscopy has the ability simultaneously to determine enantiomeric excess of the individual chiral components. Enantioselective determination of the levels of chiral volatile species in essential oils is recognised as an extremely sensitive, informative tool when seeking to establish authenticity or prove adulteration of essential oils52,54,64 and hence would be a desirable additional capability.
Photoelectron circular dichroism (PECD) measures, as an adjunct to their kinetic energy, the angular distribution of the photoelectrons. With circularly polarized radiation, PECD angular distributions become chiral enantiomer sensitive.65 A proof of principle demonstration of this chiral analytical capability made use of coincident ion-electron detection, just as here, to distinguish by mass two terpenoid compounds in a mixture and measure, simultaneously, the enantiomeric excess (ee) of each by PECD.66 An alternative analysis of ee in a two component mixture, accomplished without using coincidence methods, has recently been reported using a pulsed laser source.67 Subsequent to the earlier report, several authors have demonstrated that PECD ee determinations can be made to sub 1% precision with measurement times of as little as a few minutes.28–31
Consequently, further work is underway to develop and extend the technique presented here, in the first instance by concurrent measurement of abundance and ee. The use of an imaging electron detector for the combined measurement of photoelectron angle and energy distributions in a fully multiplex manner will allow, as a secondary benefit, upgrading the relatively inefficient threshold electron spectroscopic method used here. Indeed, rather than slowly scanning the photon energy, a complete photoelectron spectrum covering the few eV above threshold can be acquired using a single, fixed energy photon beam, with much reduced data acquisition times, albeit with a reduced electron kinetic energy resolution.
Viable alternatives to synchrotron radiation for such applications are already established with laboratory-scale lasers systems able to provide more convenient and accessible sources of the required photons. Multiphoton ionization with UV lasers brings its own advantages in an analytical context;68 alternatively harmonic generation of VUV photons at multi kHz repetition rates has been demonstrated to permit efficient electron-ion coincidence modes of data recording.61,69 Efficient harmonic generation methods can now also generate VUV photons with a high degree of circular polarization,70 including the 9–10 eV range71 convenient for these terpene studies. Taken with recent advances in high repetition rate fibre laser technology the rapid, precise enantioselective measurement of terpene mixtures in mass-filtering coincidence detection modes is now fully feasible.69 On the other hand, dispensing with coincidence mode data recording offers the prospect, in carefully selected cases, of data acquisition with significant further speed improvements, or allows the use of low repetition rate photoionization sources, at the expense of a moderate reduction in accuracy.
The DELICIOUS/SAPHIRS photoionization spectrometer42,43 employed in this study is a provided end-station at the DESIRS beamline and as such it is configured to support the varied requirements of a multitude of user experiments. In consequence it may sometimes exceed the requirements of a particular experiment. The rather mild nozzle expansion conditions adopted for the current study generate a relatively low gas load, making SAPHIRS’ second skimmer stage strictly unnecessary. Equally, a switch from synchrotron radiation to a multi-photon laser ionization source could render both the skimmer stages redundant since, apart from differential pumping, they are intended to provide a well collimated molecular beam in order to limit the spatial extent of the interaction region along the light beam direction. The small size of the effective focal volume of a tightly focussed non-linear laser interaction automatically imposes this spatial restriction without necessitating mechanical collimation. As an illustration of the minimal instrumental requirements for making laser PES and PECD measurements, Krüger et al.72 have recently presented details of a compact table top electron or ion VMI spectrometer along with an example PECD measurement. Using an unskimmed 15μm diameter gas inlet jet the vacuum pumping requirements have been reduced to two, 300 + 700 l s−1, turbo pumps (compared to the 5900 l s−1 total pumping speed of SAPHIRS).
Accordingly, all the required elements are in place to envisage how enantioselective photoelectron analysis for volatile components of natural products could become a routinely viable method.
Author contributions
Conceptualization: IP; Methodology: GAG, LN, HG, IP; Data Curation: GAG; Formal analysis: HG, RH, GAG; Software: HG, GAG, IP; Visualisation: HG, IP; Writing – original draft: ip; Review & editing: LN, GAG, HG. Investigation: All authors.Conflicts of interest
The authors declare they have no conflict of interests.Acknowledgements
This research was undertaken as part of the ASPIRE Innovative Training Network, which has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 674960. HG and RH acknowledge ESR fellowships provided by Aspire. We are grateful for the provision of beamtime by Synchrotron Soleil (beamtime Proposal No. 20151368, 20161224, 20181872) and we thank the technical staff at Soleil, and especially J.-F. Gil, for their support and for the smooth operation of the facility.References
- Handbook of Essential Oils, ed. K. H. C. Başer and G. Buchbauer, CRC Press, Boca Raton, 2020 Search PubMed.
- H. S. Elshafie and I. Camele, BioMed Res. Int., 2017, 2017, 9268468 CrossRef PubMed.
- M. Perricone, E. Arace, M. R. Corbo, M. Sinigaglia and A. Bevilacqua, Front. Microbiol., 2015, 6 DOI:10.3389/fmicb.2015.00076.
- J. R. Calo, P. G. Crandall, C. A. O'Bryan and S. C. Ricke, Food Control, 2015, 54, 111–119 CrossRef CAS.
- N. Sadgrove and G. Jones, Agriculture, 2015, 5, 48–102 CrossRef.
- N. J. Sadgrove, G. F. Padilla-Gonzalez and M. Phumthum, Plants, 2022, 11, 34 Search PubMed.
- N. Mahato, K. Sharma, R. Koteswararao, M. Sinha, E. Baral and M. H. Cho, Crit. Rev. Food Sci. Nutr., 2019, 59, 611–625 CrossRef CAS PubMed.
- Citrus Oils: Composition, Advanced Analytical Techniques, Contaminants, and Biological Activity, ed. G. Dugo and L. Mondello, CRC Press, Taylor & Francis, Boca Raton, 2011 Search PubMed.
- T. K. T. Do, F. Hadji-Minaglou, S. Antoniotti and X. Fernandez, TrAC, Trends Anal. Chem., 2015, 66, 146–157 CrossRef CAS.
- C. Bicchi, E. Liberto, M. Matteodo, B. Sgorbini, L. Mondello, B. D. Zellner, R. Costa and P. Rubiolo, Flavour Fragrance J., 2008, 23, 382–391 CrossRef CAS.
- N. Bousbia, M. A. Vian, M. A. Ferhat, B. Y. Meklati and F. Chemat, J. Food Eng., 2009, 90, 409–413 CrossRef.
- A. Arigò, M. Zoccali, D. Sciarrone, P. Q. Tranchida, P. Dugo and L. Mondello, in Handbook of Essential Oils, ed. K. H. C. Başer and G. Buchbauer, CRC Press, Boca Raton, 3rd edn, 2020, ch. 7, DOI:10.1201/9781351246460.
- P. J. Marriott, R. Shellie and C. Cornwell, J. Chromatogr. A, 2001, 936, 1–22 CrossRef CAS PubMed.
- P. Q. Tranchida, I. Bonaccorsi, P. Dugo, L. Mondello and G. Dugo, Flavour Fragrance J., 2012, 27, 98–123 CrossRef CAS.
- E. Liberto, C. Bicchi, C. Cagliero, C. Cordero, P. Rubiolo and B. Sgorbini, in Advanced Gas Chromatography in Food Analysis, ed. P. Q. Tranchida, 2020, ch. 1, vol. 17, pp. 3–37 Search PubMed.
- P. R. Chaudhary, G. Jayaprakasha and B. S. Patil, in Instrumental methods for the analysis and identification of bioactive molecules, ed. G. K. Jayprakasha, B. S. Patil and F. Pellati, ACS Publications, 2014, ch. 12, pp. 243–256, DOI:10.1021/bk-2014-1185.ch012.
- N. Reyes-Garcés, E. Gionfriddo, G. A. Gómez-Ríos, M. N. Alam, E. Boyacı, B. Bojko, V. Singh, J. Grandy and J. Pawliszyn, Anal. Chem., 2018, 90, 302–360 CrossRef PubMed.
- A. Shapira, P. Berman, K. Futoran, O. Guberman and D. Meiri, Anal. Chem., 2019, 91, 11425–11432 CrossRef CAS PubMed.
- E. Papada, A. Gioxari, C. Amerikanou, N. Galanis and A. C. Kaliora, Foods, 2020, 9, 1019 CrossRef CAS PubMed.
- L. K. Silva, M. F. Espenship, C. A. Newman, B. C. Blount and V. R. De Jesus, Environ. Sci. Technol., 2020, 54, 13861–13867 CrossRef CAS PubMed.
- M. Hernandez-Mesa, D. Ropartz, A. M. Garcia-Campana, H. Rogniaux, G. Dervilly-Pinel and B. Le Bizec, Molecules, 2019, 24, 2706 CrossRef CAS PubMed.
- R. Rodríguez-Maecker, E. Vyhmeister, S. Meisen, A. Rosales Martinez, A. Kuklya and U. Telgheder, Anal. Bioanal. Chem., 2017, 409, 6595–6603 CrossRef PubMed.
- C. L. Qiu, J. Smuts and K. A. Schug, J. Sep. Sci., 2017, 40, 869–877 CrossRef CAS PubMed.
- K. R. Strehle, P. Rösch, D. Berg, H. Schulz and J. Popp, J. Agric. Food Chem., 2006, 54, 7020–7026 CrossRef CAS PubMed.
- D. J. Daferera, P. A. Tarantilis and M. G. Polissiou, J. Agric. Food Chem., 2002, 50, 5503–5507 CrossRef CAS PubMed.
- S. Lafhal, P. Vanloot, I. Bombarda, J. Kister and N. Dupuy, Ind. Crops Prod., 2016, 80, 156–164 CrossRef CAS.
- B. Steuer, H. Schulz and E. Läger, Food Chem., 2001, 72, 113–117 CrossRef CAS.
- A. Kastner, C. Lux, T. Ring, S. Zullighoven, C. Sarpe, A. Senftleben and T. Baumert, ChemPhysChem, 2016, 17, 1119–1122 CrossRef CAS PubMed.
- A. Comby, E. Bloch, C. M. M. Bond, D. Descamps, J. Miles, S. Petit, S. Rozen, J. B. Greenwood, V. Blanchet and Y. Mairesse, Nat. Commun., 2018, 9, 5212 CrossRef CAS PubMed.
- L. Nahon, L. Nag, G. A. Garcia, I. Myrgorodska, U. Meierhenrich, S. Beaulieu, V. Wanie, V. Blanchet, R. Géneaux and I. Powis, Phys. Chem. Chem. Phys., 2016, 18, 12696–12706 RSC.
- J. Miles, D. Fernandes, A. Young, C. M. M. Bond, S. W. Crane, O. Ghafur, D. Townsend, J. Sá and J. B. Greenwood, Anal. Chim. Acta, 2017, 984, 134–139 CrossRef CAS PubMed.
- A. Stolle, C. Brauns, M. Nüchter, B. Ondruschka, W. Bonrath and M. Findeisen, Eur. J. Org. Chem., 2006, 2006, 3317–3325 CrossRef.
- A. Stolle, B. Ondruschka and H. Hopf, Helv. Chim. Acta, 2009, 92, 1673–1719 CrossRef CAS.
- C. Geron, R. Rasmussen, R. R. Arnts and A. Guenther, Atmos. Environ., 2000, 34, 1761–1781 CrossRef CAS.
- B. Larsen, T. Bomboi-Mingarro, E. Brancaleoni, A. Calogirou, A. Cecinato, C. Coeur, I. Chatzianestis, M. Duane, M. Frattoni, J. L. Fugit, U. Hansen, V. Jacob, N. Mimikos, T. Hoffmann, S. Owen, R. Perez-Pastor, A. Reichmann, G. Seufert, M. Staudt and R. Steinbrecher, Atmos. Environ., 1997, 31, 35–49 CrossRef CAS.
- M. L. R. Del Castillo, M. M. Caja, G. P. Blanch and M. Herraiz, J. Food Prot., 2003, 66, 1448–1454 CrossRef PubMed.
- M. G. Chisholm, M. A. Wilson and G. M. Gaskey, Flavour Fragrance J., 2003, 18, 106–115 CrossRef CAS.
- W. J. Zubyk and A. Z. Conner, Anal. Chem., 1960, 32, 912–917 CrossRef CAS.
- S. Hamm, E. Lesellier, J. Bleton and A. Tchapla, J. Chromatogr. A, 2003, 1018, 73–83 CrossRef CAS PubMed.
- L. Mondello, A. Casilli, P. Q. Tranchida, R. Costa, P. Dugo and G. Dugo, J. Chromatogr. Sci., 2004, 42, 410–416 CAS.
- H. Ganjitabar, R. Hadidi, G. Garcia, L. Nahon and I. Powis, J. Mol. Spectrosc., 2018, 353, 11–19 CrossRef CAS.
- G. A. Garcia, B. K. C. de Miranda, M. Tia, S. Daly and L. Nahon, Rev. Sci. Instrum., 2013, 84, 053112 CrossRef CAS PubMed.
- X. F. Tang, G. A. Garcia, J. F. Gil and L. Nahon, Rev. Sci. Instrum., 2015, 86, 123108 CrossRef PubMed.
- L. Nahon, N. D. Oliveira, G. Garcia, J. F. Gil, B. Pilette, O. Marcouille, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- M. M. Rafiee Fanood, H. Ganjitabar, G. A. Garcia, L. Nahon, S. Turchini and I. Powis, ChemPhysChem, 2018, 19, 921–933 CrossRef CAS PubMed.
- G. A. Garcia, B. Gans, X. Tang, M. Ward, S. Batut, L. Nahon, C. Fittschen and J.-C. Loison, J. Electron Spectrosc. Relat. Phenom., 2015, 203, 25–30 CrossRef CAS.
- J. C. Poully, J. P. Schermann, N. Nieuwjaer, F. Lecomte, G. Gregoire, C. Desfrancois, G. A. Garcia, L. Nahon, D. Nandi, L. Poisson and M. Hochlaf, Phys. Chem. Chem. Phys., 2010, 12, 3566–3572 RSC.
- X. Wang, S. Tong, M. Ge, W. Wang and D. Wang, Chin. Sci. Bull., 2010, 55, 4018–4025 CrossRef CAS.
- I. Novak, B. Kovač and G. Kovačević, Spectrochim. Acta, Part A, 2002, 58, 2223–2226 CrossRef PubMed.
- H. Ganjitabar, G. Garcia, L. Nahon and I. Powis, J. Chem. Phys., 2020, 153, 034302 CrossRef CAS PubMed.
- A. Leggio, V. Leotta, E. L. Belsito, M. L. Di Gioia, E. Romio, I. Santoro, D. Taverna, G. Sindona and A. Liguori, Chem. Cent. J., 2017, 11, 111 CrossRef CAS PubMed.
- M. L. R. del Castillo, M. M. Caja and M. Herraiz, J. Agric. Food Chem., 2003, 51, 1284–1288 CrossRef PubMed.
- M. J. Trujillo-Rodríguez, V. Pino, E. Psillakis, J. L. Anderson, J. H. Ayala, E. Yiantzi and A. M. Afonso, Anal. Chim. Acta, 2017, 962, 41–51 CrossRef PubMed.
- M. Guadayol, J. M. Guadayol, E. Vendrell, F. Collgrós and J. Caixach, J. Essent. Oil Res., 2018, 30, 244–252 CrossRef CAS.
- M. Sawamura, Citrus essential oils: flavor and fragrance Wiley, Hoboken, N.J., 2010 Search PubMed.
- M. Mejean, A. Giuliani, A. Brunelle and D. Touboul, Eur. J. Mass Spectrom., 2014, 20, 403–407 CrossRef PubMed.
- A. Sabljic and H. Güsten, Atmos. Environ., Part A, 1990, 24, 73–78 CrossRef.
- M. Allan, J. Dannacher and J. P. Maier, J. Chem. Phys., 1980, 73, 3114–3122 CrossRef CAS.
- A. Bodi, P. Hemberger, D. L. Osborn and B. Sztaray, J. Phys. Chem. Lett., 2013, 4, 2948–2952 CrossRef CAS.
- J. Kruger, G. A. Garcia, D. Felsmann, K. Moshammer, A. Lackner, A. Brockhinke, L. Nahon and K. Kohse-Hoinghaus, Phys. Chem. Chem. Phys., 2014, 16, 22791–22804 RSC.
- D. E. Couch, G. T. Buckingham, J. H. Baraban, J. P. Porterfield, L. A. Wooldridge, G. B. Ellison, H. C. Kapteyn, M. M. Mumane and W. K. Peters, J. Phys. Chem. A, 2017, 121, 5280–5289 CrossRef CAS PubMed.
- P. Hemberger, A. Bodi, T. Bierkandt, M. Kohler, D. Kaczmarek and T. Kasper, Energy Fuels, 2021, 35, 16265–16302 CrossRef CAS.
- X. F. Tang, X. X. Lin, G. A. Garcia, J. C. Loison, Z. Gouid, H. H. Abdallah, C. Fittschen, M. Hochlaf, X. J. Gu, W. J. Zhang and L. Nahon, Chem. Commun., 2020, 56, 15525–15528 RSC.
- I. Bonaccorsi, D. Sciarrone, A. Cotroneo, L. Mondello, P. Dugo and G. Dugo, Rev. Bras. Farmacogn., 2011, 21, 841–849 CrossRef CAS.
- M. H. M. Janssen and I. Powis, Phys. Chem. Chem. Phys., 2014, 16, 856–871 RSC.
- M. M. Rafiee Fanood, N. B. Ram, C. S. Lehmann, I. Powis and M. H. M. Janssen, Nat. Commun., 2015, 6, 7511 CrossRef PubMed.
- S. T. Ranecky, G. B. Park, P. C. Samartzis, I. C. Giannakidis, D. Schwarzer, A. Senftleben, T. Baumert and T. Schafer, Phys. Chem. Chem. Phys., 2022, 24, 2758–2761 RSC.
- T. Streibel, U. Boesl and R. Zimmermann, in Photoionization and Photo–Induced Processes in Mass Spectrometry, ed. R. Zimmermann and L. Hanley, Wiley-VCH Verlag GmbH, 2021, ch. 4, pp. 125–158, DOI:10.1002/9783527682201.ch4.
- A. Comby, E. Bloch, S. Beauvarlet, D. Rajak, S. Beaulieu, D. Descamps, A. Gonzalez, F. Guichard, S. Petit, Y. Zaouter, V. Blanchet and Y. Mairesse, J. Phys. B: At., Mol. Opt. Phys., 2020, 53, 234003 CrossRef CAS.
- A. Ferré, C. Handschin, M. Dumergue, F. Burgy, A. Comby, D. Descamps, B. Fabre, G. A. Garcia, R. Géneaux, L. Merceron, E. Mével, L. Nahon, S. Petit, B. Pons, D. Staedter, S. Weber, T. Ruchon, V. Blanchet and Y. Mairesse, Nat. Photonics, 2015, 9, 93–98 CrossRef.
- V. Svoboda, M. D. J. Waters, D. Zindel and H. J. Wörner, Opt. Express, 2022, 30, 14358–14367 CrossRef CAS PubMed.
- P. Krüger, M. Balster, B. R. Niraghatam, M. H. M. Janssen and D. A. Horke, 2023, arXiv: 2310.02893 [physics.chem-ph].
- M. Q. Cao, J. Chen, W. Z. Fang, Y. Q. Li, S. L. Ge, X. B. Shan, F. Y. Liu, Y. J. Zhao, Z. Y. Wang and L. S. Sheng, Eur. J. Mass Spectrom., 2014, 20, 419–428 CrossRef CAS PubMed.
- I. Jantan, A. S. Ahmad, A. R. Ahmad, N. A. M. Ali and N. Ayop, J. Essent. Oil Res., 1996, 8, 627–632 CrossRef CAS.
- P. Dugo, L. Mondello, G. Lamonica and G. Dugo, J. Agric. Food Chem., 1997, 45, 3608–3616 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an01448g |
| ‡ Present address: School of Mathematics, Statistics and Physics, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. |
| § Present address: Department of Chemistry, University of The Pacific, Stockton, CA 95211, USA. |
| This journal is © The Royal Society of Chemistry 2023 |