
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development of an electropolymerized, molecularly imprinted polymer (MIP) sensor for insulin determination using single-drop analysis
Tanja
Zidarič
a,
David
Majer
 b,
Tina
Maver
ac,
Matjaž
Finšgar
*b and
Uroš
Maver
ac
b,
Tina
Maver
ac,
Matjaž
Finšgar
*b and
Uroš
Maver
ac
aUniversity of Maribor, Faculty of Medicine, Institute of Biomedical Sciences, Taborska ulica 8, 2000 Maribor, Slovenia
bUniversity of Maribor, Faculty of Chemistry and Chemical Engineering, Smetanova ulica 17, 2000 Maribor, Slovenia. E-mail: matjaz.finsgar@um.si; Tel: +386 2 2294 447
cUniversity of Maribor, Faculty of Medicine, Department of Pharmacology, Taborska ulica 8, 2000 Maribor, Slovenia
First published on 24th January 2023
Abstract
An electrochemical sensor for the detection of insulin in a single drop (50 μL) was developed based on the concept of molecularly imprinted polymers (MIP). The synthetic MIP receptors were assembled on a screen-printed carbon electrode (SPCE) by the electropolymerization of pyrrole (Py) in the presence of insulin (the protein template) using cyclic voltammetry. After electropolymerization, insulin was removed from the formed polypyrrole (Ppy) matrix to create imprinting cavities for the subsequent analysis of the insulin analyte in test samples. The surface characterization, before and after each electrosynthesis step of the MIP sensors, was performed using atomic force microscopy, scanning electron microscopy, and energy-dispersive X-ray spectroscopy. The performance of the developed MIP–SPCE sensor was evaluated using a single drop of solution containing K3Fe(CN)6 and the square-wave voltammetry technique. The MIP–SPCE showed a linear concentration range of 20.0–70.0 pM (R2 = 0.9991), a limit of detection of 1.9 pM, and a limit of quantification of 6.2 pM. The rapid response time to the protein target and the portability of the developed sensor, which is considered a disposable MIP-based system, make this MIP–SPCE sensor a promising candidate for point-of-care applications. In addition, the MIP–SPCE sensor was successfully used to detect insulin in a pharmaceutical sample. The sensor was deemed to be accurate (the average recovery was 108.46%) and precise (the relative standard deviation was 7.23%).
1. Introduction
Insulin is a polypeptide hormone that regulates glucose homeostasis by controlling blood glucose levels in the body.1–3 An imbalance in insulin levels, due to either the impairment of insulin-producing beta cells or the ineffective use of insulin due to cellular resistance in metabolizing blood glucose, can cause type 1 (insulin-dependent) or type 2 (insulin-independent) diabetes.4 Direct insulin monitoring in a diabetic patient can provide insight into associated complications and improve the treatment prognosis. The normal blood insulin concentration is 50.0 pM3,5 under fasting conditions, while <50.0 pM and >70.0 pM indicate type 1 and the onset of type 2 diabetes,3,6,7 respectively. Given the vital role of insulin in living organisms, the measurement of insulin in diagnostic laboratories is important not only for the clinical diagnosis of diabetes and the evaluation of patients with chronic pancreatitis, but also for doping control in athletes and for the detection of insulinoma (tumors of the pancreas) and even breast cancer.8–11There are a variety of approaches to the laboratory determination of insulin resistance. The most commonly used analytical techniques are insulin immunoassays. However, in addition to development costs, they have several limitations. In particular, specificity is limited due to cross-reactivity with interfering structures such as proinsulin, human protein C (HPC), and insulin-like growth factors 1 and 2 (IGF1 and IGF2), which reduce the reliability of insulin measurement in a clinical setting.12,13 Capillary electrophoresis14 and high-performance liquid chromatography15,16 are successfully employed for the quantitative analysis of insulin. However, these methods are time-consuming, expensive, tedious, and have high limits of detection (LOD) for samples with nanomolar or lower concentrations.2,9,13
In recent decades, biosensing has become considerably more popular due to its recognized ability to develop selective sensing systems to determine target analytes. This is reflected in many publications, a wide range of applications, and active research ranging from environmental and food safety analysis, to pharmaceutical and clinical applications.17–20 With the growing market demand for the detection of various analytes, sensing platforms are limited to detecting small molecules, but macromolecules are also becoming more prominent targets.19,20 Bioactive macromolecules are routinely used as biomarkers and are of great interest in clinical diagnostics. Biosensors are a valuable alternative in the rapid and cost-effective detection of biomolecules in samples and/or solutions, due to their high sensitivity and specificity. These superior characteristics are achieved by means of the integrated recognition unit of the biosensor, which is usually biological, such as enzymes and antibodies.19,21–23 However, these biological recognition units face several challenges, including instability under measurement conditions, a short shelf life, and costly and complex antibody production. Surface modifications with nanomaterials or biomimetic materials are considered promising alternatives for circumventing these problems. Similar approaches have also been used for the electrochemical detection of insulin.2,3,9,11,13,24,25
Biomimetic receptor units are normally tailored by applying the principle of molecular imprinting technology. The main sensory properties of these biomimetic materials, called molecularly imprinted polymers (MIPs), include the selective recognition of the target analyte due to the specific architecture embedded in the polymer matrix. The formation of these cavities results from the polymerization of the functional monomers in the presence of the target analyte (template) and its subsequent removal.13,22,23,26–29 In the case of proteins, MIPs mimic the binding site by replacing the amino acid backbone of the target molecule with a synthetic polymer. Although the preparation of MIPs is simple for small molecules, the imprinting of high molecular weight molecules, such as proteins, is limited, mainly due to the stability problems of these macromolecules in polymerization media.2,27,28 For electrochemical detection, there are several approaches to the surface imprinting of proteins,22,23 where the target biomolecules can simply be adsorbed30,31 or covalently bound32 to the sensing platforms. The specificity and selectivity of synthetic MIP receptors in the design of insulin sensors have been reported previously.2,11,13,24,33 However, existing methods for designing MIPs are based on solid-phase synthesis combined with nanomaterials or epitope imprinting, which usually complicates manufacturing and in situ analysis due to the complexity of the systems.13 Electropolymerization overcomes several limitations of conventional bulk polymerization, as it allows polymer synthesis from an aqueous solution under mild conditions. It also offers the good reproducibility of results and the easy control of polymer thickness.22,23,28 For the success of this technique, the selection of a suitable monomer is crucial to effectively entrapping the template molecules and achieving the desired physical properties, especially the conductivity, of the thin polymer layers on the electrode surface.22
Electrode systems based on established screen-printing technology have challenged the use of conventional solid electrodes to quantify various compounds.3,34–38 Due to their low cost, availability, portability, ease of fabrication, and commercial availability, screen-printed electrodes (SPEs) can be used for the rapid and sensitive electroanalysis of many target analytes.34–37 Moreover, their application for on-site analysis is further advanced by their modification with different materials and flexibility.36,37 Three electrode systems (a working electrode, WE; a reference electrode, RE; and a counter electrode, CE) were identified as potential candidates for a point-of-care (POC) device design and applications in environmental and clinical analysis.34,37 In addition, they can also be used as disposable electrochemical cells that allow a sample drop to adhere to the sensor surface by surface tension. In this constellation, no wall is required to contain the solution and prevent its outflowing.36 As such, they are highly suitable for working with microvolumes and decentralized assays, such as a single-drop analysis.35–37,39 Although the latter simplifies on-site analysis, since no electrochemical cell or stirrer is required, there are only a few publications on this subject.3,34,35,37,40–43 But, for example, a smartphone-based cyclic voltammetry (CV) platform was designed as a portable system for single-drop determination of glucose.44 In another study, a similar smartphone-based sensory system using square-wave voltammetry (SWV) and a graphene SPE was constructed to detect norepinephrine in a sample drop.45 A single-drop analysis was also used by Singh,3 who decorated multi-walled carbon nanotubes (MWCNTs) with quantum dots (QCs) on an SPE array for the measurement of insulin in buffer solution. A concept using a single-drop analysis on a screen-printed carbon electrode (SPCE) combined with statistical analysis was presented. This combination proved to be an accurate, precise, and rapid analytical method for the individual determination of epinephrine and uric acid in a biological sample.37 In addition, the direct application of whole blood in drop form for the sequential determination of glucose and lactate was demonstrated. For this purpose, a dual enzyme-based biosensor was prepared using glucose oxidase and lactate oxidase on two working SPCEs.43
This study aimed to develop the MIP–SPCE sensor to detect insulin in one sample drop (50 μL). Although examples of protocols for the synthesis of MIPs for the electrochemical detection of insulin have been described previously,2,11,13,24 to the best of our knowledge, this is the first demonstration of an MIP-based sensor for a single-drop analysis of insulin without the use of solid-state synthesis or nanomaterials for signal amplification. In addition, by not using nanomaterials or aptamers, one preparational step in the electrosynthesis of such a sensor is excluded, making this method more straightforward, faster, and less expensive. Since the MIP is part of an electrochemical sensor, electropolymerization is the simplest method for electrosynthesizing a polymer film on the electrode surface.23 Pyrrole (Py) appears to be a good choice for building an MIP sensor surface, as evidenced by some MIP designs for detecting several important biomolecules, including the hazelnut allergen,46 cortisol,47 lactose,48 hemoglobin,49 α-amylase,22 cystatin C,50 and even the SARS-CoV-2 spike glycoprotein.51 Polypyrrole (Ppy) is a conductive polymer that can easily be electropolymerized and used as a polymeric matrix for immobilizing various biological compounds, including insulin.9 Its further advantages include biocompatibility, stability, and control over thickness. In addition, the N–H moieties in the pyrrole ring can facilitate binding to the hydroxyl group of the target molecule.52 For this purpose, Py was polymerized in a polymerization mixture with insulin using cyclic voltammetry (CV) directly on the surface of an SPCE to entrap the insulin in the polymeric backbone. After the subsequent removal of the template molecules from the Ppy matrix, the resulting MIP film could selectively recognize the target molecule upon rebinding. The electroanalytical performance of the MIP–SPCE was indirectly evaluated by square-wave voltammetry (SWV) using a K3[Fe(CN)6]/K4[Fe(CN)6] redox probe.
2. Materials and methods
2.1. Reagents and solutions
Pyrrole monomer (≥98%), potassium ferricyanide (K3[Fe(CN)6]), potassium ferrocyanide (K4[Fe(CN)6]), potassium chloride (KCl), sodium hydroxide (NaOH) solution (1.00 M), and insulin were supplied by Merck (Darmstadt, Germany). Phosphate-buffered saline (PBS) solution (0.01 M, pH 7.2) was prepared according to the manufacturer's instructions, i.e. by dissolving one PBS tablet (2005.5 mg) in 200 mL of ultra-pure water. The solution of K3[Fe(CN)6]/K4[Fe(CN)6] (5 mM) was prepared in PBS (0.01 M, pH 7.2). The insulin solution was prepared by dissolving insulin in ultra-pure water to give a final concentration of 0.58 mM. Solutions of insulin with lower concentrations were prepared by diluting the insulin solution in PBS (0.01 M, pH 7.2). All chemicals purchased were of analytical grade and used without further purification. All aqueous solutions were prepared with ultra-pure water (resistivity 18.2 MΩ cm at 25 °C) obtained with an ELGA PureLab water purification system (Veolia Water Technologies, United Kingdom).2.2. Instrumentation and electrodes
All electrochemical measurements in this study were performed using a PalmSens4 potentiostat/galvanostat (PalmSens, Houten, the Netherlands) under laboratory conditions (23 ± 2 °C). The PalmSens4 was controlled using PSTrace 5.8 software.SPCEs, model AC1.W4.R2, were supplied by BVT Technologies (Brno, Czech Republic) and used as a three-electrode electrochemical system. These SPCE sensors have a WE and CE made of carbon (the diameter of the WE was 1 mm), while the RE was made of Ag that was electrolytically oxidized to AgCl (by the supplier). All potentials (E) in this study are reported against this Ag/AgCl RE.
2.3. MIP–SPCE design
The formation of insulin-imprinted Ppy film on the WE of the SPCE was achieved by CV-based electropolymerization. For this purpose, the protocol for electrosynthesis was adapted from previous work.47 The prepared polymerization solution containing Py (0.80 M) and insulin (0.58 mM) in PBS (pH 7.2) with a monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :template (Py:insulin) ratio of 1:4 was drop-casted (50 μL) onto the SPCE so that all three electrodes were covered. Electropolymerization was performed by cycling the E from 0.000 V to 0.900 V and back to 0.000 V at a scan rate (ν) of 50 mV s−1 for 10 cycles using a step E (Estep) of 7.0 mV. For every cycle, the insulin molecule migrated toward the WE and became entrapped in the growing Ppy film. After polymerization, the entrapped insulin molecules were extracted from the conducting polymer matrix, leaving the cavities complementary to insulin in terms of shape and functionality. The template protein was removed by two different methods: electrocleaning and solvent extraction. In the electrocleaning approach, insulin molecules were removed by the overoxidation of Ppy47 by cycling the E between −0.200 V and 1.000 V for 25 cycles in PBS (0.01 M, pH 7.2) at a ν of 50 mV s−1 using an Estep of 7.0 mV. To extract the template protein using a solvent, the modified SPCE was immersed in an electrochemical cell containing a 1.00 M NaOH solution for 30 min under magnetic stirring. After removing the template (regardless of the method used), the modified SPCE was rinsed extensively with ultra-pure water to remove protein residues from the Ppy film and dried under a compressed air stream.
:template (Py:insulin) ratio of 1:4 was drop-casted (50 μL) onto the SPCE so that all three electrodes were covered. Electropolymerization was performed by cycling the E from 0.000 V to 0.900 V and back to 0.000 V at a scan rate (ν) of 50 mV s−1 for 10 cycles using a step E (Estep) of 7.0 mV. For every cycle, the insulin molecule migrated toward the WE and became entrapped in the growing Ppy film. After polymerization, the entrapped insulin molecules were extracted from the conducting polymer matrix, leaving the cavities complementary to insulin in terms of shape and functionality. The template protein was removed by two different methods: electrocleaning and solvent extraction. In the electrocleaning approach, insulin molecules were removed by the overoxidation of Ppy47 by cycling the E between −0.200 V and 1.000 V for 25 cycles in PBS (0.01 M, pH 7.2) at a ν of 50 mV s−1 using an Estep of 7.0 mV. To extract the template protein using a solvent, the modified SPCE was immersed in an electrochemical cell containing a 1.00 M NaOH solution for 30 min under magnetic stirring. After removing the template (regardless of the method used), the modified SPCE was rinsed extensively with ultra-pure water to remove protein residues from the Ppy film and dried under a compressed air stream.
The same procedure was used to form a non-imprinted polymer (NIP) modified SPCE. The only difference in the preparation was that the insulin molecules were not present during the electropolymerization process.
2.4. Surface characterization
The surface topography and roughness parameters of all of the prepared SPCEs were characterized by atomic force microscopy (AFM) in tapping mode using a Keysight 7500 AFM multimode scanning probe microscope (Keysight Technologies, Santa Barbara, CA, USA). Images were acquired after drying the samples in a dry, high-grade (99.999 wt%) nitrogen gas stream. The images were scanned using silicon cantilevers (ATEC-CN-20, Nanosensors, Wetzlar, Germany) with a resonant frequency of 210–490 kHz and a force constant of 12–110 N m−1. Images of 10 × 10 μm2 with a resolution of 2048 × 2048 pixels53 were measured for all samples. Pico Image Basic 7.2 software (Keysight Technologies, Wokingham, UK) was used to process all of the images and calculate the mean square height of the surface (Sq) and the mean surface roughness of the surface (Sa) according to ISO 25178.54The surface morphology and porosity of all fabricated SPCEs were analysed by scanning electron microscopy (SEM).55 Micrographs were taken using a field emission scanning electron microscope (FE-SEM, Supra 35 VP, Carl Zeiss, Oberkochen, Germany) operated at a low accelerating voltage (1 keV). The images were taken at magnifications of 2500 and 10000.
2.5. Electrochemical measurements
The SWV was used to characterize the electroanalytical response of the assembled MIP–SPCE sensor and to analyse real samples because it offers a low LOD and a short analysis time.22,37,56,57 The square-wave (SW) measurements were performed in the presence of a 5 mM [Fe(CN)6]3−/4− redox probe prepared in 0.01 M PBS (pH 7.2). The E in the SWV was scanned with a frequency of 10 Hz, an amplitude of 100 mV, and an Estep of 10 mV in the anodic direction from −0.800 V to a final E of 0.600 V. As soon as a drop of the solution was placed on the surface of the SPCE, the SWV voltammograms were measured (the measurement lasted 25 s). The electrochemical behaviour of the modified SPCE surfaces was investigated using the CV in an E range of −0.800 V to 0.600 V at a ν of 50 mV s−1 and Estep of 7.0 mV.2.6. Single-drop analysis of the MIP–SPCE sensor
A single-drop analysis of rebound insulin on the surface of the MIP–SPCE sensor was performed using the SWV as follows. Solutions of different insulin concentrations were prepared. One drop (5 μL) was placed on the WE of the MIP–SPCE for 15 min to allow the rebinding of insulin in the formed cavities of the Ppy matrix. Then, the surface was rinsed with ultra-pure, and the remaining water was soaked up using the paper towel without touching WE. A drop (50 μL) of [Fe(CN)6]3−/4− redox probe was then pipetted onto the surface of the MIP–SPCE sensor so that all three electrodes were covered. Immediately after the redox probe was applied, the SWV voltammogram was measured. After each measurement, the surface of the MIP–SPCE was rinsed with ultra-pure water, and the remaining water was soaked up with a paper towel (KIMTECH® Science) without touching the active WE surface. Then the surface was air dried. After this cleaning procedure, the whole procedure was repeated.For the real sample analysis, the insulin content in a cartridge (insulin aspart Novorapid, Novo Nordisk, Bagsværd, Denmark) was tested. For this purpose, insulin from a cartridge was diluted with 0.01 M PBS before analysis. A volume of 5 μL was applied to the MIP–SPCE and incubated for 15 min, followed by pipetting 50 μL of 5 mM [Fe(CN)6]3−/4− redox probe. The quantification of insulin was performed by employing the multiple standard addition method. The real sample was analysed three times, each time with a new MIP–SPCE sensor.
3. Results and discussion
3.1. Step-by-step sensor fabrication
MIPs are tailored biomimetic materials that can recognize target molecules selectively.58 As such, they can potentially complement antibodies in bioanalytics.59 The concept of molecular imprinting, in particular the electrochemical surface imprinting of proteins, was used in this study. The overall process involved three main steps: (1) the imprinting of the Ppy film with the template/target protein (insulin); (2) the removal of insulin molecules from the Ppy matrix; and (3) the rebinding of insulin (see Fig. 1). Each step resulted in changes in the electron transfer properties of the electrode surface,22,23 which could be monitored by CV and SWV techniques in conjunction with the [Fe(CN)6]3−/4− redox probe in subsequent measurements.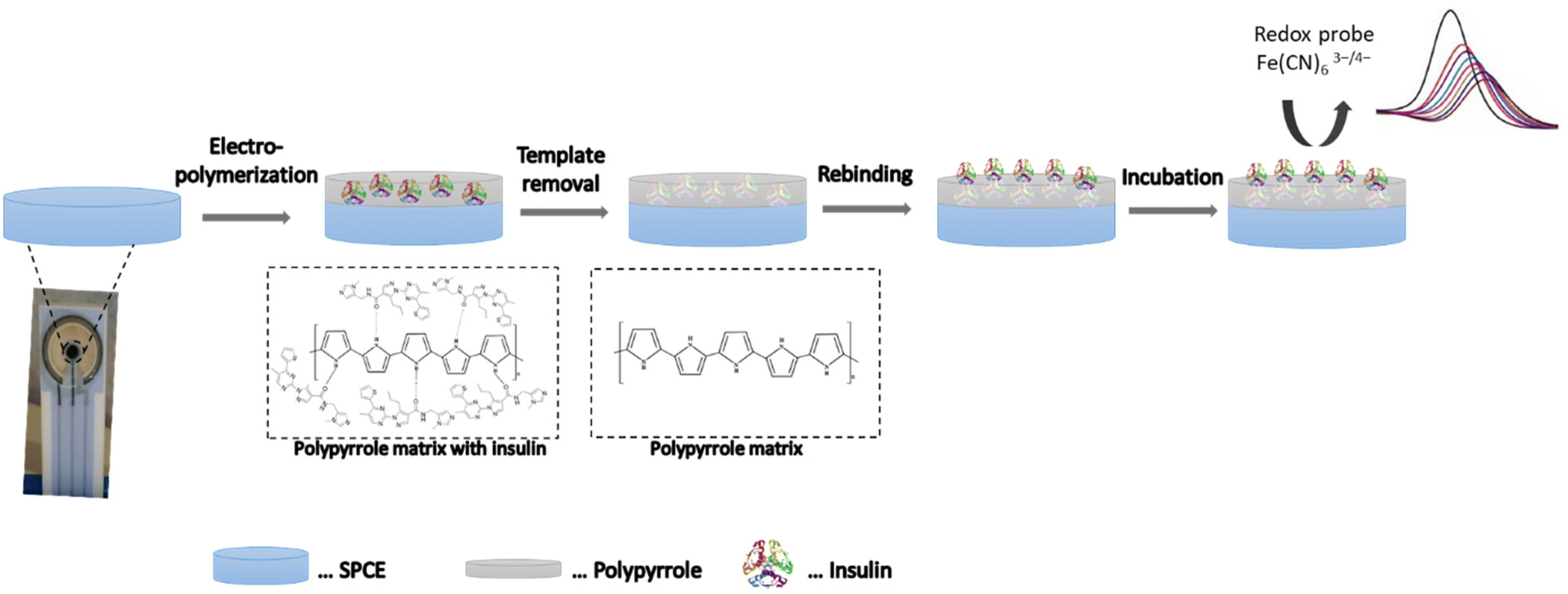 | ||
| Fig. 1 A schematic depiction of insulin imprinting on the surface of the SPCE. The overall process involved (1) the electrosynthesis of Ppy in the presence of the template molecule (insulin), (2) the removal of the target protein by electrocleaning, and (3) the determination of the rebinding ability of the formed molecular imprinted polymer film (MIP–SPCE) by using a standard redox probe and SWV. | ||
Previous studies22,46,47,50 have shown that these electropolymerization parameters appear to provide a stable 3D architecture of the MIP structure for the effective template removal and accessibility of the binding sites for rebinding a target molecule. Inspired by previous studies, the synthesis of the MIP film on the surface of the SPCE was performed according to a protocol reported in ref. 47 and was adapted to the target protein (insulin). The polymer film was formed by the electropolymerization of Py in the presence of insulin by performing 10 consecutive CV cycles at a ν of 50 mV s−1. Polymer growth was monitored by changes in the CV voltammograms. After the first cycle of the electropolymerization of Py on the SPCE surface, the CV voltammograms (see Fig. 2) show similar electrochemical behaviour for the MIP and NIP systems in terms of the position of the oxidation peaks. However, the peak heights for oxidation were different.
 | ||
| Fig. 2 Cyclic voltammograms after the first scan of the electrochemical oxidation of Py (0.80 M in 0.01 M PBS, pH 7.2) on the surface of the SPCE in the presence of insulin (MIP) and without insulin (NIP) in the polymerization solution (ν = 50 mV s−1). | ||
The electrochemical oxidation of the monomer begins with the loss of an electron from the Py unit and the formation of a cation radical, which occurs at approx. 0.800 V, initiating the electrodeposition of a thin and homogeneous Ppy film on the WE surface.22,46,61,66 The polymerization of Py is a reaction with multiple reaction pathways, in which a formed cation radical combines with another cation radical, resulting in the formation of a dimeric product. Upon subsequent oxidations, this Ppy dimer couples with other cation radicals, leading to the formation of a Ppy matrix.
The insulin has only a minor effect on the Py monomer oxidation mechanism with the change in the peak intensity and the shift in the E peak (for oxidation) to a more negative E for the MIP (Fig. 2). The latter can occur due to the formation of cavities on the MIP surface due to the incorporation of insulin into the polymer matrix, which provides an additional barrier to the diffusion of Py for its further oxidation.46 The increase in the oxidation current in subsequent cycles following the first cycle (Fig. 3) indicates the growth of the polymer film, which can be tuned by the number of cycles.47,50 In contrast, the absence of the cathodic peaks indicates the irreversibility of the reaction.23
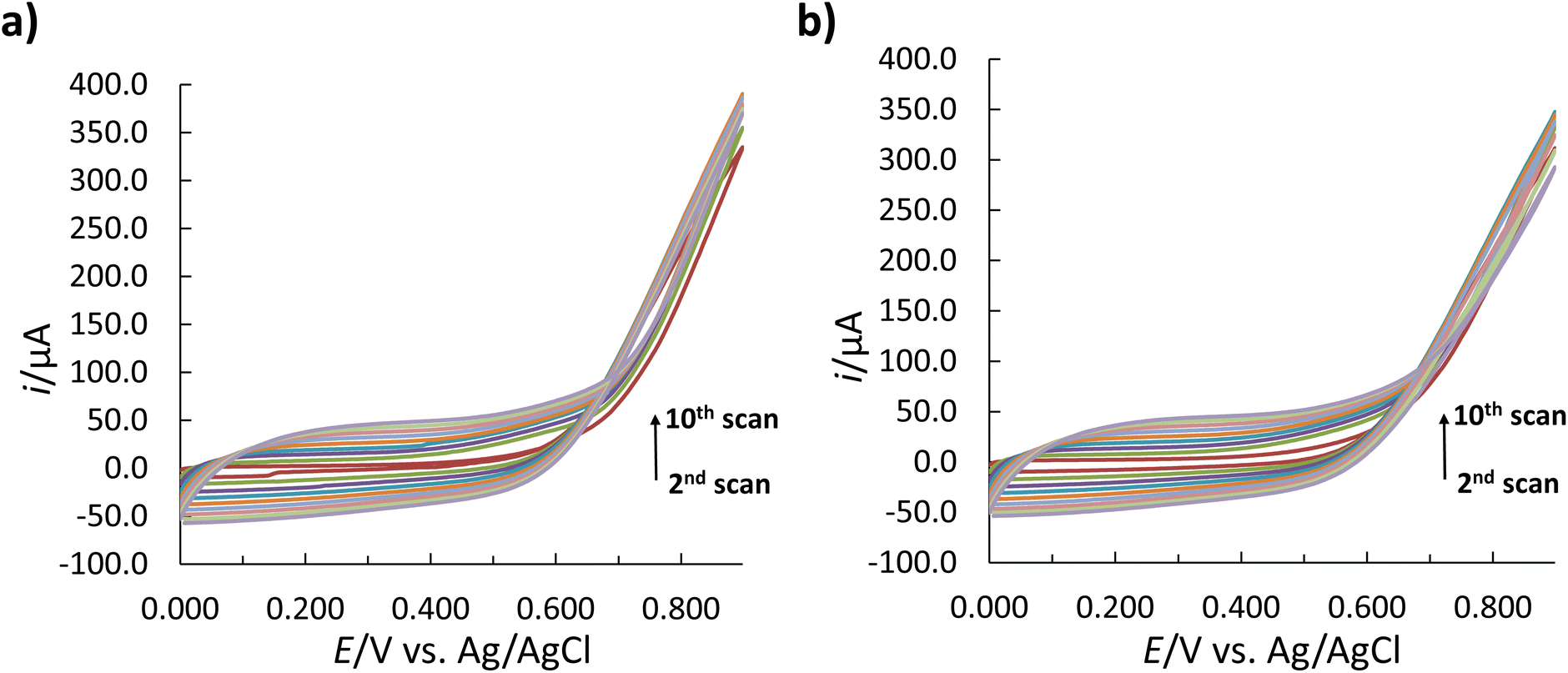 | ||
| Fig. 3 Cyclic voltammograms representing the Py electropolymerization on the surface of the SPCE; (a) with and (b) without insulin in the polymerization solution. | ||
During electropolymerization, negatively charged insulin67,68 diffuses into the electrode surface and binds to the Ppy matrix through electrostatic interactions.23,47 The insulin and Py in the inner wall of the imprinted cavities can bind through hydrogen bonding and π–π stacking.69 These molecular interactions between the template molecule and the Py units are crucial for forming imprinted cavities in the polymer matrix.48
The alkaline template removal damaged the polymer film, leading to a deterioration of the electrochemical properties of the MIP surface. This is likely due to the degradation of the polymer film, which resulted in a loss of adhesion to the electrode surface. This degradation of the polymer film could be due to the strong oxidation effect of the 1.00 M NaOH and the applied potential in the subsequent electrochemical measurements (the results are not shown herein). The latter probably caused the formation of gaseous species, facilitating the polymer film detachment.70 Therefore, the electrochemical oxidation of Ppy was used for insulin extraction from the formed polymer film. This was achieved by cycling the E between −0.200 V and 1.000 V for 25 cycles. During this step, the overoxidation of Ppy occurred at approx. 0.400 V for the first cycle (Fig. 4).47,61 With every sequential cycle, a small amount of Ppy matrix, together with the entrapped insulin, was peeled off the surface, which resulted in a decrease in the oxidation peak. In the last five cycles of the electrocleaning step, no significant difference between the CV voltammograms was observed. In addition, the absence of the oxidation peak indicated that the majority of the available Ppy film was overoxidized, resulting in the formation of carboxyl groups and the removal of insulin from the Ppy matrix (Fig. 4).
 | ||
| Fig. 4 Measured CV voltammograms during the removal of insulin (electrocleaning approach) from the formed Ppy matrix in 0.01 M PBS (pH 7.2) with a ν of 50 mV s−1. | ||
The proposed mechanism underlying the overoxidation of Ppy in aqueous solutions such as PBS suggests that the oxygen produced during water oxidation causes the overoxidation of the conductive Ppy film.61 This also leads to the formation of carboxyl groups on the Py rings, which break the links in the Ppy chain.60,61 These cleavages in the Ppy matrix facilitated the extraction of insulin molecules from the polymer film,47 while also leading to the spatial functionalization of the imprinted cavities. This, in turn, contributes to the more successful oriented rebinding of insulin through the interactions between the amine groups of the proteins and the carboxyl groups of the imprinted cavities. After the first cycle, successive cycles (only the first and the last five scans are shown in Fig. 4 for clarity reasons) showed a decrease in the background contribution, indicating the decreasing conductivity of Ppy. Although overoxidation breaks the bonds in the Ppy network, the neutral pH of PBS (7.2) prevented the degradation of the polymer film.47 The oxidation of Py in a neutral medium leads to the formation of polarons and bipolarons, which could prevent the leaching effect of the MIP film. A polaron is a charge associated with a lattice distortion that creates connections in the Ppy network to improve the electrochemical properties along with the electropolymerized film. Although the overoxidation of Ppy results in the loss of protons from the polymer matrix, neutral pH promoted the generation of polarons and bipolarons through a proton-coupled electron transfer reaction that stabilized the Ppy film.69
3.2. The electrochemical behaviour of differently imprinted SPCEs
The electrochemical behaviour of the stepwise imprinting process was investigated using the [Fe(CN)6]3−/4− redox probe on the surfaces of the coated SPCE after each modification step (Fig. 5).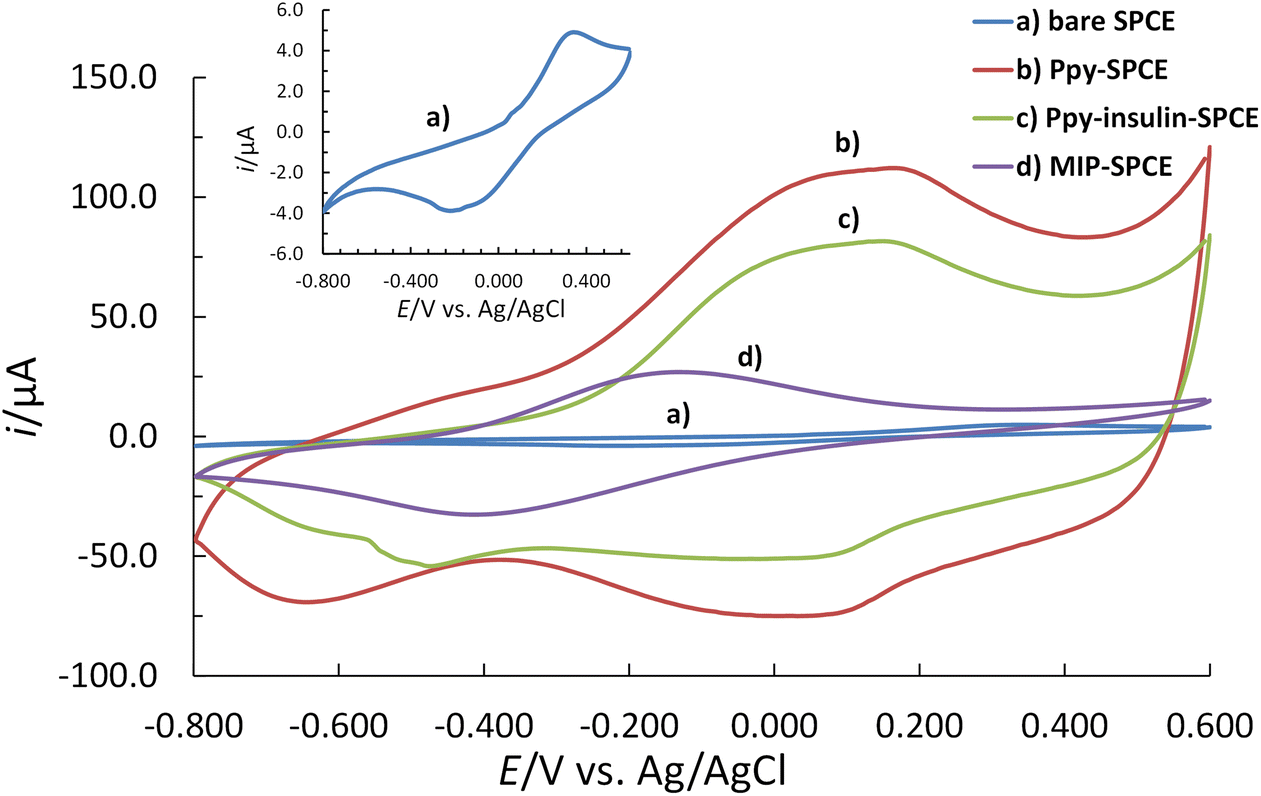 | ||
| Fig. 5 The electrochemical behaviour of the step-by-step imprinting process in 0.01 M PBS (pH 7.2) containing the 5 mM [Fe(CN)6]3−/4− redox probe using the CV: (a) the bare SPCE (blue solid line), (b) the Ppy–SPCE (red solid line), (c) the Ppy–insulin–SPCE (green solid line), and (d) the MIP–SPCE (purple solid line). The measurements were performed at a ν of 50 mV s−1. The insert shows the CV voltammogram of a bare SPCE at a lower i scale. | ||
CV voltammograms show that the oxidation of insulin at the Ppy–insulin–SPCE surface does not occur in the employed potential range (from −0.800 V to 0.600 V) since no additional peak formed compared to the Ppy–SPCE (Fig. 5). Compared to a bare SPCE, all Ppy-coated SPCEs (pure Ppy–SPCE, Ppy–insulin–SPCE, and MIP–SPCE) showed enhanced electrochemical activity due to the favourable electrochemical properties of the electropolymerized Ppy film.47,71 The higher current of the [Fe(CN)6]3−/4− redox probe measured on Ppy–SPCE, Ppy–insulin–SPCE, and MIP–SPCE compared to bare SPCE can arise due to the increase in double layer capacitance (Fig. 5). Moreover, the higher capacitance can also be attributed to the increase in the effective surface area of the conductive Ppy film on the surface of SPCE and the properties of the Ppy surface film. In addition, the presence of positively charged nitrogen atoms in Ppy72 contributes to the fast electron transfer kinetics of the [Fe(CN)6]3−/4− redox couple. Due to the electrostatic interactions, [Fe(CN)6]3−/4− can easily interact with the positively charged Ppy film,47 which resulted in increased peak current. However, the peak current response on the Ppy–insulin–SPCE surface was less intense than on the Ppy–SPCE surface. The difference in the peak current intensity may be attributed to the distribution of the nuclei formed by polymerization in the presence of an insulin molecule. The properties of conducting polymers depend strongly on their morphology and structure.71 The insulin molecule can be seen as a protrusion in the monolayer. The area around the molecule can immediately be filled with the growing polymer, leading to the formation of an uneven coating. Moreover, the negative surface charge of insulin hinders the diffusion of the [Fe(CN)6]3−/4− redox probe to the surface of the SPCE. On the contrary, pure Ppy can be deposited on the surface of an SPCE as a completely covered homogeneous and adherent film.71 This observation supports the assumption that the insulin molecules were entrapped in the Ppy backbone, which was reflected in the reduced current response of the [Fe(CN)6]3−/4− redox probe. Finally, the overoxidation of Ppy, used as a strategy to remove insulin, affected the conductivity of the composite MIP on the surface of the SPCE, as evidenced by the overall decrease in current.47 The presence of carboxyl groups in the Ppy matrix contributed to the more anionic charge of the MIP–SPCE surface,50 resulting in the observed electrochemical behaviour. Despite the reduced conductivity of the MIP–SPCE sensor, the imprinting cavities provided with carboxyl groups can promote the rebinding of insulin through the interactions between the amino group of the protein and the carboxyl group of the binding sites in the imprinted polymer.23
The resulting MIP and NIP films were also evaluated by SWV measurements in the presence of the 5 mM [Fe(CN)6]3−/4− redox probe. Fig. 6 shows that both polymer-coated SPCEs (MIP–SPCE and NIP–SPCE) showed a higher square-wave signal (and higher background current) compared to bare SPCE, with the NIP–SPCE providing a higher analytical signal than MIP–SPCE. This may be due to the more uniform distribution of the Ppy film on the surface of the SPCE, resulting in a higher current compared to the MIP (however, as described below, the NIP suffers from poor analytical performance). In addition, the differences in the electrochemical response can also be attributed to the different chemistry of the two Ppy films formed. Namely, the imprinted cavities in the MIP film, which have a spatial arrangement of the interacting groups,73 could affect the electrochemical features of the MIP–SPCE sensor. These synthetic MIP receptors do not provide the same access to the redox probe ions as NIP–SPCE (there are no imprinted cavities present on NIP–SPCE), resulting in a lower peak current. The presence of the Ppy film on both MIP–SPCE and NIP–SPCE shifted the oxidation E of the redox probe to more negative potentials compared to the bare SPCE (Fig. 6). The E shift was due to the Ppy film, which changes the overall charge of the sensor surface and thus the electrochemical properties of the surface.
 | ||
| Fig. 6 SW voltammograms measured in 0.01 M PBS (pH 7.2) containing the 5 mM [Fe(CN)6]3−/4− redox probe for different SPCEs: bare SPCE, NIP–SPCE, and MIP–SPCE. | ||
3.3. Surface characterization
The design and development of a surface-based electrochemical sensor strongly depends on the surface properties and topography of the materials. From this perspective, the improved analytical performance of a sensor is the result of many factors, including maximized target–surface interactions, reduced non-specific binding, and the avoidance of fouling on the sensor surface. Although substrate (transducer) surface modifications represent only a small portion of the total modified surface, they have an uncertain impact on sensor performance and reliability. Therefore, many researchers equate electrochemical performance with the quality of the surface modification strategy.74AFM measurements were performed before and after each step of the electrosynthesis of the MIP films on the SPCE surface to check the homogeneity and surface roughness of the SPCE surfaces. The surface topography and corresponding surface roughness parameters Sq and Sa measured at different analysed spot sizes (20 × 20 μm, 10 × 10 μm, and 1 × 1 μm) are shown in Fig. 7.
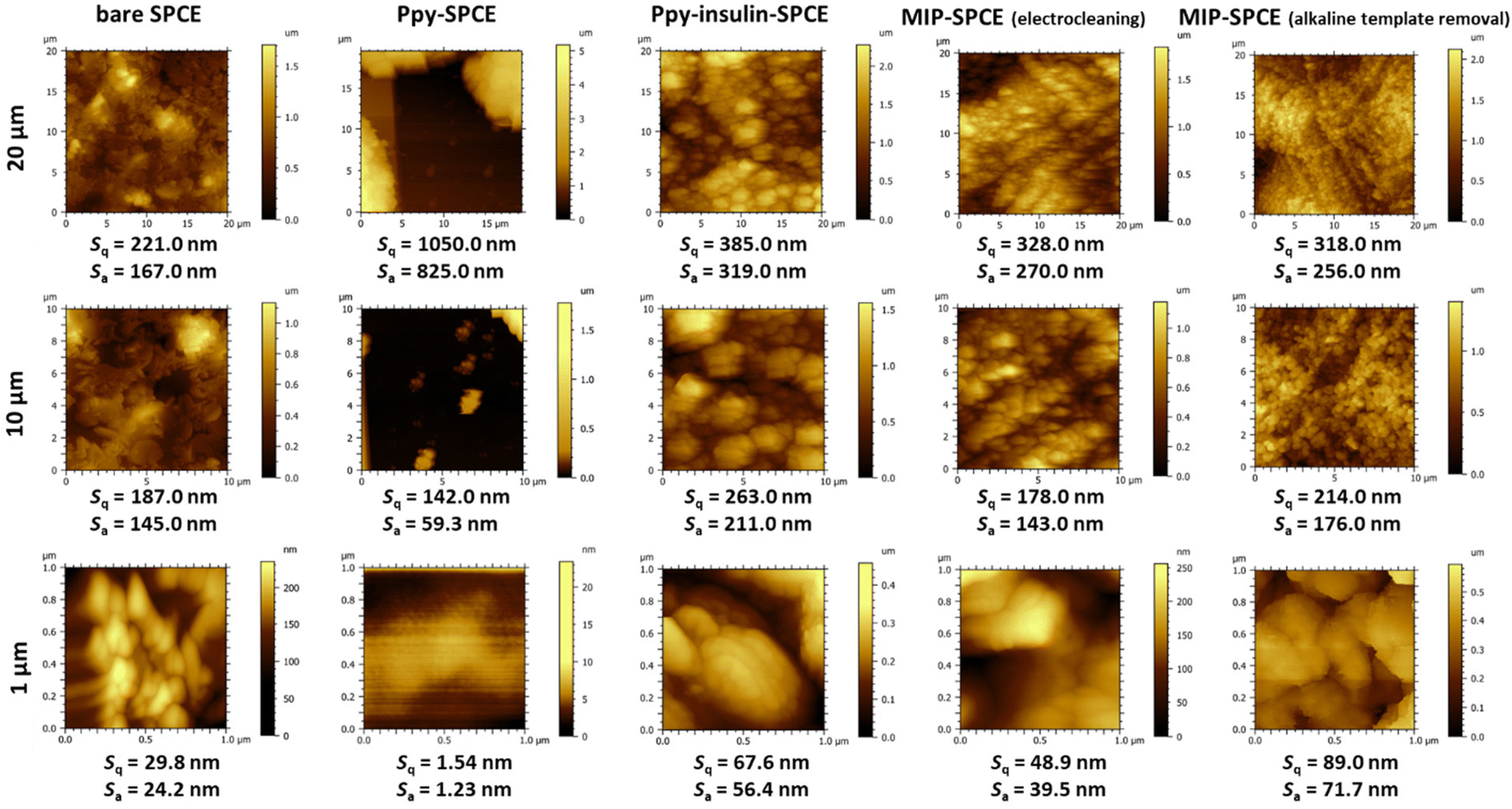 | ||
| Fig. 7 Surface topography and roughness parameters measured by AFM at the SPCE surface before and after monomer electropolymerization in the presence of the template, followed by template removal (by electrocleaning and alkaline template removal) from the Ppy matrix. | ||
The bare SPCE surface exhibited a rough surface associated with the composition of carbon ink with a higher density of edge graphite particles used for screen-printing production.75 As expected, the surface roughness of the 20 × 20 μm analysed spot increased after the electropolymerization of Py (with or without insulin), indicating the successful formation of polymer film on the bare electrode.76,77 However, for smaller scan areas (10 × 10 μm and 1 × 1 μm, respectively), the surface of the pure Ppy–SPCE sensor appeared to be more homogeneous, which was due to the obstruction of signals by the surface defects (e.g. holes and pores) on the SPCE-containing carbon with different bonding organization.78 In contrast, the trapped insulin on the Ppy–insulin SPCE surface contributed to the “swellings” in the topographic image. Regardless of the extraction method used, a decrease in surface roughness was observed, indicating the successful extraction of insulin from the polymer matrix; however, after the removal of the template, different surface patterns can be seen.76 By comparing AFM images measured at smaller scan areas, the effects of different removal methods can be observed. A rougher topography was shown on the surface of the MIP–SPCE, where insulin was removed by alkaline template removal (1.00 M NaOH solution). This increase in the roughness can probably be attributed to the abrasiveness of the chemical approach that disintegrated (damaged) the polymer matrix. These results are related to the heterogeneous conductivity of these SPCE arrays described above.
Next, SEM was used to further investigate the imprinting process's influence on the morphology of the deposited polymer films (Fig. 8).
 | ||
| Fig. 8 SEM micrographs of different SPCE arrays taken at different magnifications. | ||
The SEM analysis confirmed the formation of polymer films with different patterning. Pure Ppy deposited on a bare SPCE shows a porous morphology with small clusters. This cauliflower-like structure consists of microspherical grains formed on the defect sites of the SPCE surface after nucleation.47,71 However, the Ppy film grown in the presence of insulin shows a more granular morphology with no observed clusters. This could support the idea that insulin entrapped in the polymer leads to the swelling of the Ppy film, which is associated with a lower conductivity than in pure Ppy film.79 On the other hand, the extraction of insulin from the Ppy matrix resulted in increased porosity due to the formed imprinted cavities of insulin. Compared to the surface of the NIP–SPCE sensor, both MIP–SPCE sensors (MIP–SPCE (electrocleaning) and MIP–SPCE (alkaline template removal)) revealed a more compact morphology with the formed cavities.
The EDS analysis (Fig. 9) confirms the production of the MIP–SPCE sensor. The presence of nitrogen (N) in all modified SPCE sensors (weight concentrations between 14.70% and 16.30%) confirms the deposition of a Ppy film due to the presence of N–H components in the Py ring.52 Moreover, the highest amount (16.30%) was found in Ppy–insulin–SPCE, which is due to the contribution of the terminal –NH2 groups in the insulin molecule. In addition, traces of sulfur (S) in the EDS spectra of Ppy–insulin–SPCE and both MIP–SPCEs, likely associated with thiol groups in the insulin molecule, indicate protein imprinting in the polymer. The lower N content and the increased oxygen (O) content after the removal procedure (by electrocleaning or alkaline template removal) can be explained by the removal of the insulin molecules from the Ppy film and the formation of –COOH groups in the Ppy matrix. Comparing the amount of N and O between the two MIP–SPCE sensors (the bottom row in Fig. 9), electrocleaning is the most efficient method of removing insulin from the Ppy matrix, as evidenced by the lower N content (indicating the better removal of insulin molecules) and a slightly higher O content (due to the formation of –COOH groups). This is also reflected in the improved analytical performance of the resulting MIP–SPCE sensor.
 | ||
| Fig. 9 EDS spectra obtained on the different SPCE surfaces. | ||
3.4. Analytical response of the MIP–SPCE
After template removal, the performance of the MIP–SPCE was evaluated by the SWV technique in a single drop of solution (as described in section 2.6). The MIP–SPCE methodology was partially validated, where the LOD, limit of quantification (LOQ), linear concentration range, accuracy, and precision were determined. The linear correlation between the insulin concentration and SW signal was investigated in the range of 20.0 to 70.0 pM. For this purpose, 5 μL of insulin solutions (different concentrations) were incubated for 15 min to allow insulin adsorption before applying the [Fe(CN)6]3−/4− redox probe and measuring the electrochemical response by SWV. A longer incubation time could lead to lower LODs, but may also contribute to non-specific adsorption, resulting in poorer selectivity.50,80,81 The different insulin solutions were incubated sequentially at increasing concentrations. The measured SWV voltammograms of the 5 mM [Fe(CN)6]3−/4− redox probe with the corresponding linear calibration curve are presented in Fig. 10.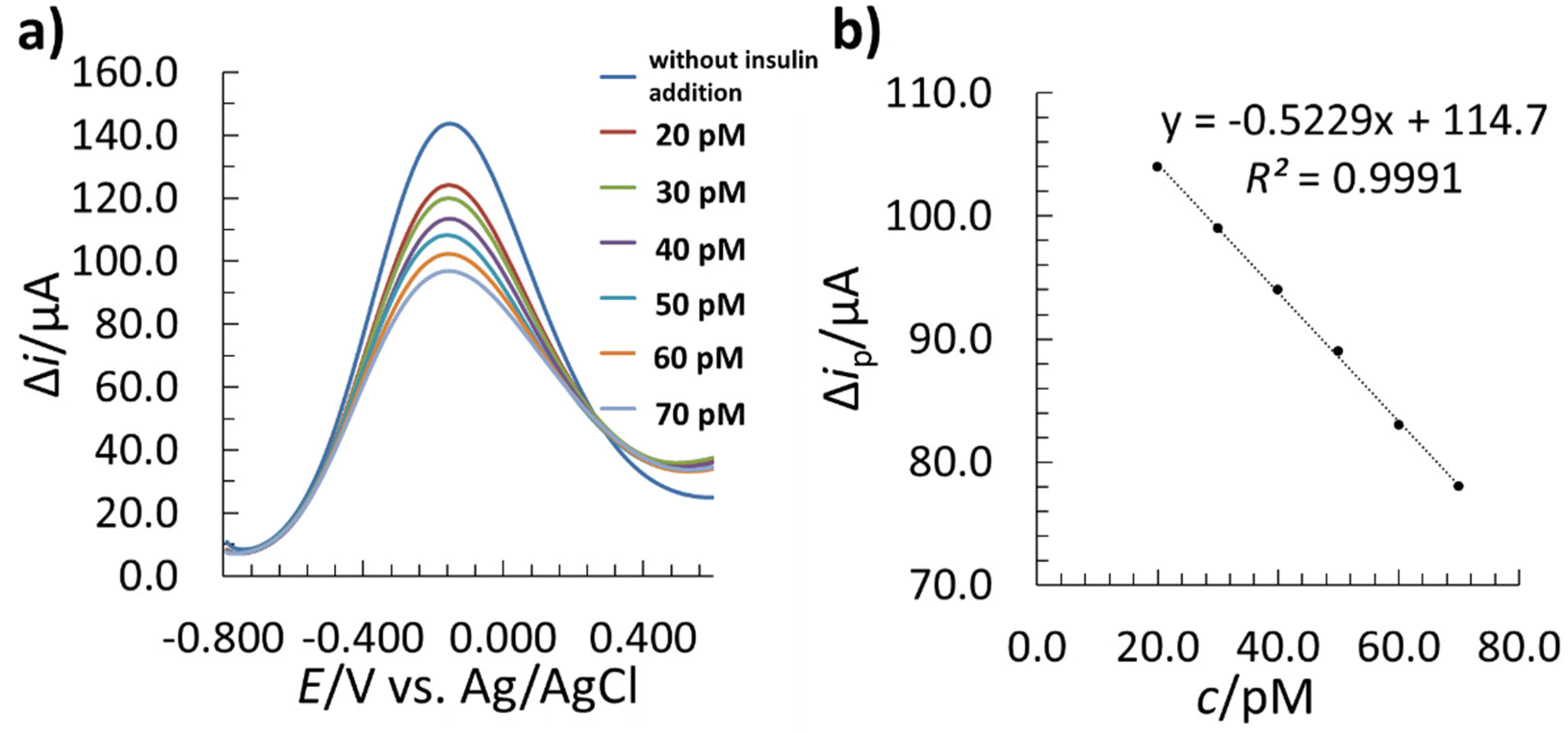 | ||
| Fig. 10 The electrochemical performance of the MIP–SPCE for rebinding insulin in a single drop: (a) SWV voltammograms of the MIP–SPCE measured in 0.01 M PBS (pH 7.2) containing the 5 mM [Fe(CN)6]3−/4− redox probe with different concentrations of insulin, (b) the corresponding linear calibration curve (Δip stands for the SW peak height). | ||
The instant after the first insulin incubation, a significant decrease in the Δip of the redox probe occurred (Fig. 10a), which was due to the re-binding of insulin to the available imprinted recognition sites on the MIP film surface, and was more pronounced with increasing insulin concentration. This event blocked the diffusion of the redox probe onto the WE surface, leading to a drop in Δip. The method showed a linear dependence between Δip and insulin concentration in a concentration range between 20.0–70.0 pM (Fig. 10b).
In the same manner, the NIP–SPCE sensor was examined to evaluate the extent of the recognition and rebinding of insulin on the imprinted sites on the MIP surface. Since the electrosynthesis of the Ppy-based NIP film was performed without insulin, the formed polymer film has no complementary recognition sites for insulin rebinding. Therefore, any interaction with insulin that might occur would be attributed to non-specific binding with the Ppy matrix. On that basis, a non-linear response of the NIP–SPCE sensor was present (Fig. 11) in the concentration range, where the MIP–SPCE showed a linear response. Therefore, the imprinting cavities on the MIP–SPCE surface allow specific recognition and rebinding.
 | ||
| Fig. 11 The electrochemical performance of the NIP–SPCE in a single drop: (a) the SWV voltammograms of the NIP–SPCE measured in 0.01 M PBS (pH 7.2) containing the 5 mM [Fe(CN)6]3−/4− redox probe with different concentrations of insulin, and (b) the corresponding linear calibration curve of Δip as a function of the insulin concentration for the electrochemical response on the NIP–SPCE sensor. | ||
The LOD and LOQ were determined based on the standard deviation of the residuals (se)82 and the slope of the calibration curve (b1). In order to obtain the LOD and LOQ, the se were multiplied by 3 and 10, respectively, and divided by the b1.37 The determined LOD was 1.9 pM, and the LOQ was 6.2 pM. Compared to other insulin detection methods reported previously (Table 1), the proposed MIP–SPCE sensor shows similar or lower LOD values. On the other hand, it must be pointed out that, unlike the other reported methods, the MIP–SPCE sensor herein allows the analysis of insulin with a low LOD in a single drop of the sample.
| Sensor | Technique | Linear concentration range | LOD | Real sample | Ref. |
|---|---|---|---|---|---|
| Key: nanoparticles (NP), nickel oxide nanoparticles (NiOx), acetylene black nanocarbon particles (AB), polypyrrole (PpY), molecular imprinted polymer (MIP), glassy-carbon electrodes (GCE), multi-walled carbon nanotubes (MWCNT), graphene electrode (GF), pencil graphite electrode (PGE), gold electrode (AuE), carbon paste electrode (CPE), screen-printed electrode (SPE), quantom dots (DC), screen-printed carbon electrode (SPCE), alternating current voltammetry (ACV), chronoamperometry (CA), differential pulse voltammetry (DPV), and differential pulse anodic stripping voltammetry (DPASV). | |||||
| Guanine/NiOx/GCE | Amperometry | 100 pM–4 μM | 22 pM | N/A | 83 |
| MWCNT/MIP | DPASV | 0.068–5.682 nM | 0.0183 nM | Blood serum, insulin injection | 2 |
| Ni(OH)2NPs/Nafion–MWCNTs/GCE | CV | 1.5–40 μM | 85 nM | Human plasma, pharmaceuticals | 84 |
| AB/CPE | DPV | 20–1000 nM | 5 nM | Insulin injection | 85 |
| Aptamer (In1-IT)-based sensor | ACV | 10–200 nM | 10 nM | N/A | 86 |
| Ppy–GF/PGE | CA | 0.225–1.235 μM | 8.65 nM | Blood serum | 9 |
| SPE/MWNT–QD | SWV | 100–5000 pM | 100 pM | N/A | 3 |
| SPCE/MWCNT/NiO1.5 | Amperometry | 600 nM–10 μM | 19.6 nM | Blood serum | 87 |
| MIP–SPCE | SWV | 20.0–70.0 pM | 1.9 pM | Insulin cartridge | This work |
Precision in terms of the repeatability and reproducibility of the MIP–SPCE sensors was evaluated using relative standard deviation (RSD) and was lower than 10.00% in both cases. Reproducibility was defined by six replicate measurements using the determined Δip on freshly prepared MIP–SPCEs, which was measured after the incubation of the MIP–SPCE sensors with 20.0 pM insulin and a 5 mM redox probe. The calculated RSD was 5.92%. The repeatability of the developed MIP–SPCE sensor was determined in the same manner as for the reproducibility, with the difference of using a single MIP–SPCE sensor ten times. The calculated RSD value was 5.13%. Both RSD values obtained indicate that the performance of the MIP–SPCE sensor is consistent with industrial precision standards (ISO-15917:2003).13
The developed MIP–SPCE sensor was also used to determine insulin in a commercial pharmaceutical test sample (a cartridge containing a fast-acting insulin analogue, insulin aspart). For this purpose, the real sample was diluted to the required concentration (which was in the linear concentration range of the method) since the declared concentration of insulin aspart in a cartridge was 100 U mL−1 (0.60 mM). The quantification of insulin was performed using a multiple standard addition method (five additions were added). The analysis was performed using three different MIP–SPCE sensors. The accuracy and precision were evaluated in terms of recovery and RSD, which had to be between 80.00–120.00% and less than 20.00%, respectively.88 The results of the insulin determination in the real sample demonstrate that the method is deemed to be precise and accurate (Table 2).
| Sample | The concentration of the diluted solution of the declared product (pM) | Determined concentration (pM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 1 | 25.0 | 25.2 | 99.42 | |
| 2 | 25.0 | 28.3 | 113.33 | 7.23 |
| 3 | 25.0 | 24.9 | 112.63 |
4. Conclusions
This work presents the development of a molecular imprinting polymer method using an electrochemical sensor to detect clinically relevant biomarker insulin in pharmaceutical samples. The first step in the development of a molecularly imprinted polymer (MIP) sensor was the electropolymerization of pyrrole in the presence of insulin using cyclic voltammetry. To obtain synthetic recognition units on a screen-printed carbon electrode (SPCE) surface for the selective determination of insulin, the insulin was successfully extracted by electrochemical oxidation of polypyrrole in 0.01 M PBS. The resulting MIP sensor enables the analysis of a single drop of solution (50 μL) containing insulin without additional nanomaterials or other complex immobilization compounds, such as aptamers, which is cost- and time-saving. At each step of MIP sensor development, the surface was characterized using atomic force microscopy, scanning electron microscopy, and energy-dispersive spectroscopy. The surface characterization confirmed the formation of the imprinted cavities in the polypyrrole (Ppy) matrix at MIP–SPCE, allowing electroanalytical performance for insulin detection. Thus, the use of pyrrole as a functional monomer provided useful electrochemical properties for the SPCE array, leading to the formation of a 3D insulin-imprinted structure. The obtained limit of detection (1.9 pM) and limit of quantification (6.2 pM) values are of interest as regards clinical diagnostics and were one of the lowest values reported in the literature. The method showed a linear response in a concentration range from 20.0–70.0 pM (R2 = 0.9991). The precision in terms of repeatability and reproducibility was evaluated using relative standard deviation (RSD) and was 5.13% and 5.92%, respectively. The developed MIP–SPCE sensor was successfully used to analyse insulin in the pharmaceutical sample (insulin cartridge). The quantification of insulin in the real sample was performed using multiple standard addition method. The accuracy in terms of the average recovery was 108.46%, and the precision in terms of RSD was 7.23%. The possibility of the analysis of the low sample volumes associated with the rapid response time offers an advantage over conventional methods. As a disposable and portable sensor system, it is also a promising candidate for point-of-care applications.Author contributions
Conceptualization: T. Z., D. M.; funding acquisition: U. M., M. F.; methodology: T. Z., D. M., T. M.; project administration: U. M., M. F.; resources: U. M., M. F.; software: T. Z., D. M., T. M.; supervision: U. M., M. F.; visualization: T. Z., D. M., T. M.; writing – original draft: T. Z., D. M.; writing – review and editing: U. M., M. F., D. M., T. Z., T. M. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support of the Ministry of Education, Science, and Sport (Grant Number: C3330-19-952027) and the Slovenian Research Agency (Grant Numbers: P3-0036, P2-0118, P2-0414, J3-2538, J3-1762, L7-4494, and J1-2470).References
- R. R. Bowsher and W. L. Nowatzke, Bioanalysis, 2011, 3, 883–898 CrossRef CAS PubMed.
- B. B. Prasad, R. Madhuri, M. P. Tiwari and P. S. Sharma, Electrochim. Acta, 2010, 55, 9146–9156 CrossRef CAS.
- V. Singh, Mater. Lett., 2019, 254, 415–418 CrossRef CAS.
- K. G. M. M. Alberti and P. Z. Zimmet, Diabetic Med., 1998, 15, 539–553 CrossRef CAS.
- G. Freckmann, S. Hagenlocher, A. Baumstark, N. Jendrike, R. C. Gillen, K. Rössner and C. Haug, J. Diabetes Sci. Technol., 2007, 1, 695–703 CrossRef PubMed.
- D. M. Muoio and C. B. Newgard, Nat. Rev. Mol. Cell Biol., 2008, 9, 193–205 CrossRef CAS PubMed.
- F. Goetz, L. French, W. Thomas, R. Gingerich and J. Clements, Metabolism, 1995, 44, 1371–1376 CrossRef CAS PubMed.
- K. V. Gobi, H. Iwasaka and N. Miura, Biosens. Bioelectron., 2007, 22, 1382–1389 CrossRef CAS PubMed.
- S. Ebrahimiasl, E. Fathi and M. Ahmad, Nanomed. Res. J., 2018, 3, 219–228 Search PubMed.
- B. Zhang, Y. Jiang, H. Kuang, C. Yao, Q. Huang, S. Xu, D. Tang and W. Fu, J. Immunol. Methods, 2008, 338, 7–13 CrossRef CAS PubMed.
- F. Kartal, D. Çimen, N. Bereli and A. Denizli, Mater. Sci. Eng., C, 2019, 97, 730–737 CrossRef CAS PubMed.
- A. Dayaldasani, M. Rodriguez Espinosa, P. Ocon Sanchez and V. Perez Valero, Ann. Clin. Biochem., 2015, 52, 312–318 CrossRef CAS PubMed.
- A. G. Cruz, I. Haq, T. Cowen, S. Di Masi, S. Trivedi, K. Alanazi, E. Piletska, A. Mujahid and S. A. Piletsky, Biosens. Bioelectron., 2020, 169, 112536 CrossRef PubMed.
- J. Grimshaw, Á. Kane, J. Trocha-Grimshaw, A. Douglas, U. Chakravarthy and D. Archer, Electrophoresis, 1994, 15, 936–940 CrossRef CAS PubMed.
- K. Van Uytfanghe, D. Rodríguez-Cabaleiro, D. Stöckl and L. M. Thienpont, Rapid Commun. Mass Spectrom., 2007, 21, 819–821 CrossRef CAS PubMed.
- Z. Chen, M. P. Caulfield, M. J. McPhaul, R. E. Reitz, S. W. Taylor and N. J. Clarke, Clin. Chem., 2013, 59, 1349–1356 CrossRef CAS PubMed.
- B. Adhikari and S. Majumdar, Prog. Polym. Sci., 2004, 29, 699–766 CrossRef CAS.
- R. Gui, H. Jin, H. Guo and Z. Wang, Biosens. Bioelectron., 2018, 100, 56–70 CrossRef CAS PubMed.
- Z. Iskierko, P. S. Sharma, K. Bartold, A. Pietrzyk-Le, K. Noworyta and W. Kutner, Biotechnol. Adv., 2016, 34, 30–46 CrossRef CAS PubMed.
- Y. Saylan, S. Akgönüllü, H. Yavuz, S. Ünal and A. Denizli, Sensors, 2019, 19, 1279 CrossRef CAS PubMed.
- S. K. Metkar and K. Girigoswami, Biocatal. Agric. Biotechnol., 2019, 17, 271–283 CrossRef.
- T. S. Rebelo, I. M. Miranda, A. T. Brandão, L. I. Sousa, J. A. Ribeiro, A. F. Silva and C. M. Pereira, Electrochemistry, 2021, 2, 427–438 CAS.
- A. Ben Hassine, N. Raouafi and F. T. Moreira, Chemosensors, 2021, 9, 238 CrossRef CAS.
- Z. Cheng-Jun, M. Xiong-Hui and L. Jian-Ping, Chin. J. Anal. Chem., 2017, 45, 1360–1366 Search PubMed.
- T. Zidarič, M. Finšgar, U. Maver and T. Maver, Biosensors, 2022, 12, 44 CrossRef PubMed.
- M. Cieplak and W. Kutner, Trends Biotechnol., 2016, 34, 922–941 CrossRef CAS PubMed.
- J. Erdőssy, V. Horváth, A. Yarman, F. W. Scheller and R. E. Gyurcsányi, TrAC, Trends Anal. Chem., 2016, 79, 179–190 CrossRef.
- F. W. Scheller, X. Zhang, A. Yarman, U. Wollenberger and R. E. Gyurcsányi, Curr. Opin. Electrochem., 2019, 14, 53–59 CrossRef CAS.
- A. Yarman and F. W. Scheller, Sensors, 2020, 20, 2677 CrossRef CAS PubMed.
- F. T. Moreira, S. Sharma, R. A. Dutra, J. P. Noronha, A. E. Cass and M. G. F. Sales, Sens. Actuators, B, 2014, 196, 123–132 CrossRef CAS.
- J. Ribeiro, C. Pereira, A. Silva and M. G. F. Sales, Anal. Chim. Acta, 2017, 981, 41–52 CrossRef CAS PubMed.
- T. S. Rebelo, C. Santos, J. Costa-Rodrigues, M. Fernandes, J. P. Noronha and M. G. F. Sales, Electrochim. Acta, 2014, 132, 142–150 CrossRef CAS.
- R. Schirhagl, D. Podlipna, P. A. Lieberzeit and F. L. Dickert, Chem. Commun., 2010, 46, 3128–3130 RSC.
- S. Knežević, M. Ognjanović, N. Nedić, J. F. Mariano, Z. Milanović, B. Petković, B. Antić, S. V. Djurić and D. Stanković, Microchem. J., 2020, 155, 104778 CrossRef.
- N. Žurga, D. Majer and M. Finšgar, Chemosensors, 2021, 9, 38 CrossRef.
- P. Noyrod, O. Chailapakul, W. Wonsawat and S. Chuanuwatanakul, J. Electroanal. Chem., 2014, 719, 54–59 CrossRef CAS.
- D. Majer and M. Finšgar, Biosensors, 2021, 11, 285 CrossRef CAS PubMed.
- M. Ognjanović, D. M. Stanković, M. Fabián, A. Vukadinović, Ž. Prijović, B. Dojčinović and B. Antić, Electroanalysis, 2018, 30, 2620–2627 CrossRef.
- O. D. Renedo, M. Alonso-Lomillo and M. A. Martínez, Talanta, 2007, 73, 202–219 CrossRef CAS PubMed.
- F. Tseliou, P. Pappas, K. Spyrou, J. Hrbac and M. I. Prodromidis, Biosens. Bioelectron., 2019, 132, 136–142 CrossRef CAS PubMed.
- D. M. Stanković, Z. Milanović, Ľ. Švorc, V. Stanković, D. Janković, M. Mirković and S. V. Đurić, Diamond Relat. Mater., 2021, 113, 108277 CrossRef.
- R. A. Couto, S. S. Costa, B. Mounssef Jr., J. G. Pacheco, E. Fernandes, F. Carvalho, C. M. Rodrigues, C. Delerue-Matos, A. A. Braga and L. M. Goncalves, Sens. Actuators, B, 2019, 290, 378–386 CrossRef CAS.
- M. Thapa, R. Sung and Y. S. Heo, Biosensors, 2021, 11, 507 CrossRef CAS PubMed.
- D. Ji, L. Liu, S. Li, C. Chen, Y. Lu, J. Wu and Q. Liu, Biosens. Bioelectron., 2017, 98, 449–456 CrossRef CAS PubMed.
- D. Ji, Z. Shi, Z. Liu, S. S. Low, J. Zhu, T. Zhang, Z. Chen, X. Yu, Y. Lu and D. Lu, Smart Mater. Med., 2020, 1, 1–9 CrossRef.
- R. Costa, J. Costa, P. Moreira, A. T. Brandão, I. Mafra, A. F. Silva and C. M. Pereira, Anal. Chim. Acta, 2021, 1191, 339310 CrossRef PubMed.
- P. Manickam, S. K. Pasha, S. A. Snipes and S. Bhansali, J. Electrochem. Soc., 2016, 164, B54 CrossRef.
- J. L. da Silva, E. Buffon, M. A. Beluomini, L. A. Pradela-Filho, D. A. G. Araújo, A. L. Santos, R. M. Takeuchi and N. R. Stradiotto, Anal. Chim. Acta, 2021, 1143, 53–64 CrossRef CAS PubMed.
- X. Kan, Z. Xing, A. Zhu, Z. Zhao, G. Xu, C. Li and H. Zhou, Sens. Actuators, B, 2012, 168, 395–401 CrossRef CAS.
- R. S. Gomes, B. A. Gomez-Rodríguez, R. Fernandes, M. G. F. Sales, F. T. Moreira and R. F. Dutra, Biosensors, 2021, 11, 175 CrossRef CAS PubMed.
- V. Ratautaite, R. Boguzaite, E. Brazys, A. Ramanaviciene, E. Ciplys, M. Juozapaitis, R. Slibinskas, M. Bechelany and A. Ramanavicius, Electrochim. Acta, 2022, 403, 139581 CrossRef CAS PubMed.
- S. A. Gupta and J. S. Singh, J. Sci. Res., 2021, 65, 110–115 Search PubMed.
- U. Maver, L. Gradišnik, D. M. Smrke, K. S. Kleinschek and T. Maver, Appl. Surf. Sci., 2019, 489, 485–493 CrossRef CAS.
- M. Milojević, G. Harih, B. Vihar, J. Vajda, L. Gradišnik, T. Zidarič, K. Stana Kleinschek, U. Maver and T. Maver, Pharmaceutics, 2021, 13, 564 CrossRef PubMed.
- T. Maver, T. Mastnak, M. Mihelič, U. Maver and M. Finšgar, Materials, 2021, 14, 1464 CrossRef CAS PubMed.
- S. J. Cobb and J. V. Macpherson, Anal. Chem., 2019, 91, 7935–7942 CrossRef CAS PubMed.
- J. J. Otarola, A. K. C. Solis, M. E. Farias, M. Garrido, N. M. Correa and P. G. Molina, Colloids Surf., A, 2020, 606, 125396 CrossRef CAS.
- J. Zhang, X.-T. Guo, J.-P. Zhou, G.-Z. Liu and S.-Y. Zhang, Mater. Sci. Eng., C, 2018, 91, 696–704 CrossRef CAS PubMed.
- A. Yarman, K. J. Jetzschmann, B. Neumann, X. Zhang, U. Wollenberger, A. Cordin, K. Haupt and F. W. Scheller, Chemosensors, 2017, 5, 11 CrossRef.
- V. Ratautaite, U. Samukaite-Bubniene, D. Plausinaitis, R. Boguzaite, D. Balciunas, A. Ramanaviciene, G. Neunert and A. Ramanavicius, Int. J. Mol. Sci., 2021, 22, 5032 CrossRef CAS PubMed.
- Y. Li and R. Qian, Electrochim. Acta, 2000, 45, 1727–1731 CrossRef CAS.
- J. E. Contreras-Naranjo and O. Aguilar, Biosensors, 2019, 9, 15 CrossRef CAS PubMed.
- W. B. S. Machini, N. V. Marques and A. M. Oliveira-Brett, J. Electroanal. Chem., 2019, 851, 113251 CrossRef CAS.
- L. Kong, X. Jiang, Y. Zeng, T. Zhou and G. Shi, Sens. Actuators, B, 2013, 185, 424–431 CrossRef CAS.
- P. K. Sharma, G. Gupta, V. V. Singh, B. Tripathi, P. Pandey, M. Boopathi, B. Singh and R. Vijayaraghavan, Synth. Met., 2010, 160, 2631–2637 CrossRef CAS.
- B. Lu, J. Xia, Z. Wang, F. Zhang, M. Yang, Y. Li and Y. Xia, RSC Adv., 2015, 5, 82930–82935 RSC.
- A. M. Elsayed, Recent Adv. Novel Drug Carrier Syst., 2012, 281–314 CAS.
- R. Sariri, Pharmacologyonline, 2011, 2, 1003–1012 Search PubMed.
- M. Shamsipur, N. Moradi and A. Pashabadi, J. Solid State Electrochem., 2018, 22, 169–180 CrossRef CAS.
- A. Turco, S. Corvaglia and E. Mazzotta, Biosens. Bioelectron., 2015, 63, 240–247 CrossRef CAS PubMed.
- G. Bolat, Y. T. Yaman, F. Kuralay and S. Abaci, J. Appl. Polym. Sci., 2020, 137, 49313 CrossRef CAS.
- X. Zhang and R. Bai, Langmuir, 2003, 19, 10703–10709 CrossRef CAS.
- G. Ozcelikay, S. Kurbanoglu, X. Zhang, C. Kosak Soz, U. Wollenberger, S. A. Ozkan, A. Yarman and F. W. Scheller, Polymers, 2019, 11, 1970 CrossRef CAS PubMed.
- D. Bizzotto, I. J. Burgess, T. Doneux, T. Sagara and H.-Z. Yu, ACS Sens., 2018, 3, 5–12 CrossRef CAS PubMed.
- P. Fanjul-Bolado, D. Hernández-Santos, P. J. Lamas-Ardisana, A. Martín-Pernía and A. Costa-García, Electrochim. Acta, 2008, 53, 3635–3642 CrossRef CAS.
- Y. Aghoutane, A. Diouf, L. Österlund, B. Bouchikhi and N. El Bari, Bioelectrochemistry, 2020, 132, 107404 CrossRef CAS PubMed.
- A. Waffo, C. Yesildag, G. Caserta, S. Katz, I. Zebger, M. Lensen, U. Wollenberger, F. Scheller and Z. Altintas, Sens. Actuators, B, 2018, 275, 163–173 CrossRef CAS.
- Y. L. Su, C. Y. Tai and J. M. Zen, Electroanalysis, 2013, 25, 2539–2546 CrossRef CAS.
- G. Moro, D. Cristofori, F. Bottari, E. Cattaruzza, K. De Wael and L. M. Moretto, Sensors, 2019, 19, 4433 CrossRef CAS PubMed.
- R. S. Gomes, F. T. Moreira, R. Fernandes and M. G. F. Sales, PLoS One, 2018, 13, e0196656 CrossRef PubMed.
- J. Ribeiro, C. Pereira, A. Silva and M. G. F. Sales, Biosens. Bioelectron., 2018, 109, 246–254 CrossRef CAS PubMed.
- E. C. Alexopoulos, Hippokratia, 2010, 14, 23–28 CAS.
- A. Salimi, A. Noorbakhash, E. Sharifi and A. Semnani, Biosens. Bioelectron., 2008, 24, 792–798 CrossRef CAS PubMed.
- E. Martinez-Perinan, M. Revenga-Parra, M. Gennari, F. Pariente, R. Mas-Ballesté, F. Zamora and E. Lorenzo, Sens. Actuators, B, 2016, 222, 331–338 CrossRef CAS.
- C. Guo, C. Chen, Z. Luo and L. Chen, Anal. Methods, 2012, 4, 1377–1382 RSC.
- J. Y. Gerasimov, C. S. Schaefer, W. Yang, R. L. Grout and R. Y. Lai, Biosens. Bioelectron., 2013, 42, 62–68 CrossRef CAS PubMed.
- J. Shepa, I. Šišoláková, M. Vojtko, L. Trnková, G. Nagy, I. Maskaľová, A. Oriňak and R. Oriňaková, Sensors, 2021, 21, 5063 CrossRef CAS PubMed.
- U. N. O. o. Drugs, C. Laboratory and S. Section, Guidance for the Validation of Analytical Methodology and Calibration of Equipment Used for Testing of Illicit Drugs in Seized Materials and Biological Specimens: A Commitment to Quality and Continuous Improvement, United Nations Publications, 2009 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |