
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic binary charge-transfer compounds of 2,2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :6′,2′′:6′′,6-trioxotriphenylamine and a pyrene-annulated azaacene as donors†
:6′,2′′:6′′,6-trioxotriphenylamine and a pyrene-annulated azaacene as donors†
Rajorshi Dasa,
Michael Linseisa,
Stefan M. Schuppb,
Franciska S. Gogescha,
Lukas Schmidt-Mende b and
Rainer F. Winter*a
b and
Rainer F. Winter*a
aFachbereich Chemie, Universität Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
bFachbereich Physik, Universität Konstanz, Universitätsstrasse 10, 78457 Konstanz, Germany
First published on 30th January 2023
Abstract
Three binary charge-transfer (CT) compounds resulting from the donor 2,2′![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′,2′′:6′′,6-trioxotriphenylamine (TOTA) and the acceptors F4TCNQ and F4BQ and of a pyrene-annulated azaacene (PAA) with the acceptor F4TCNQ are reported. The identity of these CT compounds are confirmed by single-crystal X-ray diffraction as well as by IR, UV-vis-NIR and EPR spectroscopy. X-ray diffraction analysis reveals a 1:1 stoichiometry for TOTA·F4TCNQ, a 2:1 donor:acceptor ratio in (TOTA)2·F4BQ, and a rare 4:1 stoichiometry in (PAA)4·F4TCNQ, respectively. Metrical parameters of the donor (D) and acceptor (A) constituents as well as IR spectra indicate full CT in TOTA·F4TCNQ, partial CT in (TOTA)2·F4BQ and only a very modest one in (PAA)4·F4TCNQ. Intricate packing motifs are present in the crystal lattice with encaged, π-stacked (F4TCNQ−)2 dimers in TOTA·F4TCNQ or mixed D/A stacks in the other two compounds. Their solid-state UV-vis-NIR spectra feature CT transitions. The CT compounds with F4TCNQ are electrical insulators, while (TOTA)2·F4BQ is weakly conducting.
:6′,2′′:6′′,6-trioxotriphenylamine (TOTA) and the acceptors F4TCNQ and F4BQ and of a pyrene-annulated azaacene (PAA) with the acceptor F4TCNQ are reported. The identity of these CT compounds are confirmed by single-crystal X-ray diffraction as well as by IR, UV-vis-NIR and EPR spectroscopy. X-ray diffraction analysis reveals a 1:1 stoichiometry for TOTA·F4TCNQ, a 2:1 donor:acceptor ratio in (TOTA)2·F4BQ, and a rare 4:1 stoichiometry in (PAA)4·F4TCNQ, respectively. Metrical parameters of the donor (D) and acceptor (A) constituents as well as IR spectra indicate full CT in TOTA·F4TCNQ, partial CT in (TOTA)2·F4BQ and only a very modest one in (PAA)4·F4TCNQ. Intricate packing motifs are present in the crystal lattice with encaged, π-stacked (F4TCNQ−)2 dimers in TOTA·F4TCNQ or mixed D/A stacks in the other two compounds. Their solid-state UV-vis-NIR spectra feature CT transitions. The CT compounds with F4TCNQ are electrical insulators, while (TOTA)2·F4BQ is weakly conducting.
Introduction
Ever since the first report on the charge-transfer compound TTF+·TCNQ− (TTF = tetrathiafulvalene, TCNQ = tetracyanoquinodimethane) with metal-like conductivity,1 organic charge-transfer (CT) systems Dδ+·Aδ−, composed of a donor (D) and an acceptor (A), have attracted a great deal of attention.2–6 Compounds of this type can act as good electrical conductors or components of ambipolar semiconductors3 and may show other intriguing properties including strong electron–phonon coupling,7,8 photoelectricity,9–13 ferromagnetism,14 antiferromagnetism,15 or luminescence.16–19 The degree of charge-transfer between the D and A components tends to increase with a decreasing energy difference between the lowest unoccupied molecular orbital LUMO of the acceptor A and the highest occupied molecular orbital HOMO of the donor D. Such information can be retrieved by quantum chemical calculations20 or, on an experimental basis, from their electrochemical reduction or oxidation potentials, respectively.3 These data therefore provide useful guidelines to adjust the likely degree of CT and aid in the purposeful design of CT compounds. For example, the combination of a strong donor with a strong acceptor usually leads to the formation of a CT compound D+·A− with a full transfer of charge (type-I, Fig. 1). This class of compounds may show strong ferromagnetism, but is often associated with only poor charge-transport capabilities.15,21,22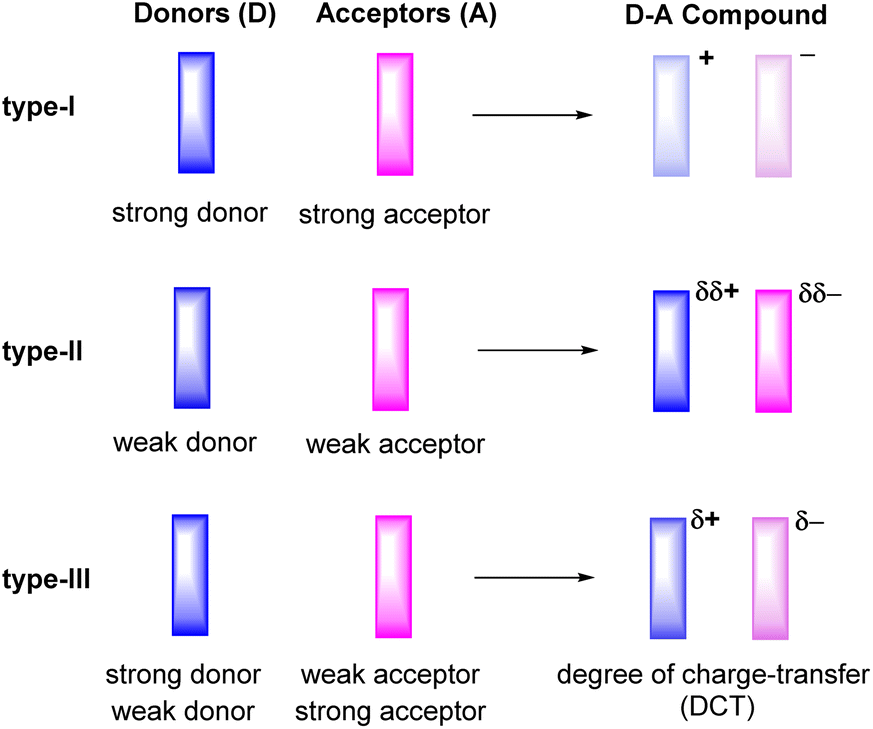 | ||
| Fig. 1 Degrees of charge-transfer for varieties of donor–acceptor (D–A) based organic CT compounds. | ||
The properties of D–A-based CT compounds do, however, not only depend on the degree of charge-transfer, but also on the stoichiometric ratio D:A and on intermolecular D/D, A/A and D/A interactions in the crystal lattice. The various modes of π-stacking play a particularly important role in this respect.3–5 From the wealth of previous studies it has emerged that D–A based CT compounds where the donors and the acceptors form segregated stacks (motif II in Fig. 2) show often higher charge mobilities than CT compounds where the donors and the acceptors form alternating (1:1 ratio) or mixed stacks (n:1 ratios) (motif I in Fig. 2). Illustrative examples are provided by various CT compounds assembled from TTF or its derivatives and TCNQ that crystallize in segregated D and A stacks and show conductivities of as high as 200 to 1000 S·cm−1.3,21 In contrast, CT compounds of tetramethoxyselenanthrenes and TCNQ form an alternately mixed-stacked structure [D⋯A]∞ of type A–I and exhibit poor conductivities of 4 × 10−10 S cm−1.3,21
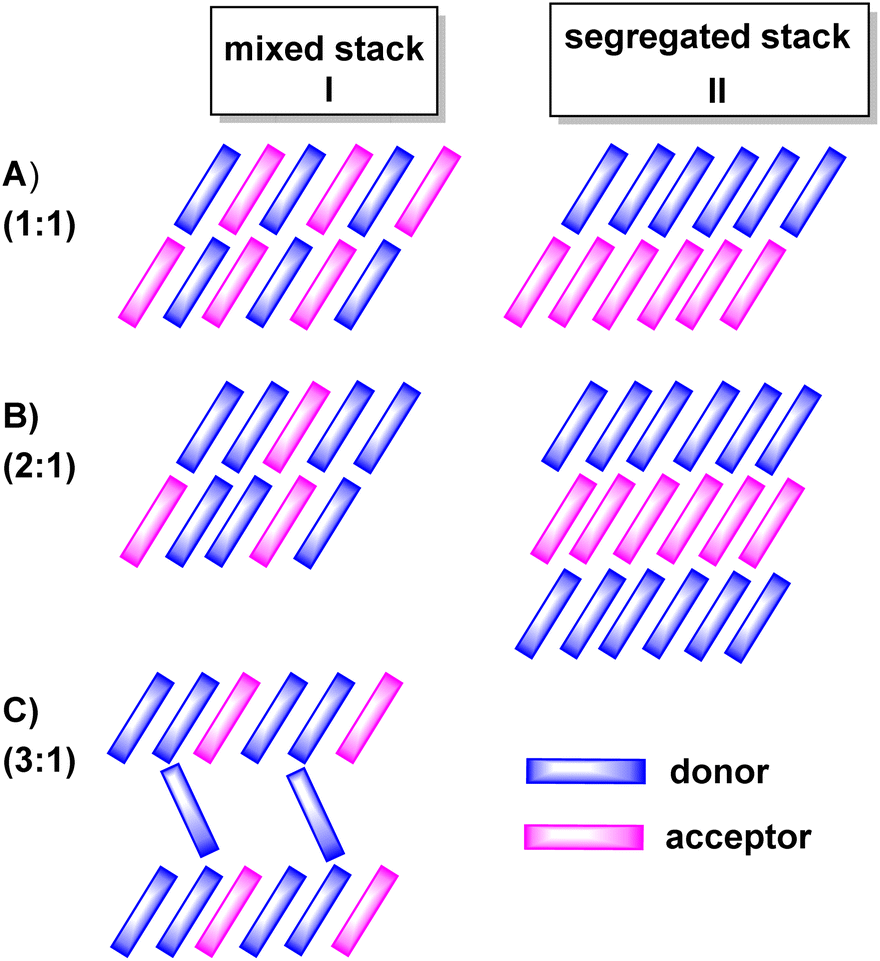 | ||
| Fig. 2 Different stoichiometric ratios (top to bottom) and stacking motifs (left to right) commonly observed in charge-transfer (CT) compounds. (A) 1:1, (B) 2:1 and (C) 3:1 donor/acceptor or acceptor/donor ratios. Column I displays structures with mixed stacks and column II structures with segregated stacks. | ||
In this work, we have studied D–A compounds formed by combining the donors 2,2′:6′,2′′:6′′,6-trioxotriphenylamine (TOTA, Fig. 3) and a pyrene-annulated azaacene (PAA) with five different acceptors, namely the 2,3,5,6-tetrahalogeno-p-benzoquinones X4BQ (X = F, Cl, Br), 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) and 7,7,8,8-tetracyanoquinodimethane (TCNQ). The donors constitute extended, planar π-systems and hence should be well-suited for π-stacking,23–25 but also allow for interdonor hydrogen bonding involving the O or N heteroatoms and for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N or C–F π-hole tetrel interactions.26–30 TOTA is a nonplanar, electron-rich molecule with a low half-wave potential E1/20/+ of 110 mV (in CH2Cl2/NBu4PF6 against the ferrocene/ferrocenium standard couple FcH/FcH+ = 0 mV) for its first one-electron oxidation. In contrast, PAA shows its first oxidation potential at Eox0/+ = 1205 mV under the same experimental conditions and is hence a much weaker donor (Table 1, vide infra). The acceptors were chosen in order to cover a wider range of half-wave potentials for their first one-electron reduction, ranging from E1/20/− = 153 mV for F4TCNQ to −464 mV for Cl4BQ in the order F4TCNQ > TCNQ > F4BQ ≈ Cl4BQ ≈ Br4BQ (Table 1). We show that the degree of CT in the TOTA compounds varies considerably from the strong acceptor F4TCNQ to the weaker acceptor F4BQ as it is, inter alia, manifested by a structural change of the TOTA donor from bowl-shaped to planar with an increasing degree of CT. Almost no CT to F4TCNQ was observed for the CT compound with the weaker PAA donor.
N or C–F π-hole tetrel interactions.26–30 TOTA is a nonplanar, electron-rich molecule with a low half-wave potential E1/20/+ of 110 mV (in CH2Cl2/NBu4PF6 against the ferrocene/ferrocenium standard couple FcH/FcH+ = 0 mV) for its first one-electron oxidation. In contrast, PAA shows its first oxidation potential at Eox0/+ = 1205 mV under the same experimental conditions and is hence a much weaker donor (Table 1, vide infra). The acceptors were chosen in order to cover a wider range of half-wave potentials for their first one-electron reduction, ranging from E1/20/− = 153 mV for F4TCNQ to −464 mV for Cl4BQ in the order F4TCNQ > TCNQ > F4BQ ≈ Cl4BQ ≈ Br4BQ (Table 1). We show that the degree of CT in the TOTA compounds varies considerably from the strong acceptor F4TCNQ to the weaker acceptor F4BQ as it is, inter alia, manifested by a structural change of the TOTA donor from bowl-shaped to planar with an increasing degree of CT. Almost no CT to F4TCNQ was observed for the CT compound with the weaker PAA donor.
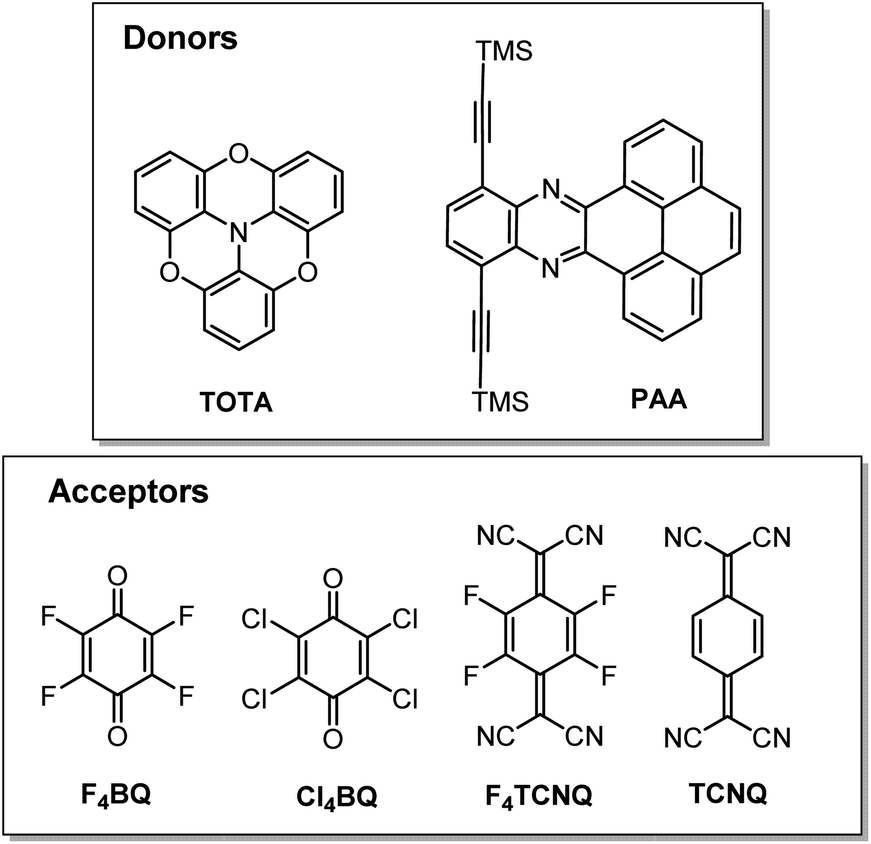 | ||
| Fig. 3 The donors and acceptors used in this study. | ||
| E1/20/+ | E1/2+/2+ | E1/20/− | E1/2−/2- | |
|---|---|---|---|---|
| a All data in millivolts versus FcH/FcH+ in CH2Cl2/NBu4PF6 at r. t. and at ν = 100 mV s−1.b Peak potential of the forward peak of a chemically irreversible anodic wave. | ||||
| TOTA | 110 | 1250 | — | — |
| PAA | 1205b | — | −1590 | — |
| F4TCNQ | — | — | 153 | −484 |
| TCNQ | — | — | −270 | −850 |
| F4BQ | — | — | −448 | −1336 |
| Cl4BQ | — | — | −452 | −1236 |
| Br4BQ | −464 | −1224 | ||
Results and discussion
Synthesis of the charge-transfer compounds
The D–A compounds formed on dissolving equimolar quantities of the respective pair of a donor and an acceptor in CH2Cl2 in a small vial. The vial was loosely capped by a screwcap and the solvent was allowed to slowly evaporate. After one or two weeks, dark purple crystals of a CT compound of TOTA and F4TCNQ, dark orange crystals of a TOTA/F4BQ and dark green crystals of a PAA/F4TCNQ D–A compound were obtained. Other combinations of one of these donors and another acceptor as well as using toluene as the solvent did not result in crystalline or phase-pure materials, although the formation of some pale-greenish crystals of a CT adduct of TOTA and TCNQ and of few yellow-green crystals from the PAA/TCNQ mixture were observed. These crystals did however not diffract well and could not be isolated in sufficient quantities and purity from the cocrystallized donor or acceptor to allow for their further investigation. Photographs of the three CT compounds discussed in this study and their precursors are provided in Fig. S1 in the ESI.†As discussed in the introduction, the electron donating and accepting abilities of all donors and acceptors were investigated by cyclic voltammetry. Representative cyclic voltammograms are provided in Fig. S2 to S4 in the ESI†; relevant data are listed in Table 1.
The TOTA-based CT compounds (TOTA)2·(F4TCNQ)2·CH2Cl2 and (TOTA)2·(F4BQ)
2,2′:6′,2′′:6′′,6-Trioxotriphenylamine (TOTA) is slightly bowl-shaped and can be easily oxidized to a stable, planar radical cation.31,32 Its first oxidation potential E1/20/+ of 110 mV against the ferrocene/ferrocenium standard (FcH/FcH+) in CH2Cl2/nBu4PF6 (nBu4PF6 = tetrabutylammonium hexafluorophosphate) is close to the reported value of 140 mV in DMF.31 Its reaction with equimolar quantities of the strong acceptor F4TCNQ accordingly yielded the 1:1 salt (TOTA)2·(F4TCNQ)2·CH2Cl2 with an essentially full transfer of charge between the donor and the acceptor (vide infra). The compound crystallized in the monoclinic space group P21/c with two crystallographically distinct molecules of the donor and the acceptor each and one CH2Cl2 solvent molecule in the unit cell. The structure is shown in Fig. 4.
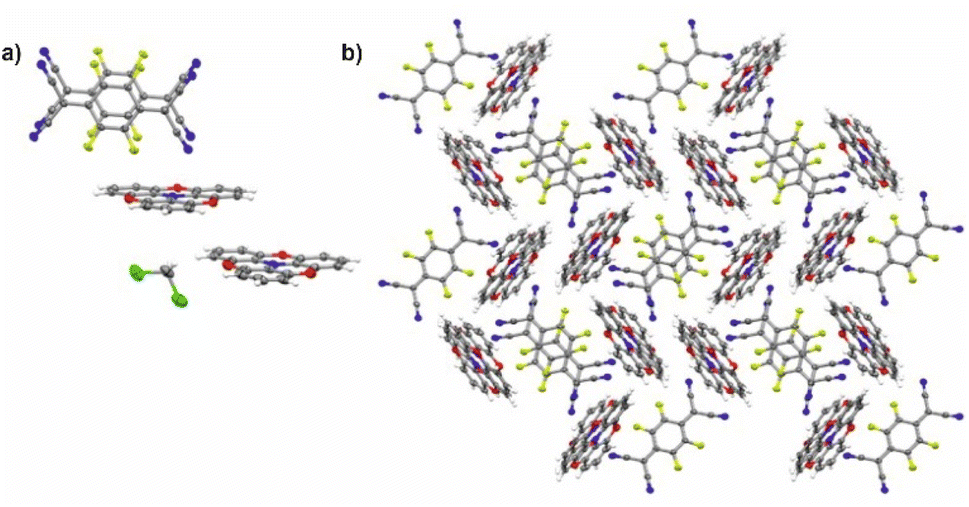 | ||
| Fig. 4 X-ray structure of (TOTA)2·(F4TCNQ)2·CH2Cl2. (a) Asymmetric unit showing the (F4TCNQ−)2 dimer and the two crystallographically different TOTA+ cations. (b) Packing motif of the donor and acceptor constituents in the crystal lattice; the CH2Cl2 solvent molecules are removed for clarity reasons. Colour codes: H, white; C, grey; N, blue; F, yellow-green; O, red; Cl, green. | ||
The bond parameters of F4TCNQ and, to a lesser extent, of TOTA are sensitive to their oxidation state. Neutral F4TCNQ has a quinoid structure with pronounced short-long-short bond length alternation (see Fig. 3 and Table 2). One-electron reduction increases the aromaticity of the central ring and renders the intracyclic CC bonds more similar while lengthening the exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. This bond lengthening appears to constitute the most indicative structure change in tetracyanoquinodimethanes concomitant with reduction, which complies with the notion that the cyano groups are the primary electron acceptors.46–48 On the other hand, bond length changes on oxidation of TOTA are diluted over the entire polycyclic π-system so that the most indicative structural changes are the shortening of the N–C bonds and the flattening of the cone at the amine N atom from 10° to fully planar (see Fig. 5).
C bonds. This bond lengthening appears to constitute the most indicative structure change in tetracyanoquinodimethanes concomitant with reduction, which complies with the notion that the cyano groups are the primary electron acceptors.46–48 On the other hand, bond length changes on oxidation of TOTA are diluted over the entire polycyclic π-system so that the most indicative structural changes are the shortening of the N–C bonds and the flattening of the cone at the amine N atom from 10° to fully planar (see Fig. 5).
|
||||||
|---|---|---|---|---|---|---|
| a | b | c | d | e | ref. | |
| a Average values from different structures in the provided references. For F4TCNQ−, the data for the various crystallized NBu4+ salts and for TOTA+ the data for the PF6−, ClO4− and ReO4− salts were used.b Average values for two crystallographically different donor and acceptor molecules in the unit cell. | ||||||
| F4TCNQa | 1.337 | 1.439 | 1.372 | 1.437 | 33–36 | |
| F4TCNQ−a | 1.358 | 1.417 | 1.418 | 1.430 | 1.385 | 37 and 38 |
| TOTA | 1.408 | 1.392 | 1.388 | 1.384 | 1.385 | 31 |
| TOTA+a | 1.376 | 1.375 | 1.394 | 1.378 | 1.378 | 31 and 32 |
| F4BQa | 1.339 | 1.477 | 1.213 | — | — | 39–41 |
| Cl4BQ | 1.344 | 1.489 | 1.211 | — | — | 42 |
| Cl4BQ− | 1.360 | 1.448 | 1.248 | — | — | 43 |
| TOTA·F4TCNQb | This work | |||||
| TOTA | 1.376 | 1.375 | 1.397 | 1.397 | 1.389 | |
| F4TCNQ | 1.358 | 1.417 | 1.410 | 1.423 | — | |
| (PAA)4·F4TCNQ | This work | |||||
| 1.341 | 1.445 | 1.381 | 1.443 | — | ||
| (TOTA)2·F4BQ | This work | |||||
| TOTA | 1.402 | 1.390 | 1.389 | 1.386 | 1.393 | |
| F4BQ | 1.336 | 1.472 | 1.219 | — | — | |
| TTF-F4BQ | 1.328 | 1.470 | 1.212 | — | — | 44 |
| 2 TMIQ-F4BQ | 1.316 | 1.470 | 1.210 | — | — | 45 |
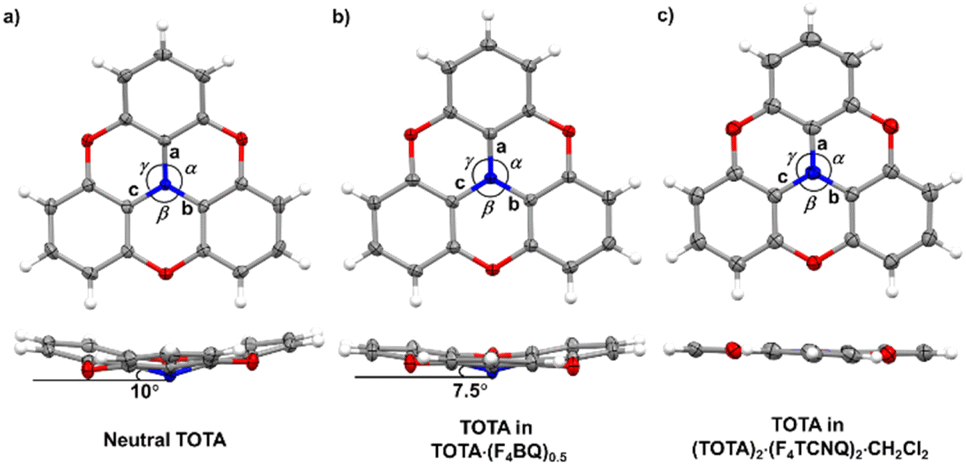 | ||
| Fig. 5 X-ray structures and metric parameters of (a) neutral TOTA,31 (b) the TOTA constituent in (TOTA)2·F4BQ and (c) the TOTA+ cation. | ||
Table 2 summarizes pertinent bond lengths of reference compounds F4TCNQ, F4TCNQ−, TOTA, TOTA+, F4BQ, Cl4BQ, Cl4BQ− and the D–A compounds of the present study. As can be seen from the data in Table 2, the metrics of the TOTA and the F4TCNQ constituents in (TOTA)2·(F4TCNQ)2·CH2Cl2 agree with those of the TOTA+ cation in the PF6−, ClO4− and ReO4− salts and of the F4TCNQ− anion in NBu4+ F4TCNQ−, respectively.37,38 In particular, the TOTA constituent has completely flattened out (Fig. 5c). This characterizes (TOTA)2·(F4TCNQ)2·CH2Cl2 as a true CT salt with full ionicity.
In the crystal lattice, the F4TCNQ− anions associate to pairs of nearly parallel, ecliptically arranged molecules with a tilt angle of 1.60° between their ring planes and a rather small interplanar distance of 3.215 Å (Fig. 4 and 6). The formation of FnTCNQ− (n = 0, 4) dimers has been observed on previous occasions and is associated with an antiferromagnetic alignment of their unpaired spins.38,49–51 Individual (F4TCNQ−)2 dimers are separated by two CH2Cl2 solvent molecules and arrange in columns that run parallel to the a axis of the unit cell. F4TCNQ− ions of neighbouring columns are nearly coplanar with a modest tilt of their ring planes by 6.7° and rotated by almost 90°.
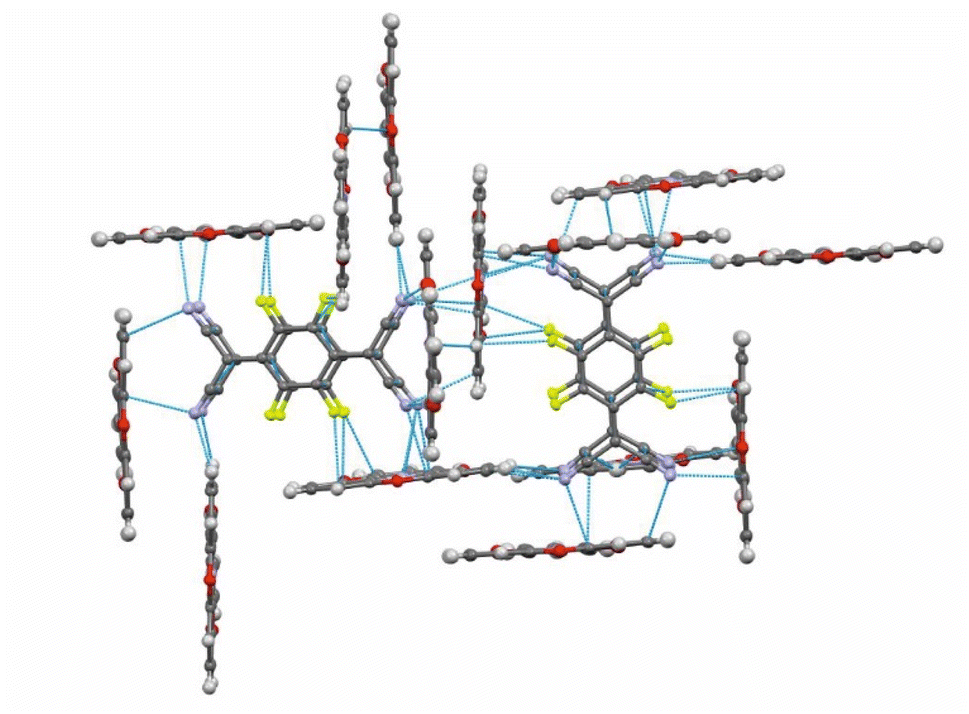 | ||
| Fig. 6 Intermolecular π-stacking, CN⋯ and C–F⋯π-hole tetrel and H-bonding interactions in (TOTA)2·(F4TCNQ)2·CH2Cl2, distances shorter than the sum of the van der Waals radii are indicated by blue dotted lines. | ||
The F4TCNQ− columns are separated by sheets that are formed by surrounding TOTA+ cations. Like the F4TCNQ− anions, TOTA+ polycycles that belong to different sheets adopt nearly orthogonal orientations with interplanar angles of 85.8° and 88.0° between their ring planes. As is shown in Fig. 4b, the F4TCNQ− dimers are encaged by six TOTA+ cations and every TOTA cation is in turn surrounded by three F4TCNQ− dimers. This rather curious packing arrangement is established by various CN⋯ and C–F⋯π-hole tetrel bonds26–30 as well as by C–H⋯NC hydrogen bonds and one weak CH⋯F interaction of 2.640 Å. Fig. 6 provides a view of two neighbouring (F4TCNQ−)2(TOTA+)6 cages with two additional weakly associated TOTA+ cations and the ensuing network of noncovalent interactions. The CN⋯π-hole tetrel interactions range from 2.998 Å to 3.183 Å, while the C–F⋯π-hole contacts measure 2.977 Å to 3.136 Å; CH⋯N interactions cover a range from 2.500 Å to 2.701 Å. Adjacent cages weakly associate by pairwise CH⋯O contacts of 2.697 Å between parallel displaced and laterally offset TOTA+ cations. When viewed along the b-axis of the unit cell (see the horizontal rows in Fig. 4b), an alternating arrangement of (F4TCNQ−)2 π-dimers and two coplanar TOTA+ cations emerges. The D+⋯D+ π–π interactions of 3.388–3.445 Å are notably weaker than the A−⋯A− π–π interactions of 3.108–3.195 Å (Fig. 4 and 6).
Single crystals of (TOTA)2·F4BQ were grown by slow evaporation of a CH2Cl2 solution of their equimolar mixture. The asymmetric unit cell contains one TOTA donor molecule and half a F4BQ acceptor molecule. In the crystal lattice, each F4BQ acceptor molecule is surrounded by two slightly bowl-shaped TOTA donors to provide a centrosymmetric arrangement D⋯A⋯D of nearly coplanar TOTA and F4BQ molecules with interplanar angles of 3.6° between their planes as defined by the three oxygen atoms at the TOTA ether straps or the central C6 ring of F4BQ. The N atom of the TOTA donor is displaced by 0.25 Å from the TOTA ring plane and points towards the F4BQ acceptor to provide a N⋯F4BQcentr. distance of 2.851 Å. These D⋯A⋯D arrays stack into infinite columns that run along the c-axis of the unit cell. Fig. 7b provides a view of two such D⋯A⋯D triples; an extended view over several unit cells down the c axis can be found as Fig. S5 of the ESI.† Neighbouring donors have an interplanar distance of 3.791 Å between the centroids as defined by their oxygen atoms and form pairwise contacts C5⋯C15 of 3.238 Å. Within the ab-plane of the unit cell, every F4BQ acceptor associates with six coplanarly arranged TOTA donors through a total of eight C–F⋯H–C hydrogen bonds of 2.485 Å to 2.643 Å and four O⋯H–C hydrogen bonds of 2.567 Å and 2.614 Å, respectively (see Fig. S6 of the ESI†). The TOTA molecules that surround the F4BQ acceptors connect through pairwise O⋯H–C hydrogen bonds of 2.656 Å, respectively. In turn, every TOTA molecule is surrounded by alternately arranged TOTA donors and F4BQ acceptors. Fig. 7a provides a view of the molecule arrangement and the resulting H-bonding network.
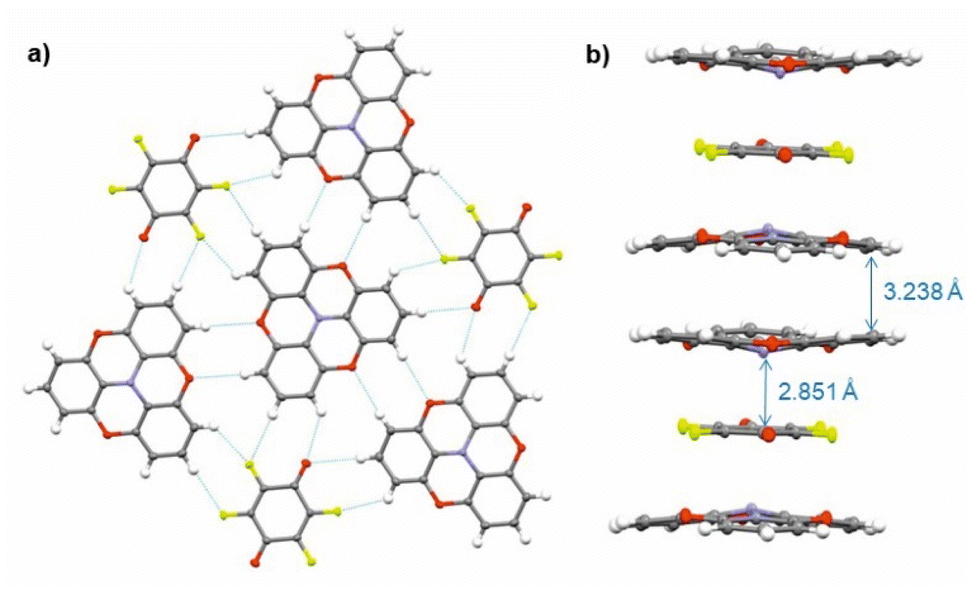 | ||
| Fig. 7 X-ray molecular structure of (TOTA)2 F4BQ. (a) Packing diagram within the ab plane with intermolecular H-bonding interactions indicated by blue dotted lines. (b) One-dimensional [D⋯A⋯D]∞ columns that run along the c axis of the unit cell. Colour codes: H, white; C, grey; N, blue; F, yellow-green; O, red. | ||
The bonding parameters of both, the F4BQ acceptor and the TOTA donor argue for an only limited degree of charge transfer, but do not allow for a quantitative assessment. Since no X-ray data for an authenticated F4BQ− anion seem to be available in the literature, we resort to its chloro-substituted analogue Cl4BQn− (n = 0, 1) for comparison.43 One-electron reduction of Cl4BQ causes a lengthening of the intracycle CC bonds and, by a larger margin, the external CO bonds while the former C–C bonds contract. The bond parameters of the F4BQ constituent of (TOTA)2·F4BQ are close to that of F4BQ itself or to those of its 1:1 TTF or its 1:2 TMIQ CT compounds, which also show only a fair degree of CT (TMIQ represents the 1,4-phenylene-bridged ditopic bis-carbazole donor shown on the right of the header of Table 2).44,45 In further agreement with an only modest degree of CT, the donor constituents retain the domed, non-planar structure of neutral TOTA, albeit with a smaller cone angle of 7.5° at the N atom and slightly wider Cph–N–Cph bond angles α, β, and γ of 116.57(10)°, 116.86(10)° and 117.00(10)° as compared to the values of 115.3(2)°, 115.6(2)° and 115.7(2)° for neutral TOTA (Fig. 5).31 As already mentioned, TOTA+ is planar with angles α, β, and γ close to 120° (e.g. 119.7(5)°, 119.9(5)° and 120.3(5)° in the perrhenate salt).27 The N–Cphenyl and O–Cphenyl bond lengths in (TOTA)2·F4BQ are nearly identical to those of pristine, neutral TOTA and significantly longer than in (TOTA)2·(F4TCNQ)2·CH2Cl2 or other salts with authenticated TOTA+ cations.31,32
The donor–acceptor compound (PAA)4·F4TCNQ
As suggested by the high anodic peak potential of 1205 mV, the pyrene-annulated azaacene PAA (Fig. 3) is only a very poor donor. Hence, the formation of a binary D–A compound was only observed with the strongest acceptor F4TCNQ. The dark green crystals obtained by slow evaporation of CH2Cl2 from a 1:1 solution of PAA and F4TCNQ turned out to assume a rather unusual 4:1 D:A stoichiometry (PAA)4·F4TCNQ. The compound crystallized in the monoclinic space group C2/c. The asymmetric unit consists of two molecules of the PAA donor and half a molecule of the F4TCNQ acceptor. In the crystal lattice, repeat units of four D and one A molecules form one-dimensional infinite columns [⋯D⋯D⋯A⋯D⋯D⋯]∞ that run along alternately the a or the b axis of the unit cell (Fig. 8). The neighbouring, crystallographically unique PAA donors PAA1 and PAA2 of an A⋯D⋯D⋯ sequence align in a quasi-centrosymmetric fashion in order to minimize steric repulsion between the bulky trimethylsilyl substituents. As is shown in the left panel of Fig. 8, these building blocks repeat in a centrosymmetric manner to generate columns. With interplane angles F4TCNQ–PAA1 of 2.30° and PAA1–PAA2 of 1.96°, the individual donor and the acceptor molecules arrange in a nearly coplanar fashion. Separations of 3.324 Å (F4TCNQ–PAA1), 3.328 Å (PAA1–PAA2) and 3.378 Å (PAA2–PAA2) are all smaller than 3.5 Å, which indicates π-stacking interactions.52
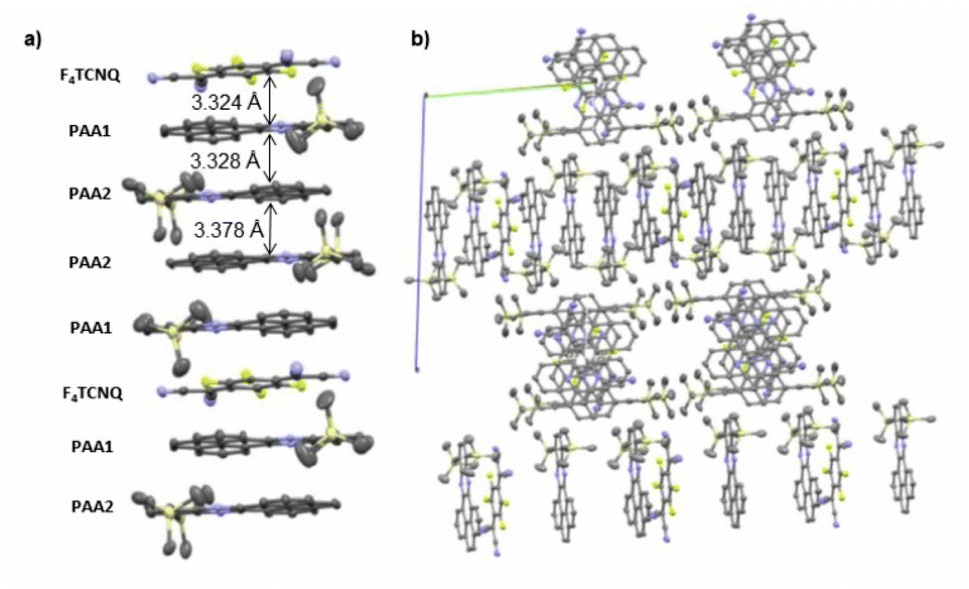 | ||
| Fig. 8 (a) Molecular packing of (PAA)4·F4TCNQ viewed along the a axis of the unit cell. (b) Alignment of the rows in the crystal lattice. Solvent molecules are removed for clarity reasons. | ||
The close match between the bond lengths of the A constituent of this compound to those of neutral F4TCNQ evidences the lack of any substantial degree of CT from PAA. The bonding parameters of the crystallographically distinct PAA donors in (PAA)4·F4TCNQ differ slightly (see Fig. S7 of the ESI†), but are in line with those in neutral, pyrene-appended azaacene derivatives and other bis(trialkylsilyl) derivatives of diethynyl-substituted azaacenes.23,24
As a bottom line, crystallographic data indicate full CT in (TOTA)2·(F4TCNQ)2, an only moderate degree of CT in (TOTA)2·F4BQ, and the absence of any significant degree of CT in (PAA)4·F4TCNQ, which complies with the difference of oxidation potentials of the respective donor and the reduction potential of the acceptor.
IR, UV-vis-NIR and EPR spectroscopy
In addition to structure data, infrared (IR) and UV-vis-NIR spectroscopy have proven highly instructive for quantifying the extent of charge-transfer in D–A compounds. Of particular diagnostic value are the redox state-dependent band positions of the nitrile CN ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (CN) or the quinone CO stretching modes (CO) of the acceptor. Their shift with respect to the neutral and the reduced forms of the respective acceptor scales linearly with the fraction of charge ρ (in units of the elementary charge e) transferred from the donor to the acceptor.53,54 These band shifts provide a widely applied criterion for assessing the degree of CT.6,21,55–60 Arene stretching and bending vibrations of the D component may provide additional information.
(CN) or the quinone CO stretching modes (CO) of the acceptor. Their shift with respect to the neutral and the reduced forms of the respective acceptor scales linearly with the fraction of charge ρ (in units of the elementary charge e) transferred from the donor to the acceptor.53,54 These band shifts provide a widely applied criterion for assessing the degree of CT.6,21,55–60 Arene stretching and bending vibrations of the D component may provide additional information.
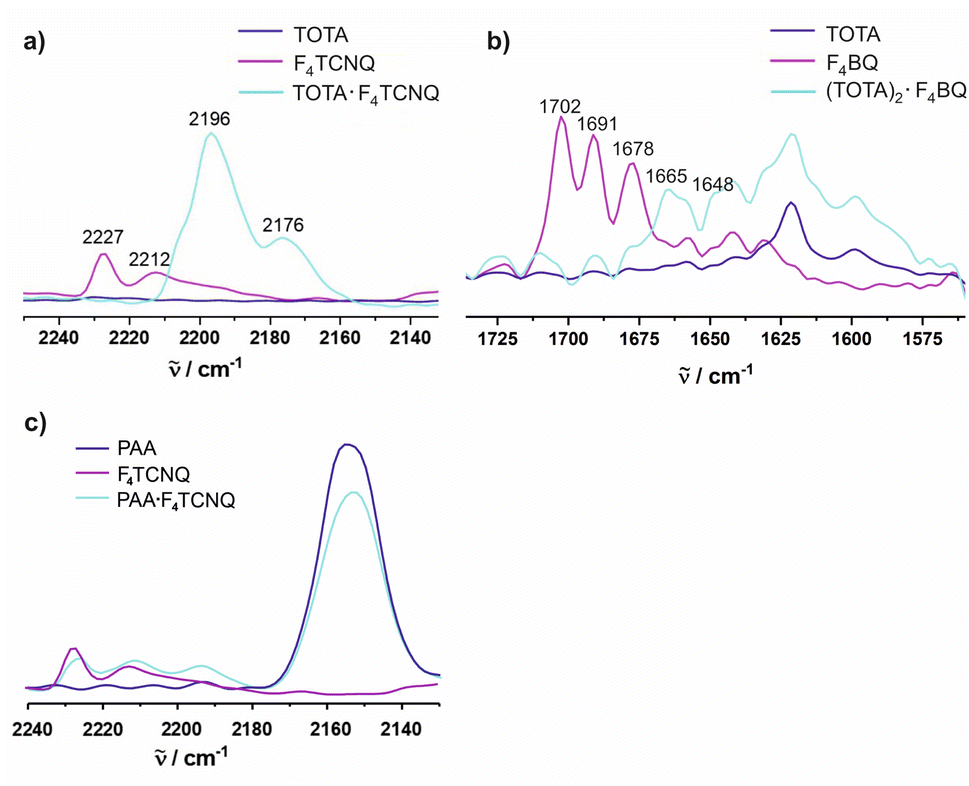
The CN stretches of the F4TCNQ acceptor in TOTA·F4TCNQ at = 2196 cm−1 and 2176 cm−1 are significantly red-shifted from their positions of 2227 cm−1 (the stronger b1u mode) and 2212 cm−1 (the weaker b2u mode) in pristine F4TCNQ (Fig. 9a)61 and closely resemble those of the F4TCNQ− anion of 2194 cm−1 and 2172 cm−1.6,61,62 The degree of charge-transfer ρ to F4TCNQ can be calculated according to eqn (1),
|
ρ = (2·Δ/0)·(1−−12/02)−1 (ref. 6 and 54)
| (1) |
0 and -1 represent the wavenumber of the more prominent CN stretching vibration in neutral F4TCNQ (0) or its associated radical anion (-1), and Δ is the difference between the band position in neutral F4TCNQ and the charge transfer compound, 0 − CT, respectively. Applying eqn (1) to TOTA·F4TCNQ yields ρ = 0.95 in agreement with essentially full electron transfer. IR data for the D constituent agree with this view. Hence, IR(KBr) spectra of TOTA·F4TCNQ feature donor bands at 1590, 1334, 1277, 1074 and 1031 cm−1 (see Fig. S8 of the ESI†). These values are nearly identical to those of 1589, 1333, 1275, 1072 and 1028 cm−1 reported in the literature for the TOTA+ radical cation and differ from those of pristine TOTA (1621, 1480, 1336, 1318, 1265, 1067 and 1018 cm−1).63
 | ||
| Fig. 9 Spectroscopic changes in the mid IR region (CN stretching vibrations) of F4TCNQ in (a) (TOTA)2·(F4TCNQ)2·CH2Cl2, (b) (TOTA)2·F4BQ, and (c) (PAA)4·F4TCNQ. | ||
The formation of D–A compounds is also evident from UV-vis-NIR spectroscopy. UV-vis-NIR spectra of solid samples of neutral TOTA and F4TCNQ and of the CT compound TOTA·F4TCNQ were recorded in an integrating sphere in order to diminish intensity losses due to scattering and reflection. As shown in Fig. 10, the spectra of the neutral precursors display intense bands at 380 and 525 nm and 405 and 455 nm, respectively, whereas TOTA·F4TCNQ shows discernible peaks at 595, 650, 770 and at ca. 1060 nm.
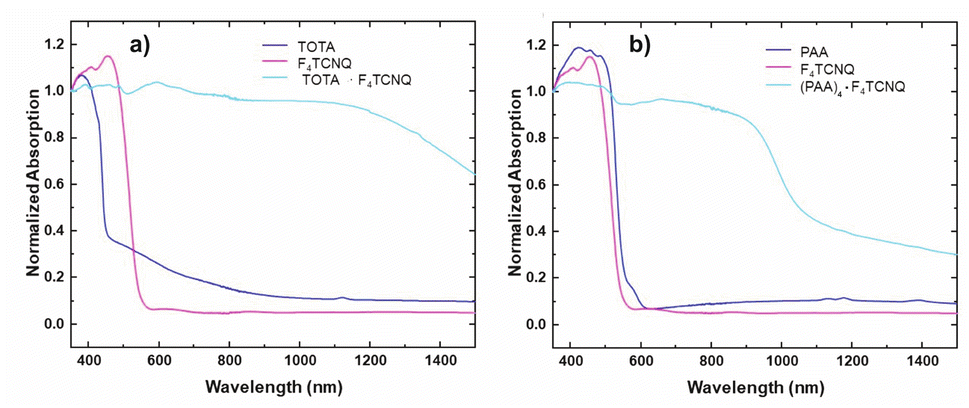 | ||
| Fig. 10 Monitoring the CT compound formation by UV-vis-NIR spectroscopy. The spectra of CT compounds are plotted with their parent donor and acceptor constituents. (a) (TOTA)2·(F4TCNQ)2·CH2Cl2 and (b) (PAA)4(F4TCNQ). | ||
EPR spectroscopy provides a highly sensitive probe of paramagnetic species resulting from charge transfer.62,64,65 Solid TOTA·F4TCNQ (Fig. 11a) shows accordingly an intense EPR resonance at a g value of 1.9990, whose intensity increases on cooling. This agrees with the T-dependent Boltzmann distribution as given in eqn (2)66,67 and the high ionicity in the ground state of this compound. One should note here that the observed EPR resonance in the solid state is likely due to exclusively the TOTA+ cation, as the F4TCNQ− anions associate to diamagnetic dimers.
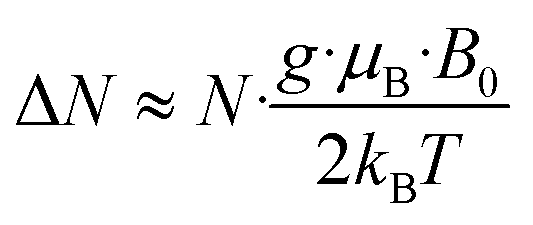 | (2) |
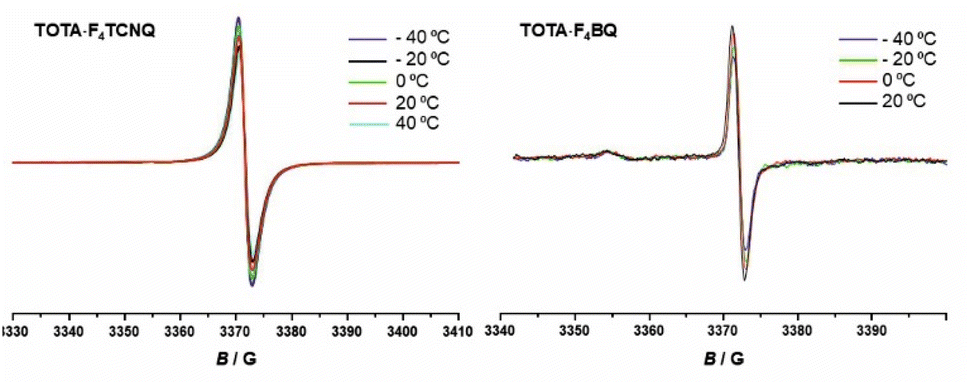 | ||
| Fig. 11 T-dependent EPR spectra of the CT compounds TOTA·F4TCNQ (left) and (TOTA)2·F4BQ (right). | ||
For F4BQ, the energies of the ν(CO) stretching vibrations serve the same purpose as the CN bands in XnTCNQ derivatives (X = Hal, n = 0–4) such that eqn (1) applies accordingly. νCO bands of F4BQ in (TOTA)2·F4BQ are found at 1665 and 1648 cm−1, whereas they are located at 1702, 1691 and 1678 cm−1 in neutral F4BQ (see Fig. 9b). The data are to be compared with literature values of 1705, 1693 and 1686 cm−1 for F4BQ68 and 1556 and 1502 cm−1 for the sodium salt of F4BQ−.69,70 Using eqn (1), ρ was calculated as 0.26. Hence, (TOTA)2·F4BQ seems to exhibit a fair degree of CT which was not so evident from the structure data. There are only minor shifts in the IR bands of the TOTA constituents (Fig. S9 of the ESI†). One should, however, note that the extent of charge loss from an individual TOTA donor is only half of that which is accumulated at the F4BQ acceptor. In the solid state, (TOTA)2·F4BQ exhibits a weak, broad CT absorption at low energy (Fig. S10 of the ESI†). As shown in Fig. 11b, solid TOTA·F4BQ is also EPR active, but shows an opposite T dependence to TOTA·F4TCNQ, i. e. the signal intensity decreases on lowering the temperature. This suggests that CT in these compounds is a thermally activated process. In summary, IR, UV-vis-NIR and EPR data on the solid samples are consistent with the notion of (nearly) quantitative CT from the donor to the acceptor in TOTA·F4TCNQ and a more modest one in (TOTA)2·F4BQ. The degree of CT ρ as quantified by the shift of the CN stretching vibrations of the donor amounts to 0.95 and 0.26, respectively.
In contrast, the IR data of (PAA)4(F4TCNQ) resemble those of the pristine, neutral constituents closely with only a small shift of the νCN band by ca. 1.5 cm−1 (Fig. 9c) which translates into ρ ≈ 0.05, thus indicating a very modest degree of CT (see also Fig. S11 of the ESI† for the arene bands). In agreement with the small degree of CT, (PAA)4(F4TCNQ) shows only a very weak EPR resonance signal (see Fig. S12 of the ESI†). Nevertheless, the compound absorbs strongly in the solid state over the entire UV and vis range down to 1100 nm as shown in Fig. 10b. We note that accounts of compounds showing prominent CT bands despite small degrees of CT have appeared in the literature.19
In order to assess their conductive properties, single crystals of all three isolated CT compounds were placed on a gold plate or a conductive Cu-tape and contacted with two closely spaced nanoprobes, which served as electrodes (for details to the experimental setup, see the Materials and methods section and Fig. S13 of the ESI†). Even on applying a maximum voltage of 20 V, no detectable current flow was observed for TOTA·F4TCNQ and for (PAA)4·F4TCNQ, even at a tip distance as small as ca. 10 μm. Their insulating behaviour (Fig. S14 and S15 of the ESI†) is a direct consequence of their structures with very weakly interacting (TOTA+)8(F4TCNQ−)2 cages or [⋯D⋯D⋯A⋯D⋯D⋯]∞ columns with negligible charge transfer. (TOTA)2·F4BQ is also nearly insulating with a resistance per unit length of 5–10 GΩ μm−1 for different specimen, which is close to the detection limit of our experimental setup (Fig. S16–S19 of the ESI†).
Conclusions
We describe two charge-transfer compounds of the 2,2′:6′,2′′:6′′,6-trioxotriphenylamine donor and tetrafluoro-tetracyano-p-quinodimethane (F4TCNQ) or tetrafluoro-p-benzoquinone (F4BQ) as the acceptor component and a donor–acceptor complex of a much less electron-rich pyrene-annulated azaacene (PAA) with F4TCNQ. Although in each case equimolar amounts of the donor D and the acceptor A were used for their synthesis, only (TOTA)2·(F4TCNQ)2·CH2Cl2 adopted a D:A stoichiometry of 1:1. Crystals isolated with F4BQ as the acceptor provided a 2:1 ratio between the donor and the acceptor constituents instead. The combination of PAA and F4TCNQ resulted even in a rare 4:1 composition, yielding (PAA)4·(F4TCNQ). X-ray diffraction analysis indicated full charge transfer from the donor to the acceptor in (TOTA)2·(F4TCNQ)2·CH2Cl2 by virtue of the bond parameters of the F4TCNQ acceptor and the planarization of the TOTA constituent. These findings are also supported by IR and UV-vis-NIR spectroscopy. In the crystalline state, π-stacked F4TCNQ− dimers are encaged by six TOTA+ counterions with only weak interactions between these structural entities.
For the other two D/A combinations, X-ray structure analysis revealed mixed stacking patterns with [D⋯A⋯D]∞ or [D⋯D⋯A⋯D⋯D]∞ packing motifs. Monitoring the shifts of the CN and CO stretching frequencies in the CT compounds with respect to the neutral and monoreduced acceptors revealed an essentially complete charge-transfer in (TOTA)2·(F4TCNQ)2·CH2Cl2, a moderate degree of charge-transfer in (TOTA)2·(F4BQ) (ρ = 0.26) and an only very modest degree of CT in (PAA)4·(F4TCNQ). Nevertheless, a fairly intense CT band was found in solid state UV-vis-NIR spectra. T-dependent EPR spectra recorded in the solid state agree with a (nearly) complete CT in the ground state of the TOTA·F4TCNQ compound, while CT in (TOTA)2·(F4BQ) is thermally activated and (PAA)4·(F4TCNQ) shows only a very weak EPR signal. All CT compounds are non-conductive or, in the case of (TOTA)2·(F4BQ) very weakly conductive in the solid state.
Data availability
Details to the methods and materials, characterization data and additional figures can be found in the ESI†. CCDC reference numbers for the reported crystallographic structures are 2220394 ((PAA)4·F4TCNQ), 2220397 ((TOTA)2·F4BQ) and 2220398 ((TOTA)2·(F4TCNQ)2·CH2Cl2).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are indebted to the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) for financial support of this work through grant Wi1262/17-1.Notes and references
- J. Ferraris, D. O. Cowan, J. V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- H. Bassler and A. Kohler, Top. Curr. Chem., 2012, 312, 1–65 Search PubMed.
- K. P. Goetz, D. Vermeulen, M. E. Payne, C. Kloc, L. E. McNeil and O. D. Jurchescu, J. Mater. Chem. C, 2014, 2, 3065–3076 RSC.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef CAS PubMed.
- W. Wang, L. Luo, P. Sheng, J. Zhang and Q. Zhang, Chem.–Eur. J., 2021, 27, 464–490 CrossRef CAS PubMed.
- B. Mahns, O. Kataeva, D. Islamov, S. Hampel, F. Steckel, C. Hess, M. Knupfer, B. Büchner, C. Himcinschi, T. Hahn, R. Renger and J. Kortus, Cryst. Growth Des., 2014, 14, 1338–1346 CrossRef CAS.
- L. Zhu, H. Geng, Y. Yi and Z. Wei, Phys. Chem. Chem. Phys., 2017, 19, 4418–4425 RSC.
- J. Singleton, J. Solid State Chem., 2002, 168, 675–689 CrossRef CAS.
- J. Zhang, H. Geng, T. S. Virk, Y. Zhao, J. Tan, C.-a. Di, W. Xu, K. Singh, W. Hu, Z. Shuai, Y. Liu and D. Zhu, Adv. Mater., 2012, 24, 2603–2607 CrossRef CAS PubMed.
- T. Wakahara, P. D'Angelo, K. i. Miyazawa, Y. Nemoto, O. Ito, N. Tanigaki, D. D. C. Bradley and T. D. Anthopoulos, J. Am. Chem. Soc., 2012, 134, 7204–7206 CrossRef CAS PubMed.
- W. Yu, X.-Y. Wang, J. Li, Z.-T. Li, Y.-K. Yan, W. Wang and J. Pei, Chem. Commun., 2013, 49, 54–56 RSC.
- H.-D. Wu, F.-X. Wang, Y. Xiao and G.-B. Pan, J. Mater. Chem. C, 2014, 2, 2328–2332 RSC.
- S. J. Kang, J. B. Kim, C.-Y. Chiu, S. Ahn, T. Schiros, S. S. Lee, K. G. Yager, M. F. Toney, Y.-L. Loo and C. Nuckolls, Angew. Chem., Int. Ed., 2012, 51, 8594–8597 CrossRef CAS PubMed.
- P.-M. Allemand, K. C. Khemani, A. Koch, F. Wudl, K. Holczer, S. Donovan, G. Grüner and J. D. Thompson, Science, 1991, 253, 301–302 CrossRef CAS PubMed.
- T. Enoki and A. Miyazaki, Chem. Rev., 2004, 104, 5449–5477 CrossRef CAS PubMed.
- Y. L. Lei, Y. Jin, D. Y. Zhou, W. Gu, X. B. Shi, L. S. Liao and S. T. Lee, Adv. Mater., 2012, 24, 5345–5351 CrossRef CAS PubMed.
- Y. L. Lei, L. S. Liao and S. T. Lee, J. Am. Chem. Soc., 2013, 135, 3744–3747 CrossRef CAS PubMed.
- W. Zhu, R. Zheng, X. Fu, H. Fu, Q. Shi, Y. Zhen, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2015, 54, 6785–6789 CrossRef CAS PubMed.
- Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS PubMed.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- H. Jiang, P. Hu, J. Ye, K. K. Zhang, Y. Long, W. Hu and C. Kloc, J. Mater. Chem. C, 2018, 6, 1884–1902 RSC.
- R. J. Walwyn, B. Chan, P. M. Usov, M. B. Solomon, S. G. Duyker, J. Y. Koo, M. Kawano, P. Turner, C. J. Kepert and D. M. D'Alessandro, J. Mater. Chem. C, 2018, 6, 1092–1104 RSC.
- P. Biegger, S. Stolz, S. N. Intorp, Y. Zhang, J. U. Engelhart, F. Rominger, K. I. Hardcastle, U. Lemmer, X. Qian, M. Hamburger and U. H. F. Bunz, J. Org. Chem., 2015, 80, 582–589 CrossRef CAS PubMed.
- X. Yu, J. Wan, C. Yuan, N. Guo, Y. Shen and J. Li, Dyes Pigm., 2019, 161, 130–136 CrossRef CAS.
- B. D. Lindner, Y. Zhang, S. Höfle, N. Berger, C. Teusch, M. Jesper, K. I. Hardcastle, X. Qian, U. Lemmer, A. Colsmann, U. H. F. Bunz and M. Hamburger, J. Mater. Chem. C, 2013, 1, 5718–5724 RSC.
- S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 5702–5717 RSC.
- A. M. Montana, ChemistrySelect, 2017, 2, 9094–9112 CrossRef CAS.
- S. J. Grabowski, Molecules, 2021, 26, 4939 CrossRef CAS PubMed.
- S. M. Nashre-ul-Islam, K. K. Borah, F. E. Ozturkkan, M. A. Raza, A. Frontera and D. M. Gil, J. Mol. Struct., 2022, 1268, 133686 CrossRef CAS.
- S. Scheiner, Molecules, 2020, 25, 4495 CrossRef CAS PubMed.
- M. Kuratsu, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2005, 44, 4056–4058 CrossRef CAS PubMed.
- Y. Nakano, H. Yamochi, G. Saito, M. Kuratsu and K. Okada, J. Phys.: Conf. Ser., 2008, 132, 012024 CrossRef.
- T. J. Emge, M. Maxfield, D. O. Cowan and T. J. Kistenmacher, Mol. Cryst. Liq. Cryst., 1981, 65, 161–178 CrossRef CAS.
- Y. Krupskaya, M. Gibertini, N. Marzari and A. F. Morpurgo, Adv. Mater., 2015, 27, 2453–2458 CrossRef CAS PubMed.
- T. Salzillo, M. Masino, G. Kociok-Köhn, D. Di Nuzzo, E. Venuti, R. G. Della Valle, D. Vanossi, C. Fontanesi, A. Girlando, A. Brillante and E. Da Como, Cryst. Growth Des., 2016, 16, 3028–3036 CrossRef CAS.
- R. Shukla, C. Ruzie, G. Schweicher, A. R. Kennedy, Y. H. Geerts, D. Chopra and B. Chattopadhyay, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 71–78 CrossRef CAS PubMed.
- J. S. Park, J. Park, Y. J. Yang, T. T. Tran, I. S. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 7598–7604 CrossRef CAS PubMed.
- S. A. O'Kane, R. Clérac, H. Zhao, X. Ouyang, J. R. Galán-Mascarós, R. Heintz and K. R. Dunbar, J. Solid State Chem., 2000, 152, 159–173 CrossRef.
- R. Shukla and D. Chopra, CrystEngComm, 2018, 20, 3308–3312 RSC.
- K. Hagen, D. G. Nicholson and L. J. Saethre, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1959–1961 CrossRef.
- A. Meresse, C. Courseille and B. C. Nguyen, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1974, 30, 524–526 CrossRef CAS.
- K. J. Van Weperen and G. J. Visser, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1972, 28, 338–342 CrossRef CAS.
- J.-M. Lü, S. V. Rosokha, I. S. Neretin and J. K. Kochi, J. Am. Chem. Soc., 2006, 128, 16708–16719 CrossRef PubMed.
- J. J. Mayerle, J. B. Torrance and J. I. Crowley, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1979, 35, 2988–2995 CrossRef.
- Z. Wang, F. Yu, J. Xie, J. Zhao, Y. Zou, Z. Wang and Q. Zhang, Chem.–Eur. J., 2020, 26, 3578–3585 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 1820–1833 CrossRef CAS.
- P. Coppens, T. N. Guru Row, P. Leung, E. D. Stevens, P. J. Becker and Y. W. Yang, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1979, 35, 63–72 CrossRef.
- P. Coppens, Phys. Rev. Lett., 1975, 35, 98–100 CrossRef CAS.
- R. H. Harms, H. J. Keller, D. Noethe, D. Wehe, N. Heimer, R. M. Metzger, D. Gundel and H. Sixl, Mol. Cryst. Liq. Cryst., 1982, 85, 1639–1645 CrossRef CAS.
- A. L. Sutton, B. F. Abrahams, D. M. D'Alessandro, T. A. Hudson, R. Robson and P. M. Usov, CrystEngComm, 2016, 18, 8906–8914 RSC.
- O. J. Dautel and M. Fourmigué, New J. Chem., 2001, 25, 834–838 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- J. S. Chappell, A. N. Bloch, W. A. Bryden, M. Maxfield, T. O. Poehler and D. O. Cowan, J. Am. Chem. Soc., 1981, 103, 2442–2443 CrossRef CAS.
- A. Salmerón-Valverde, J. G. Robles-Martínez and A. Zehe, Cryst. Res. Technol., 1994, 29, 703–706 CrossRef.
- M. Rudloff, K. Ackermann, M. Huth, H. O. Jeschke, M. Tomic, R. Valenti, B. Wolfram, M. Bröring, M. Bolte, D. Chercka, M. Baumgarten and K. Müllen, Phys. Chem. Chem. Phys., 2015, 17, 4118–4126 RSC.
- A. Morherr, S. Witt, A. Chernenkaya, J.-P. Bäcker, G. Schönhense, M. Bolte and C. Krellner, Phys. Rev. B, 2016, 496, 98–105 CrossRef CAS.
- P. Hu, K. Du, F. Wei, H. Jiang and C. Kloc, Cryst. Growth Des., 2016, 16, 3019–3027 CrossRef CAS.
- P. Hu, H. Li, Y. Li, H. Jiang and C. Kloc, CrystEngComm, 2017, 19, 618–624 RSC.
- O. Kataeva, M. Nohr, K. Ivshin, S. Hampel, B. Büchner and M. Knupfer, Cryst. Growth Des., 2021, 21, 471–481 CrossRef CAS.
- N. R. Goud and A. J. Matzger, Cryst. Growth Des., 2016, 17, 328–336 CrossRef.
- M. Meneghetti and C. Pecile, J. Chem. Phys., 1986, 84, 4149–4162 CrossRef CAS.
- R. Das, M. Linseis, S. M. Schupp, L. Schmidt-Mende and R. F. Winter, Chem.–Eur. J., 2022, 28, e202104403 CAS.
- M. Kuratsu, S. Suzuki, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Inorg. Chem., 2007, 46, 10153–10157 CrossRef CAS PubMed.
- R. Das, M. Linseis, L. Senft, I. Ivanović-Burmazović and R. F. Winter, Inorganics, 2022, 10, 82 CrossRef CAS.
- R. Das, M. Linseis, S. Scheerer, K. Zoller, L. Senft, I. Ivanović-Burmazović and R. F. Winter, Inorg. Chem., 2022, 61, 12662–12677 CrossRef CAS PubMed.
- G. R. Eaton, S. S. Eaton, D. Barr and R. T. Weber, Quantitative EPR: A Practitioner's Guide, Springer Verlag, Vienna, 1st edn, 2010 Search PubMed.
- C. Corvaja, Electron Paramagnetic Resonance - A Practitioner's Toolkit, Wiley VCH Verlag GmbH&Co, Hoboken, New Jersey, 2009 Search PubMed.
- A. Girlando and C. Pecile, J. Chem. Soc., Faraday Trans. 2, 1975, 71, 689–698 RSC.
- S. E. Boesch and R. A. Wheeler, J. Phys. Chem. A, 1997, 101, 8351–8359 CrossRef CAS.
- G. Balakrishnan, P. Mohandas and S. Umapathy, J. Phys. Chem. A, 2001, 105, 7778–7789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2220394, 2220397 and 2220398. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ra07322f |
| This journal is © The Royal Society of Chemistry 2023 |