
Multi-state photochromism of bis-tetraarylethene luminogens modulated through oligosilane linkages†
Chuanting
Zhou
a,
Zhikuan
Zhou
 *a,
Fuhuan
Yu
a,
Wei
Xie
a,
Wenjun
Zhang
b,
Qiaomei
Yang
b,
Xiaodong
Xu
b,
Lizhi
Gai
a and
Hua
Lu
*a
*a,
Fuhuan
Yu
a,
Wei
Xie
a,
Wenjun
Zhang
b,
Qiaomei
Yang
b,
Xiaodong
Xu
b,
Lizhi
Gai
a and
Hua
Lu
*a
aCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Key Laboratory of Organosilicon Material Technology, Zhejiang Province, Hangzhou Normal University, No. 2318, Yuhangtang Road, Hangzhou, 311121, P. R. China. E-mail: zkzhou@hznu.edu.cn; hualu@hznu.edu.cn
bBeili Technologies (Chongqing) Company Limited, 2 Huanan 8th Branch Road, Changshou District, Chongqing, P. R. China
First published on 24th November 2022
Abstract
Controllable photochromic molecules both in solutions and solid states (amorphous, films, and crystals) are valuable nascent optical materials. In this work, we report a facial route to design multi-state photochromic molecules by incorporating oligosilane chains into photoactive triphenylvinylthiophene (TPT) luminogens. The flexibility, bulkiness, and unique σ–π conjugation brought by silane chains enable different intramolecular interactions between two TPT units. A longer silicon chain bridge facilitates efficient electron delocalization and reduced structural constraint, thus resulting in reversible photochromism even in crystals with high-contrast color differences. This work provides a general method for developing novel multi-state photochromic materials for application in data encryption.
Introduction
Photochromism represents a reversible color change process of compounds upon photoirradiation with different wavelengths. This phenomenon mainly originates from the transformation between E/Z isomers, chemical bond rearrangement, and light-induced radical generation.1 Over the last few decades, various organic photochromic systems such as azobenzene, spiropyran, furylfulgide, and diarylethene have been designed and widely used as sensors and optical switches, and in data encryption.2 As the most studied family, diarylethene possesses many merits such as structural diversity, thermal stability of both isomers, rapid response, high sensitivity and fatigue resistance. Most diarylethenes exhibit active photochromism in solution, and only a few thienyl-based diarylethenes exhibit active photochromism based on the intramolecular cyclization reaction in the crystalline state.3 These conformational constraints were thought to be the reason for inactive photochromism in the solid state. Meanwhile, the solid-state photoluminescence (PL) of these diarylethenes is often strongly quenched, which impedes these intelligent materials from being used in practical solid surfaces.Aggregation-induced emission (AIE) luminogens offer fascinating tools to suppress the detrimental quenching effect in the solid state, and thus are suitable for fabricating multifunctional fluorescent materials.4 A general strategy to enhance the solid-state fluorescence of photochromic molecules is to integrate typical AIEgens covalently into them.5 Recently, another promising alternative approach has been proposed based on the rational modification of multiarylethene molecular skeletons. Considering tetraphenylethene (TPE) as an example, the replacement of one or more phenyl groups by a thienyl unit yields photochromic active tetraarylethene which exhibits photoinduced coloration and solid-state luminescence.6 Despite several excellent works, the development of compounds featuring multi-state photochromism with fast response and high contrast is still challenging.5a,7 Solid-state (especially the crystalline state) photochromism has extensive application values in practice.2c,3 Thus, exploiting multi-state photochromic systems based on versatile structure design is highly valuable.
Organosilicon compounds have attracted significant research attention in organic synthesis and materials science due to the unique features of silicon.8 In contrast to monosilanes, oligosilanes containing Si–Si bonds are of interest due to their characteristic σ-delocalization.9 Organic conjugated compounds that covalently combine a Si–Si σ bond with an aromatic ring give rise to σ–π conjugation, which results in enhanced dual state photoluminescence (PL),10 electron-transporting properties,11 and stimuli-responsive behavior.12 In our previous work, we employed oligosilane chains as linkages to design TPE-based AIEgens and found that they exhibit enhanced PL in solid states with multifunctional applications.13 We propose that introducing a bulky, flexible oligosilane chain into photochromic active tetraarylethene would enhance the PL intensity in the solid-state and control the photochromic behaviors conformationally and electronically. Herein, we designed a series of bis-triphenylvinylthiophene derivatives with oligosilane chains (Scheme 1, silicon number from one to four), and investigated their distinct reversible photochromic behaviours in multi-state situations. The color of the four compounds BTPTSin (n = 1–4, Scheme 1) changes from colorless to yellowish-brown in solution after UV irradiation (365 nm). This procedure is reversible upon visible light irradiation. In the powder form, BTPTSi1 shows weak photochromism, while BTPTSi2 is almost photochromic inactive. However, longer silicon chain bridged BTPTSi3 and BTPTSi4 exhibit fast and high-contrast color change upon UV irradiation in the powder form. It is noteworthy that BTPTSi4 exhibits reversible photochromism in the single crystal state. The structural analysis combined with theoretical calculations revealed the critical influence of molecular conformation on the photochromic activity in powder and crystalline forms. The use of these compounds in solid-state photoswitchable patterning was also demonstrated.
 | ||
| Scheme 1 The synthetic routes and chemical structures of BTPTSin (n = 1–4). | ||
Results and discussion
Synthesis and photophysical properties
Triphenylvinylthiophene (TPT) was selected as a photoactive unit since the non-substituted thiophene α position is easier to undergo photocyclization like the classic photochromic dithienylethene.2a It was prepared according to literature-reported procedures.6d In the target compounds BTPTSin (n = 1–4), oligosilane linkages were introduced through salt elimination reactions reported in our previous works.13b,14 Lithiation of TPT, followed by the reaction with dichlorodimethylsilane or dichlorotetramethyldisilane, yields BTPTSi1 and BTPESi2, respectively. Triflate end-capped trisilane and tetrasilane reacted with lithiated TPT to generate BTPTSi3 and BTPTSi4, respectively. All these oligosilane bridged compounds are white to light yellow crystalline solids. Their chemical structures were unambiguously elucidated by 1H and 13C NMR spectroscopy, high-resolution mass spectrometry, and single crystal X-ray diffraction.These oligosilane-bridged compounds exhibit similar absorption spectral profiles to that of TPT with maximum absorption at 315 nm (Fig. S1, ESI†), indicating their excellent optical transparency. They are not fluorescent in a good solvent such as THF. As shown in Fig. 1, BTPTSi1, BTPTSi2, and BTPTSi3 exhibit blue emissions with maxima at 446 nm, 462 nm, and 451 nm in powder forms, and the quantum yields (QYs) are 28%, 61%, and 7%, respectively. However, BTPTSi4 is almost non-fluorescent with a blue-shifted emission maximum at 424 nm and a QY as low as 1%. In the film state, BTPTSi2, BTPTSi3, and BTPTSi4 exhibit the same emission profile with maxima at 467–473 nm and QYs of 6%, 5%, and 6%, respectively. However, BTPTSi1 exhibited blue-shifted emission with a maximum centered at 433 nm and a QY of 11% (Fig. S1 and Table S1, ESI†). As for BTPTSi4, the loose intermolecular packing facilitates non-radiative decay and the powder sample is almost non-fluorescent. While in the film state, the intermolecular packing is close so that the sample emits stronger due to reduced non-radiative decay. Thus, the Si–Si bridges should significantly impact the intermolecular interaction in the solid state. All four compounds possess aggregation-induced emission in THF/water mixtures (Fig. S2, ESI†). Considering BTPTSi2 as an example, when the water fraction increased above 60%, enhanced emission intensity and bright bluish-green emission color were observed. Interestingly, when the water fraction was 90% and 95%, novel broad emission peaks emerged at around 465 nm, indicating a complicated aggregation process. The high energy emission peaks around 380 nm originated from the solution emission, while the lower energy emission peaks around 460 nm originated from the TPT aggregate emission.15
 | ||
| Fig. 1 (a) Normalized PL spectra of powder samples (λex = 320 nm). (b) PL spectra of BTPTSi2 in THF (1 × 10−4 M) with increasing water fractions from 0% to 95% (λex = 300 nm). | ||
Photophysical properties
The four compounds showed clear-cut photochromism in THF solution when irradiated by a hand-held UV lamp of 10 W (Fig. 2a–d and Fig. S3, ESI†). After UV irradiation, new absorption peaks for BTPTSin (n = 1–4) emerged with maxima of 475 nm, 477 nm, 479 nm, and 478 nm, which red-shifted 18–22 nm compared with TPT. Their colors changed from the initial colorless to yellowish-brown. The absorbance of these newly generated peaks increased gradually along with the irradiation time. The time of reaching the absorbance maxima is 90 s (BTPTSi1 and BTPTSi2) and 120 s (BTPTSi3 and BTPTSi4). These results indicate that short silicon chain bridged compounds are much more photoactive than the longer silicon chain bridged compounds in the solution state. | ||
| Fig. 2 Time-dependent UV-vis absorption spectra of (a) BTPTSi1, (b) BTPTSi2, (c) BTPTSi3 and (d) BTPTSi4 in THF (5 × 10−4 M), and time-dependent UV-vis absorption spectra of (e) BTPTSi1, (f) BTPTSi2, (g) BTPTSi3 and (h) BTPTSi4 in the solid state with different periods of UV-light irradiation. | ||
The photochromic behaviors were further examined in solid states (powder forms), and different coloration processes were observed. As shown in Fig. 2e–h and Fig. S3 (ESI†), after UV irradiation, the solids of BTPTSi1 and BTPTSi4 changed from the same pale yellow to light orange and deep purple, respectively. BTPTSi3 changed from white to light purple. However, disilane bridged BTPTSi2 showed an extremely weak color change when observed with the naked eye. A series of slightly raised absorption bands around 500 nm were detected on its UV-vis spectra. Novel absorption peaks at 480 nm, 520 nm, and 521 nm appeared on the time-dependent UV-vis spectra of solids BTPTSi1, BTPTSi3, and BTPTSi4, respectively. The absorption maxima of photochromic BTPTSi3 and BTPTSi4 red-shifted 14 nm and 15 nm compared with that of the TPT solid, indicating the efficient σ–π conjugation brought by the Si–Si bond linkage for electron delocalization. Notably, the irradiation time required for a complete color change for BTPTSi4 is 10 s, which is the shortest among those of all the compounds. These different color changes in solids can be attributed to the formation of different molecular aggregates modulated by the silane chains. This will be discussed in detail in the following crystal analysis section.
The photochromism of compounds in solids is accompanied by varying degrees of fluorescence quenching. As shown in Table S1 and Fig. S4 (ESI†), after 2 min of UV irradiation, the QYs of BTPTSi1, BTPTSi2 and BTPTSi3 descended from 28%, 61%, and 7% to 18%, 53%, and 2%, respectively. The QYs of BTPTSi4 before and after UV irradiation is 1%, indicating its weak emission character. These results demonstrated that photochromism and fluorescence are competitive processes, while the introduction of a silane chain would regulate these two processes and achieve better balance.
Wide-angle X-ray diffraction and single crystal analysis
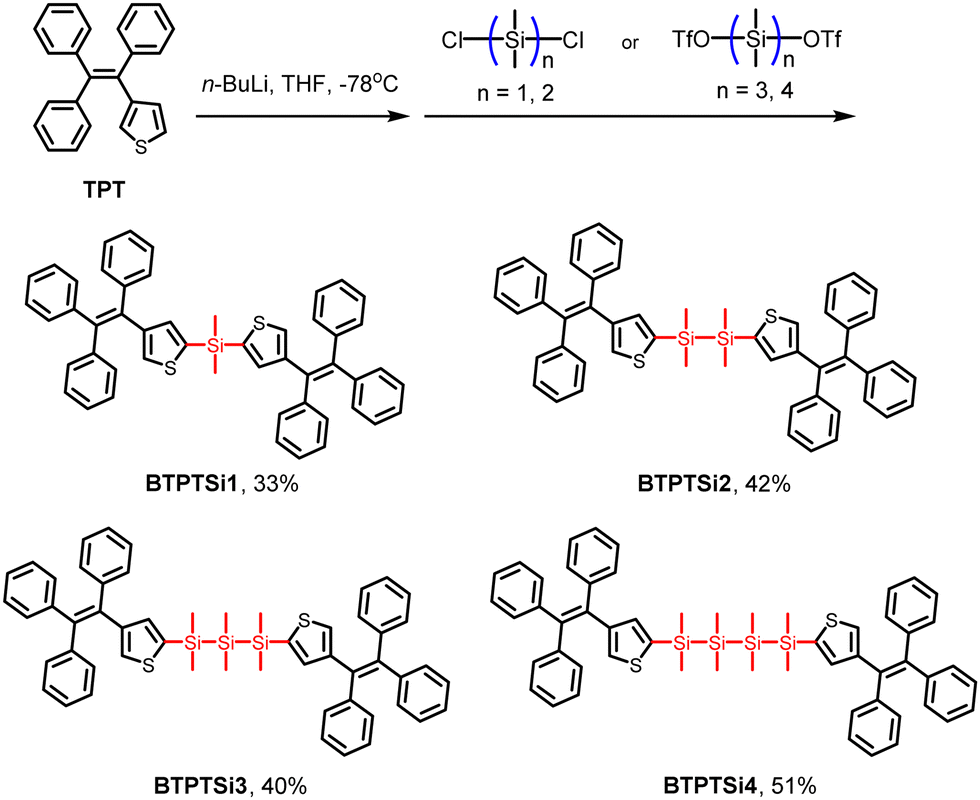
To gain deep insights into the mechanism of solid-state photochromism, we tried to cultivate single crystals. To our surprise, during the growth of single crystals of BTPTSi3, two different kinds of crystals were found showing distinct photochromic behaviors. As shown in Fig. 3a, the transparent colorless single crystal shows no color change upon UV irradiation, but exhibit strong blue emission. However, the adjacent polycrystals change from colorless to red when irradiated with UV light. This phenomenon can be well repeated when a large amount of samples are used. As shown in Fig. 3b, the mixture of polycrystals and powder samples exhibit clear-cut photochromism, while that of the single crystal sample does not. Upon UV irradiation, the QY of the powder sample decreased drastically from 7% to 2%, while the single crystals remained unchanged with a QY of 59%.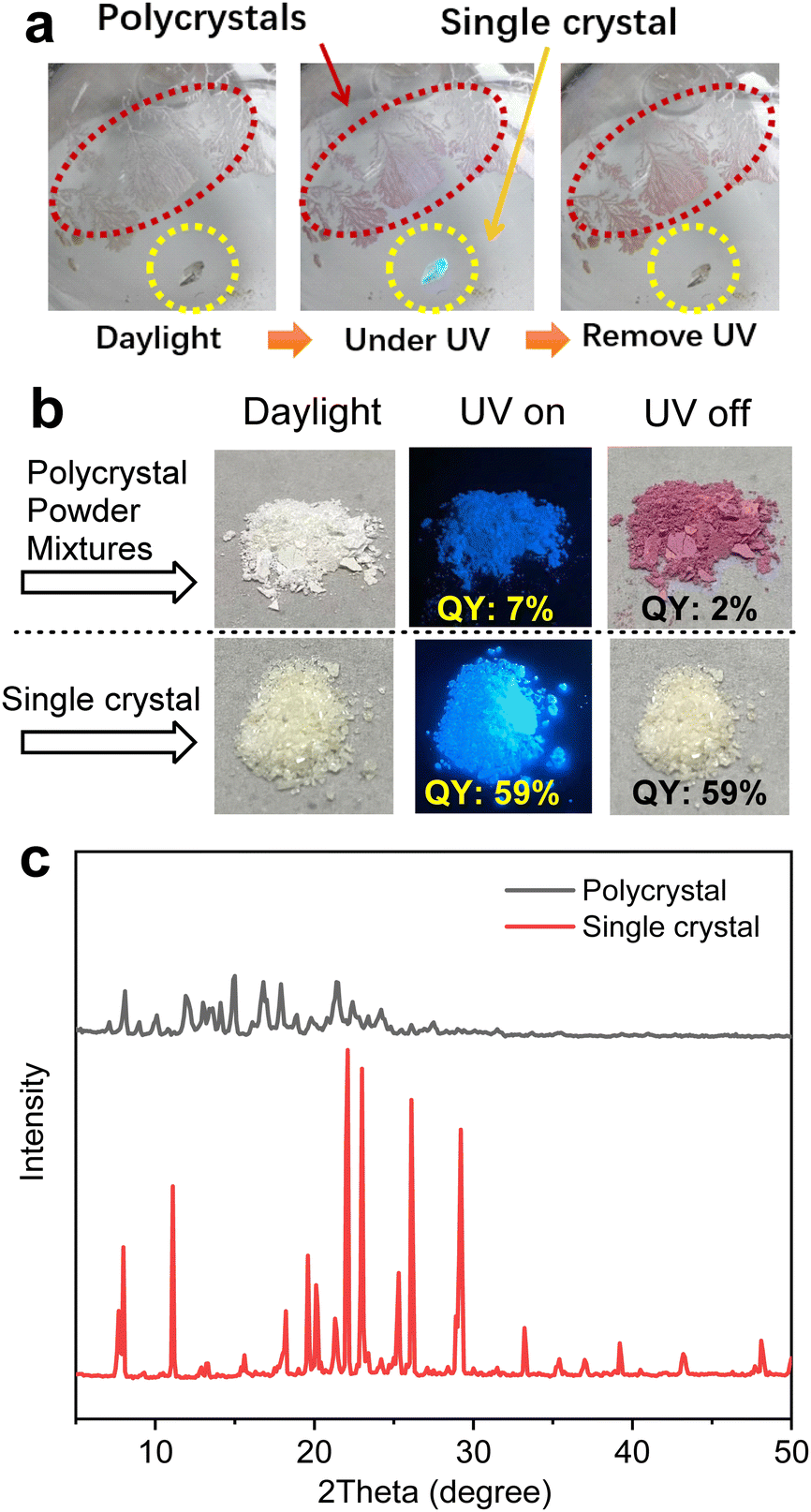 | ||
| Fig. 3 (a and b) Photographs of BTPTSi3 polycrystals and single crystals upon UV irradiation. (c) XRD patterns of BTPTSi3 in polycrystal and single crystal states. | ||
Wide-angle X-ray diffraction was employed to examine the structural difference between powder and single crystal samples (Fig. 3c). The single crystals of BTPTSi3 showed multiple sharp scattering peaks with strong intensity, evidencing their high crystallinity. However, the peaks of the powder sample weakened and the full width at half maxima (FWHM) broadened, suggesting the disordered molecular geometry. We can conclude that in the single crystalline state, molecular conformation is highly constrained, while the powder form is usually prone to possess more diverse molecular conformation.
Single crystals of BTPTSi1, BTPTSi2, and BTPTSi4 were obtained by the slow diffusion of n-hexane into their CH2Cl2 solutions. In contrast, those of BTPTSi3 were obtained by the slow evaporation of its CH2Cl2 solutions at room temperature. Their structures are shown in Fig. 4. Detailed crystallographic data are summarized in Tables S2–S5 (ESI†). The single crystal of BTPTSi1 belongs to monoclinic with the space group C2/c, while that of the other three compounds belong to triclinic with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The silane unit in BTPTSi1 adopts typical tetrahedron conformation. Disilane and tetrasilane bridged BTPTSi2 and BTPTSi4 adopt fully extended, all-trans structures, while trisilane bridged BTPTSi3 adopts a folded conformation. Generally, photochromic reactions rarely occur in crystals because a large geometrical structure change is prohibited.2a To our surprise, BTPTSi4 is the only compound that exhibits reversible photochromism in the single crystalline state (Fig. 4e). This phenomenon indicates that the aryl groups of TPT units in the four compounds adopt different conformations. Phenyl and thienyl groups in the cis position of ethene constitute the photocyclization unit, which have two possible conformers, A and B (Fig. 4e). In single crystals of BTPTSi1, BTPTSi2, and BTPTSi3, phenyl and thienyl groups adopt conformer A, in which photoinactive thiophene β carbon is close to the phenyl group. The distances between unsubstituted thiophene α carbon and photoactive phenyl carbon of TPT units in BTPTSi1 and BTPTSi2 are 4.298 Å and 4.000 Å, while those in BTPTSi3 have different values of 4.115 Å and 4.193 Å. These distances are so large that the photocyclization reaction does not take place in single crystals.2a In contrast, TPT units in BTPTSi4 adopt conformer B, in which the photoactive carbons are close to each other with a distance of 3.262 Å. This distance is favorable for intramolecular cyclization, and the crystals of BTPTSi4 change from pale yellow to dark red under UV irradiation (Fig. 4e).
. The silane unit in BTPTSi1 adopts typical tetrahedron conformation. Disilane and tetrasilane bridged BTPTSi2 and BTPTSi4 adopt fully extended, all-trans structures, while trisilane bridged BTPTSi3 adopts a folded conformation. Generally, photochromic reactions rarely occur in crystals because a large geometrical structure change is prohibited.2a To our surprise, BTPTSi4 is the only compound that exhibits reversible photochromism in the single crystalline state (Fig. 4e). This phenomenon indicates that the aryl groups of TPT units in the four compounds adopt different conformations. Phenyl and thienyl groups in the cis position of ethene constitute the photocyclization unit, which have two possible conformers, A and B (Fig. 4e). In single crystals of BTPTSi1, BTPTSi2, and BTPTSi3, phenyl and thienyl groups adopt conformer A, in which photoinactive thiophene β carbon is close to the phenyl group. The distances between unsubstituted thiophene α carbon and photoactive phenyl carbon of TPT units in BTPTSi1 and BTPTSi2 are 4.298 Å and 4.000 Å, while those in BTPTSi3 have different values of 4.115 Å and 4.193 Å. These distances are so large that the photocyclization reaction does not take place in single crystals.2a In contrast, TPT units in BTPTSi4 adopt conformer B, in which the photoactive carbons are close to each other with a distance of 3.262 Å. This distance is favorable for intramolecular cyclization, and the crystals of BTPTSi4 change from pale yellow to dark red under UV irradiation (Fig. 4e).
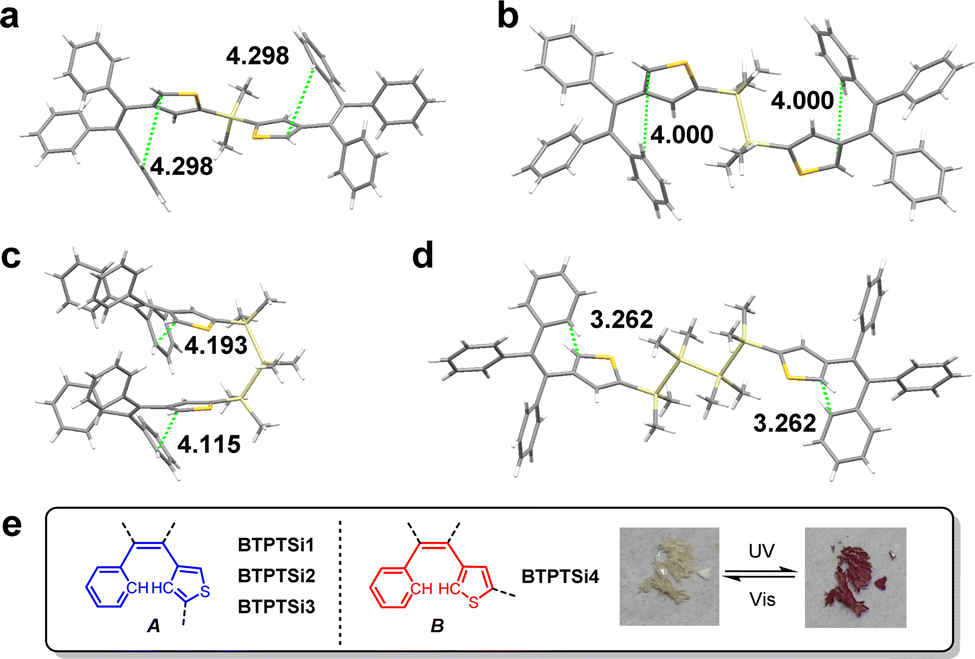 | ||
| Fig. 4 Single crystal structures of (a) BTPTSi1, (b) BTPTSi2, (c) BTPTSi3, and (d) BTPTSi4. (e) Structures of two different photoinactive conformer A and photoactive conformer B in the four crystals. The inset shows the photographs of the coloration of BTPTSi4 single crystals. CCDC No. for BTPTSi1: 2204233, BTPTSi2: 2204234, BTPTSi3: 2204235, and BTPTSi4: 2204236. | ||
It has been reported that orthogonal torsion angles between the Si–Si σ-bond axis and the terminal aryl ring plane favor the most effective σ–π conjugation.16 In the crystals of BTPTSi2, BTPTSi3, and BTPTSi4, the torsion angles are 55.25°, 64.48°, and 72.22°, suggesting that the σ–π conjugation in BTPTSi4 is more effective. This result is consistent with the experimental data that BTPTSi4 exhibited the most redshifted absorption in the solid state after photocyclization. Time-dependent density functional theory (TD-DFT) calculations were performed on the TPT and BTPTSin (n = 1–4). All the HOMOs and LUMOs are mainly located at TPT units, while in BTPTSi2 and BTPTSi4, part of HOMOs spread on the Si–Si σ bond, indicating the existence of σ–π conjugation (Fig. S5 and S6, ESI†).
Mechanistic investigation
As shown in Fig. 5a, conformer B is undoubtedly photoactive according to the reported paper,17 which undergoes chemical bond rearrangement upon UV irradiation to yield isomer C. The changes in the geometrical as well as electronic structure are the cause of color change. C may undergo photodegradation and a dehydrogenation reaction upon prolonged UV irradiation time. We examined the cyclization–dehydrogenation reaction of BTPTSi1 as an example. As shown in Fig. S7 (ESI†), before UV irradiation, the signals of four thienyl H atoms appear at 6.97 ppm and 6.59 ppm on the 1H NMR spectrum of BTPTSi1 in CDCl3. After 365 nm light irradiation for 10 s, the solution turned brownish yellow, which is reversible under visible light irradiation (Fig. 5b). After UV irradiation, the photocyclization product and a small amount of impurities formed. In Fig. S8 (ESI†), on magnifying the aromatic region and high-field region, several new signals were clearly seen. Hydrogens a and b (Fig. 5a, structure C) appeared at 2.50 ppm and 3.15 ppm. Hydrogens c–f appeared at 6.74 ppm and 7.43–8.28 ppm. From the integral we can estimate that the ratio of cyclization product C is less than 5%, indicating the low conversion ratio of the photocyclization reaction in solution. As a result, in the IR spectra of compounds before and after UV irradiation, we cannot find obvious differences (Fig. S9 in the ESI†). After UV light irradiation for 5 h in air using oxygen as an oxidant, the yellowish solution turned colorless. We tried to isolate the final dehydrogenation product D, and the yield was less than 1% since most of the compounds in solution decomposed under long-time UV irradiation. As can be seen in the 1H NMR spectrum (Fig. S10 in the ESI†), the original two groups of thienyl H signals disappeared, and no methine H signals were observed. Several new signals emerged in the range of 7.3–8.3 ppm. The molecular ion peak missing four H was found in the MALDI-TOF-MS spectrum, which further proved the formation of the cyclization–dehydrogenation product (Fig. S11 in the ESI†). Due to the extremely low conversion ratio from C to D, the solution exhibited light yellow color, and it cannot turn back to colorless upon visible light irradiation.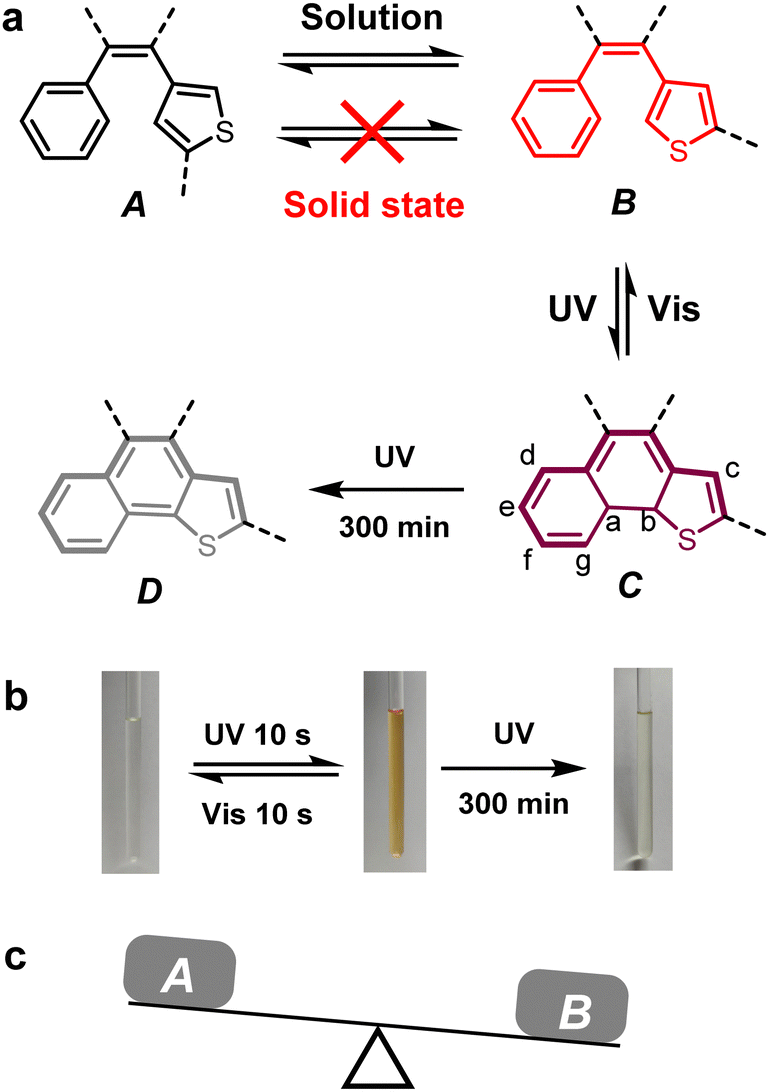 | ||
| Fig. 5 (a) Conformation change of the photoactive unit under different conditions. (b) Color change of the BTPTSi1 solution (CDCl3) under irradiation. (c) Schematic representation of the balance between conformers A and B. | ||
In solution and solid-state samples, photoinactive conformer A and photoactive conformer B may exist simultaneously.18 The ratio of these two isomers determines whether the photocyclization reaction will happen in the bulk sample. In the solution state, the thienyl group can rotate freely along the C–C single bond connected to the central ethene. It is easy to obtain conformation B and make it cyclized by UV irradiation. Thus, all the four compounds exhibit photochromism in THF solution with almost the same color change procedure, which originated from the ring-close reaction of TPT units. In single crystals, it is evident that BTPTSi1, BTPTSi2, and BTPTSi3 molecules uniformly adopt A conformation, while BTPTSi4 adopts B conformation (Fig. 4a and d). As a result, BTPTSi4 is the only compound that exhibits photochromism in the single crystal state. In amorphous and polycrystalline states, the molecular conformations become complicated. From the single crystal structures of BTPTSi1 and BTPTSi3, we can conclude that the intramolecular interactions in these molecules are weak. Both A and B conformations probably coexist in amorphous and polycrystalline states, and these two compounds exhibit photochromism. However, the single crystal structure of BTPTSi2 revealed relatively strong intramolecular C–H⋯π interactions, and thus conformer A will dominate in the solid state. As a result, we observed the weakest solid-state color change for compound BTPTSi2 after UV irradiation.
Photoswitchable application
The introduction of oligosilane chains into the bis-TPT molecule enables the modulation of molecules with reversible photochromism and solid-state luminescence. BTPTSi3 was chosen to demonstrate its application in photoswitchable patterning for information storage medium on a filter paper. The BTPTSi3 molecules were homogenously loaded on a filter paper by immersing the filter paper into BTPTSi3 solution. As shown in Fig. 6, the resulting paper is in white color similar to its original state. It emits sky blue color after instant UV irradiation. In the control experiment, when removing the UV light, red color appeared. After visible light irradiation, the red paper turns back to white entirely. The coloration and decoloration processes can be reversible at least for 100 times. In the photo-controlled patterning experiment, the filter paper is covered by a steel mask carved with a symbol “Si”. On irradiating the mask by UV light for 10 s and removing the mask, the “Si” symbol can be clearly visualized with quenched fluorescence, while the margin area emits sky blue color. After turning off the UV light, the letter becomes red and the margin area is slightly red in daylight. The figure of the Si symbol disappears entirely after irradiation of visible light, and the filter paper recovers the white color. These photoswitchable patterning cycles demonstrate the utilization of organosilicon compounds in solid-state information storage with dual signals of fluorescence and coloration. | ||
| Fig. 6 Photo-controlled patterning application of BTPTSi3 on filter paper. Top: Writing “Si” with a mask. Bottom: Control experiment. | ||
Conclusions
In summary, four bis-TPT derivatives (BTPTSin, n = 1–4) are designed and synthesized to investigate their multi-state photochromic properties. All the compounds exhibit reversible photochromism in solution. BTPTSi1 with monosilane as the bridge unit exhibits weak photochromism in powder form. Disilane bridged BTPTSi2 showed almost no color change upon UV irradiation in powder form. In contrast, longer silicon chain bridged BTPTSi3 and BTPTSi4 exhibit solid-state photochromism with a short response time, high-contrast coloration, and excellent fatigue resistance. Furthermore, BTPTSi4 exhibits photochromism in the single crystal state. According to the crystallographic data analysis, the intramolecular interaction of the photoactive unit is modulated by the conformation of oligosilane chains. Longer silane chains impart structural flexibility to bis-TPT and enables photochromism in multi-state forms. These compounds can be utilized as rewritable solid-state optical recording media. This work offers a general strategy for fabricating novel solid-state emissive molecules with multi-state photochromic behaviors, which is of great value for the development of multifunctional photochromic materials.Experimental section
Materials and instruments
All reagents and solvents were purchased from commercial suppliers and used without further purification unless otherwise indicated. All air and moisture-sensitive reactions were carried out under an argon atmosphere in glassware that was dried in an oven at 120 °C and cooled under a stream of inert gas before use. Tetrahydrofuran was refluxed with sodium and distilled out immediately before use. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX400 spectrometer and referenced to the residual proton signals of deuterated solvents (CDCl3: 7.26 ppm). HR-MS data were recorded on a Bruker Daltonics microTOF-Q II spectrometer. All solvents used for the spectroscopic measurements were of UV spectroscopic grade (Aldrich). Diffraction data were collected on a Bruker Smart Apex II CCD diffractometer or Bruker AXS Apex II diffractometer with graphite-monochromated Mo-Kα (λ = 0.71073 Å).Synthesis and characterization
Synthesis of BTPTSi1. To a solution of 3-(1,2,2-triphenylvinyl)thiophene (1.69 g, 5.0 mmol) in THF (80 mL) was added n-butyllithium (7.5 mmol, 4.7 mL, 1.6 M solution in hexane) dropwise for over 20 min at −78 °C. Upon completion of addition, the reaction was stirred for 3 h. To the suspension in THF was added dichlorodimethylsilane (Cl–Si–Cl) (1.1 mL, 2.5 mmol, dilute with 20 mL THF) dropwise for over 30 min at −78 °C. Upon completion of addition, the reaction mixture was allowed to warm to room temperature and stirred for 6 h. The reaction mixture was evaporated, extracted by dichloromethane, and dried over Na2SO4. The residue was subjected to silica gel column chromatography to obtain 0.63 g (33% yield) of BTPTSi1 as white solids.1H NMR (400 MHz, CDCl3) δ 7.13 (dd, J = 12.9, 3.8 Hz, 16H), 7.08 (d, J = 6.9 Hz, 10H), 7.03–6.98 (m, 4H), 6.97 (s, 2H), 6.59 (s, 2H), 0.25 (s, 6H).
13C NMR (101 MHz, CDCl3) δ 145.49 (s), 143.83 (s), 143.28 (s), 143.22 (s), 140.24 (s), 138.32 (s), 135.18 (s), 135.14 (s), 131.90 (d, J = 5.7 Hz), 131.04 (s), 130.76 (s), 130.67 (d, J = 3.7 Hz), 127.75 (s), 127.45 (s), 127.41 (s), 126.57 (s), 126.39 (s), 126.11 (s), −0.88 (s).
HRMS-ESI: m/z: calcd [C50H40S2Si + Na]+ 755.2239, found 755.2215.
Compounds BTPTSi2, BTPTSi3, and BTPTSi4 were obtained as white solids by using a similar procedure to that of BTPTSi1 in yields of 42%, 40%, and 51%, respectively. The silane starting materials used in these reactions are 1,2-dichloro-1,1,2,2-tetramethyldisilane (Cl–Si2–Cl) and silyl triflates.
BTPTSi2: 1H NMR (400 MHz, CDCl3) δ 7.11 (t, J = 9.6 Hz, 26H), 7.01–6.95 (m, 4H), 6.95 (s, 2H), 6.56 (s, 2H), 0.11 (s, 12H).
13C NMR (101 MHz, CDCl3) δ 145.88 (s), 144.25 (s), 143.67 (s), 140.49 (s), 137.66 (s), 136.20 (s), 135.58 (s), 131.78 (s), 131.35 (d, J = 5.7 Hz), 130.99 (s), 128.09 (s), 127.72 (d, J = 3.7 Hz), 126.85 (s), 126.64 (s), 126.39 (s), −2.88 (s).
HRMS-ESI: m/z: calcd [C52H46S2Si2 + Na]+ 813.2477, found 813.2483.
BTPTSi3: 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 5.8 Hz, 6H), 7.12–7.04 (m, 20H), 7.01–6.96 (m, 4H), 6.94 (s, 2H), 6.56 (s, 2H), 0.14 (s, 12H), −0.09 (s, 6H).
13C NMR (101 MHz, CDCl3) δ 145.63 (s), 144.11 (s), 143.51 (d, J = 2.6 Hz), 140.28 (s), 137.15 (d, J = 6.9 Hz), 135.44 (s), 131.44 (s), 131.18 (d, J = 5.3 Hz), 130.81 (s), 127.95 (s), 127.55 (d, J = 3.8 Hz), 126.57 (d, J = 19.7 Hz), 126.21 (s), −2.08 (s), −6.81 (s).
HRMS-ESI: m/z: calcd [C54H52S2Si3 + Na]+ 871.2716, found 871.2725.
BTPTSi4: 1H NMR (400 MHz, CDCl3) δ 7.16 (t, J = 5.0 Hz, 6H), 7.12–7.05 (m, 20H), 6.99 (dd, J = 7.2, 2.4 Hz, 4H), 6.95 (s, 2H), 6.57 (s, 2H), 0.21 (s, 12H), −0.04 (s, 12H).
13C NMR (101 MHz, CDCl3) δ 145.61 (s), 144.12 (s), 143.51 (d, J = 3.2 Hz), 140.26 (s), 137.23 (d, J = 11.6 Hz), 135.44 (s), 131.41 (s), 131.18 (d, J = 4.4 Hz), 130.80 (s), 127.95 (s), 127.54 (d, J = 3.5 Hz), 126.56 (d, J = 19.7 Hz), 126.20 (s), −1.87 (s), −5.79 (s).
HRMS-ESI: m/z: calcd [C56H58S2Si4 + Na]+ 929.2955, found 929.2966.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grant awarded from National Natural Science Foundation of China (Fund No. 21871072) to HL, and from Hangzhou leading innovation and entrepreneurship team project (Fund No. TD2020015) to ZZ.Notes and references
- (a) Y. Xie, Z. Wang, D. Wang, Y. Zhou, Y. Lei, W. Gao, M. Liu, X. Huang and H. Wu, Mater. Chem. Front., 2021, 5, 3413–3421 RSC; (b) L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044–1097 RSC.
- (a) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed; (b) X. Yu, H. Zhang and J. Yu, Aggregate, 2021, 2, 20–34 CrossRef; (c) C. Huang, R. Huang, S. Zhang, H. Sun, H. Wang, B. Du, Y. Xiao, T. Yu and W. Huang, Research, 2021, 2021, 9816535 CAS.
- K. Uchida, R. Nishimura, E. Hatano, H. Mayama and S. Yokojima, Chem. – Eur. J., 2018, 24, 8491–8506 CrossRef CAS PubMed.
- (a) L. Wang, L. Wang, X. Yang, W. Li, L. Chen, J. Tang, W. Cong, R. Hu, M. Tebyetekerwa and B. Tang, Prog. Org. Coat., 2022, 163, 106633 CrossRef CAS; (b) L. Wang, L. Wang, J. Wu, L. Wang, W. Cong, X. Wang, R. Hu, W. Li, M. Tebyetekerwa and B. Z. Tang, Prog. Org. Coat., 2021, 159, 106448 CrossRef CAS; (c) W. Li, Y. Ding, M. Tebyetekerwa, Y. Xie, L. Wang, H. Li, R. Hu, Z. Wang, A. Qin and B. Z. Tang, Mater. Chem. Front., 2019, 3, 2491–2498 RSC; (d) W. Yao, M. Tebyetekerwa, X. Bian, W. Li, S. Yang, M. Zhu, R. Hu, Z. Wang, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2018, 6, 12849–12857 RSC.
- (a) G. Huang, Q. Xia, W. Huang, J. Tian, Z. He, B. S. Li and B. Z. Tang, Angew. Chem., Int. Ed., 2019, 58, 17814–17819 CrossRef CAS PubMed; (b) Y. Yang, J. Yang, M. Fang and Z. Li, Chem. Res. Chin. Univ., 2021, 37, 598–614 CrossRef CAS.
- (a) S. Zhou, S. Guo, W. Liu, R. Ding, H. Sun, J. Chen, Z. Qian and H. Feng, J. Mater. Chem. C, 2021, 9, 8249–8257 RSC; (b) H. Zhang, W. Li, Q. Huang, G. Huang, Z. Chi and B. S. Li, Dyes Pigm., 2021, 187, 109128 CrossRef CAS; (c) H.-X. Yu, J. Zhi and J.-L. Wang, J. Mater. Chem. C, 2021, 9, 3882–3891 RSC; (d) S. Zhou, S. Guo, W. Liu, Q. Yang, H. Sun, R. Ding, Z. Qian and H. Feng, J. Mater. Chem. C, 2020, 8, 13197–13204 RSC; (e) S. Guo, S. Zhou, J. Chen, P. Guo, R. Ding, H. Sun, H. Feng and Z. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 42410–42419 CrossRef CAS PubMed.
- (a) X. Zhang, Z. Ma, X. Li, C. Qian, Y. Liu, S. Wang, X. Jia and Z. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 40986–40994 CrossRef PubMed; (b) F. Khan, A. Ekbote, G. Singh and R. Misra, J. Mater. Chem. C, 2022, 10, 5024–5064 RSC; (c) F. Khan, A. Ekbote and R. Misra, New J. Chem., 2019, 43, 16156–16163 RSC.
- (a) F. Ye, Z. Xu and L.-W. Xu, Acc. Chem. Res., 2021, 54, 452–470 CrossRef CAS PubMed; (b) S. Feng, Z. Qu, Z. Zhou, J. Chen, L. Gai and H. Lu, Chem. Commun., 2021, 57, 11689–11692 RSC; (c) R.-H. Tang, Z. Xu, Y.-X. Nie, X.-Q. Xiao, K.-F. Yang, J.-L. Xie, B. Guo, G.-W. Yin, X.-M. Yang and L.-W. Xu, iScience, 2020, 23 Search PubMed; (d) D. Sun, Z. Ren, M. R. Bryce and S. Yan, J. Mater. Chem. C, 2015, 3, 9496–9508 RSC.
- T. Karatsu, J. Photochem. Photobiol., C, 2008, 9, 111–137 CrossRef CAS.
- (a) T. Usuki, M. Shimada, Y. Yamanoi, T. Ohto, H. Tada, H. Kasai, E. Nishibori and H. Nishihara, ACS Appl. Mater. Interfaces, 2018, 10, 12164–12172 CrossRef CAS PubMed; (b) X. Zheng, W. Du, L. Gai, X. Xiao, Z. Li, L. Xu, Y. Tian, M. Kira and H. Lu, Chem. Commun., 2018, 54, 8834–8837 RSC; (c) S. Wang, H. Lu, Y. Wu, X. Xiao, Z. Li, M. Kira and Z. Shen, Chem. – Asian J., 2017, 12, 561–567 CrossRef CAS PubMed.
- (a) J. Zhou, C. P. Folster, S. K. Surampudi, D. Jimenez, R. S. Klausen and A. E. Bragg, Dalton Trans., 2017, 46, 8716–8726 RSC; (b) T. A. Su, H. Li, R. S. Klausen, N. T. Kim, M. Neupane, J. L. Leighton, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Acc. Chem. Res., 2017, 50, 1088–1095 CrossRef CAS PubMed.
- H. Miyabe, M. Ujita, M. Nishio, T. Nakae, T. Usuki, M. Ikeya, C. Nishimoto, S. Ito, M. Hattori, S. Takeya, S. Hayashi, D. Saito, M. Kato, H. Nishihara, T. Yamada and Y. Yamanoi, J. Org. Chem., 2022, 87, 8928–8938 CrossRef CAS PubMed.
- (a) H. Feng, Z. Zhou, A. K. May, J. Chen, J. Mack, T. Nyokong, L. Gai and H. Lu, J. Mater. Chem. C, 2021, 9, 6470–6476 RSC; (b) X. Xiang, Z. Zhou, H. Feng, S. Feng, L. Gai, H. Lu and Z. Guo, CCS Chem., 2020, 2, 329–336 CrossRef CAS.
- S. Feng, Z. Zhou, X. Xiang, H. Feng, Z. Qu and H. Lu, ACS Omega, 2020, 5, 19181–19186 CrossRef CAS PubMed.
- T. Tao, Y. Gan, Y. Zhao, J. Yu, Q. Huang, Z. Yang, M. Chen and W. Huang, J. Mater. Chem. C, 2019, 7, 3765–3771 RSC.
- M. Kira, T. Miyazawa, H. Sugiyama, M. Yamaguchi and H. Sakurai, J. Am. Chem. Soc., 1993, 115, 3116–3124 CrossRef CAS.
- A. V. Yadykov, A. G. Lvov, M. M. Krayushkin, A. V. Zakharov and V. Z. Shirinian, J. Org. Chem., 2021, 86, 10023–10031 CrossRef CAS PubMed.
- C. V. Raman, Nature, 1922, 109, 138–139 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2204233–2204236. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2tc04095f |
| This journal is © The Royal Society of Chemistry 2022 |