
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing chain alignment and preserving the pristine structure of single-ether based PBTTT helps improve thermoelectric properties in sequentially doped thin films†
Huiyan
Zeng
a,
Pablo
Durand
b,
Shubhradip
Guchait
a,
Laurent
Herrmann
a,
Céline
Kiefer
c,
Nicolas
Leclerc
 *b and
Martin
Brinkmann
*a
*b and
Martin
Brinkmann
*a
aUniversité de Strasbourg, CNRS, ICS UPR 22, F-67000 Strasbourg, France. E-mail: martin.brinkmann@ics-cnrs.unistra.fr
bUniversité de Strasbourg, CNRS, ICPEES UMR 7515, F-67087 Strasbourg, France. E-mail: leclercn@unistra.fr
cUniversité de Strasbourg, CNRS, IPCMS UMR 7504, F-67087 Strasbourg, France
First published on 28th September 2022
Abstract
Doped polymer semiconductors are of central interest in material science for their interesting charge transport and thermoelectric properties. Polymer alignment and crystallization are two means to enhance thermoelectric parameters (charge conductivity σ and Seebeck coefficient S) in the polymer chain direction. In this study, we focus on the thermoelectric properties of a PBTTT polymer semiconductor bearing linear heptyl-oxy-butyl (C7OC4) side chains (PBTTT-8O) in thin oriented films aligned by high-temperature rubbing. Both the degree of in-plane orientation and the preferential contact plane of the polymer are determined by the rubbing temperature that can be tuned in a large 25–240 °C range. Optimal TE properties with high charge conductivities in the 2–5 × 104 S cm−1 range and power factors >2.0 mW m−1 K−2 are obtained in the chain direction only when the film orientation is optimized (dichroic ratio >20). Contrary to other dopants such as FeCl3, the structure of PBTTT-8O is little modified by the inclusion of F6TCNNQ dopant molecules in the layers of disordered side chains. UV-vis-NIR spectroscopy shows that dimer formation of the radical anion F6TCNNQ˙− explains this behavior. Overall, this study demonstrates that the highest thermoelectric performances for a given polymer/dopant system can only be uncovered when the optimal conditions for polymer semiconductor growth/alignment and sequential doping are found. In addition, the S–σ scaling laws along and perpendicular to the chain direction are (i) independent of the chemical nature of side chains and dopants and (ii) essentially determined by the level of chain alignment and molecular packing of PBTTT backbones.
I. Introduction
Polymer semiconductors (PSCs) are highly versatile materials widely used for the design of numerous electronic devices such as Organic Field Effect Transistors (OFETs), Organic Light Emitting Diodes (OLEDs) and organic solar cells.1 More recently, remarkable thermoelectric properties were evidenced for conducting polymers, doped PSCs and hybrid materials.2–5 Thermoelectric materials are of interest as they have the ability to convert a temperature gradient into an electric voltage thanks to the Seebeck effect. To be effective, a thermoelectric material must have a high charge conductivity σ and a high Seebeck coefficient S as well as a moderate thermal conductivity κ. Polymeric TE materials have multiple advantages related to their flexibility, ease of processing and versatility thanks to the large palette of molecular structures accessible via adequate chemical engineering.4,5 Intrinsically, PSCs show a low thermal conductivity that is only moderately increased upon doping and/or alignment.6 In order to become conductive and efficient for TE applications, polymer semiconductors must be doped in a controlled manner.7 This is why doping PSCs is a central step in the fabrication of TE materials in order to tune the charge density and hence, charge conductivity and Seebeck coefficient. The precise localization of dopants in the polymer is pivotal since it determines the magnitude of Coulombic interactions between the charge carriers on the polymer backbones (polarons and bipolarons) and the ionized dopant molecules hosted in the PSC crystals as well as the polymer packing order.8–10 From that perspective, sequential doping of PSCs proved to be a very effective processing strategy towards conducting polymers of high conductivity since it allows a high crystallinity of the pristine polymer films to be preserved.11–16 It has also been shown that introduction of short ethylene glycol side chains can be an effective means to enhance the TE properties of the PSC without modifying the packing of its backbone.17 Recently, it was reported that the use of PSCs bearing alkyl side chains comprising a single ether group helped reach very high conductivities of up to 5 × 104 S cm−1 and TE power factors of 2.9 mW m−1 K−2 in aligned thin films.18Control of chain orientation and crystallinity allows further enhancement of TE properties as both thermopower S and charge conductivity σ can be enhanced in the direction of chain alignment.19–26 Dopant intercalation in the host polymer impacts the structure of the doped polymer and can induce disorder that is detrimental in some cases for charge transport properties. The dopant position and the way it is intercalated in the crystals of the polymer semiconductors depend on the dopant dimensions and the length of alkyl side chains.20–24 Yet, no work has focused on the role of the dominant contact plane of the crystals on TE performances. For most PSCs, the processing conditions (temperature, solvent, film preparation method) determine the dominant contact planes of the crystals on a substrate.26–30 For P3HT and PBTTT, different crystal orientations on a substrate are observed depending on the type of solvent used, the temperature of an annealing step or the molecular weight of the polymer. In this work, we have studied the way the growth conditions (rubbing temperature TR) of oriented PSC thin films affect the resulting anisotropic TE properties in the doped thin films. Indeed, for polymer semiconductors such as P3HT or PBTTT, it has been shown that the temperature at which a thin film is oriented by rubbing impacts its orientation and structure.31,32 In particular, for P3HT, the size of crystalline lamellae and the total crystallinity are controlled by TR and determine the obtained TE properties in films doped with F4TCNQ.
In this study, we focus on the previously published PBTTT-8O bearing n-C7–O–C4 side chains and we used 1,3,4,5,7,8-hexafluoro-tetracyanonaphtho-quinodimethane (F6TCNNQ) as the dopant.16 Interestingly, the use of single-ether side chains makes it possible to align the PBTTT polymer in a large temperature range (100–240 °C), and thus to prepare aligned films with different contact planes of the crystals on the substrate. Sequential doping with F6TCNNQ was used and in particular the increasing concentration doping (ICD) method was implemented. In this method, for a given final doping level, the sample is progressively doped at multiple and increasing concentrations of dopants in an orthogonal solvent (acetonitrile).33 In this way, the dopant molecules are introduced progressively in the crystal lattice of the polymer, reducing structural damages by fast and uncontrolled intercalation. When oriented at 170 °C, thin films of PBTTT-8O, doped by ICD, can show remarkable TE properties with charge conductivities that can reach 2–5 × 104 S cm−1 and TE PF of 2.9 mW m−1 K−2. The enhanced TE performances have been attributed to (i) the higher in-plane orientation achieved over PBTTT with linear side chains such as PBTTT-C12 and (ii) to the random orientation of intercalated F6TCNNQ dopants in the disordered layers of n-C7–O–C4 side chains that help screen more effectively the polaron-anion Coulombic interactions and help thus achieve larger charge carrier mobilities.
In this contribution, we propose to follow the influence of the alignment conditions on the TE properties in PBTTT-8O films doped with F6TCNNQ. F6TCNNQ has been chosen as it is as rather stable conjugated dopant with and electron affinity EA = 5.37 eV that can readily oxidize PBTTT-8O and leads to rather high stability of the doped system under inert atmosphere.34 In particular, we focus on the impact of the rubbing temperature TR on the orientation and contact plane of the PBTTT-8O crystals and how these two parameters determine the TE properties of the films. It is demonstrated that there is an optimal TR value around 170 °C that helps reach the highest TE performances that are essentially determined by the level of in-plane orientation rather than the crystal contact plane of PBTTT-8O.
2. Results
2.1. Orientation, polymorphism and contact plane as a function of rubbing temperature TR in oriented PBTTT-8O
Before turning to the doped films of PBTTT-8O, we first investigated the microstructure of the films aligned by high-T rubbing at different rubbing temperatures. Alignment of PSCs such as P3HT and PBTTT is readily obtained by using the method of high temperature rubbing.31,32 A rotating cylinder is applied on a heated PSC thin film surface at a given temperature and pressure (Fig. 1a). Adjusting the temperature during film rubbing is essential as it helps modulate the thermomechanical properties of the PSC films. Melting the alkyl side chains of a PSC imparts higher mobility to the conjugated backbone that aligns more readily along the rubbing direction. For polymers such as P3HT or PBTTT, the rubbing temperature controls both film crystallinity and in-plane orientation.31,32,35TR impacts the level of in-plane chain orientation that can be quantified by extracting the 3D order parameter OP from the dichroic ratio (DR) of the main absorption band of the aligned polymer thin films.31Fig. 2c shows the evolution of the orientation in PBTTT-8O films versus TR. The dichroic ratio DR goes through a maximum for a rubbing temperature close to 170 °C with DR = 22 corresponding to an order parameter OP = 0.86 (OP = (DR − 1)/(DR + 2)) (measured at the maximum of absorption in the range 525–540 nm). A progressive decrease of DR is observed beyond that temperature as TR approaches the melting temperature (Tm = 250 °C). TEM ED shows that this reduction in DR coincides with the progressive change of the contact plane from face-on to edge-on (Fig. 3). Importantly, we observe a clear decrease in the OP for TR ≥ 230 °C and for TR = 240 °C we obtain OP = 0.64. The fact that alignment tends to decrease beyond a given TR is a general observation (also seen for P3HT).31,35 As noted in our earlier work, the observed alignment depends on both TR and the Mn distribution of the polymer.35 We attribute the decrease in orientation for TR > 170 °C to the normal sample dispersity (Mn = 30 kDa and Đ = 1.8). Polymer chains of increasing length require larger rubbing temperature to be aligned. When approaching the melting temperature, longer chains might align but not the short chains that are molten and lead to non-oriented fraction of PBTTT-8O in the films once cooled to RT. The fraction of such non-oriented and relaxed chains increases when TR approaches the melting temperature whereby reducing the order parameter of the films. | ||
| Fig. 1 (a) Chemical structures of the polymer PBTTT-8O and the dopant F6TCNNQ and (b) method of alignment by high temperature rubbing and incremental concentration doping (ICD) used to prepare highly oriented PBTTT-8O thin films. The arrow noted R corresponds to the rubbing direction. | ||
 | ||
| Fig. 2 Evolution of the normalized UV-vis-NIR spectrum for POL‖R (a) and POL⊥R (b) as a function of the rubbing temperature for aligned PBTTT-8O films. (c) Dichroic ratio DR measured at 540 nm (black dots) and corresponding order parameter (red dots) defined as (DR − 1)/(DR + 2) as a function of the rubbing temperature TR for thin films of PBTTT-8O. | ||
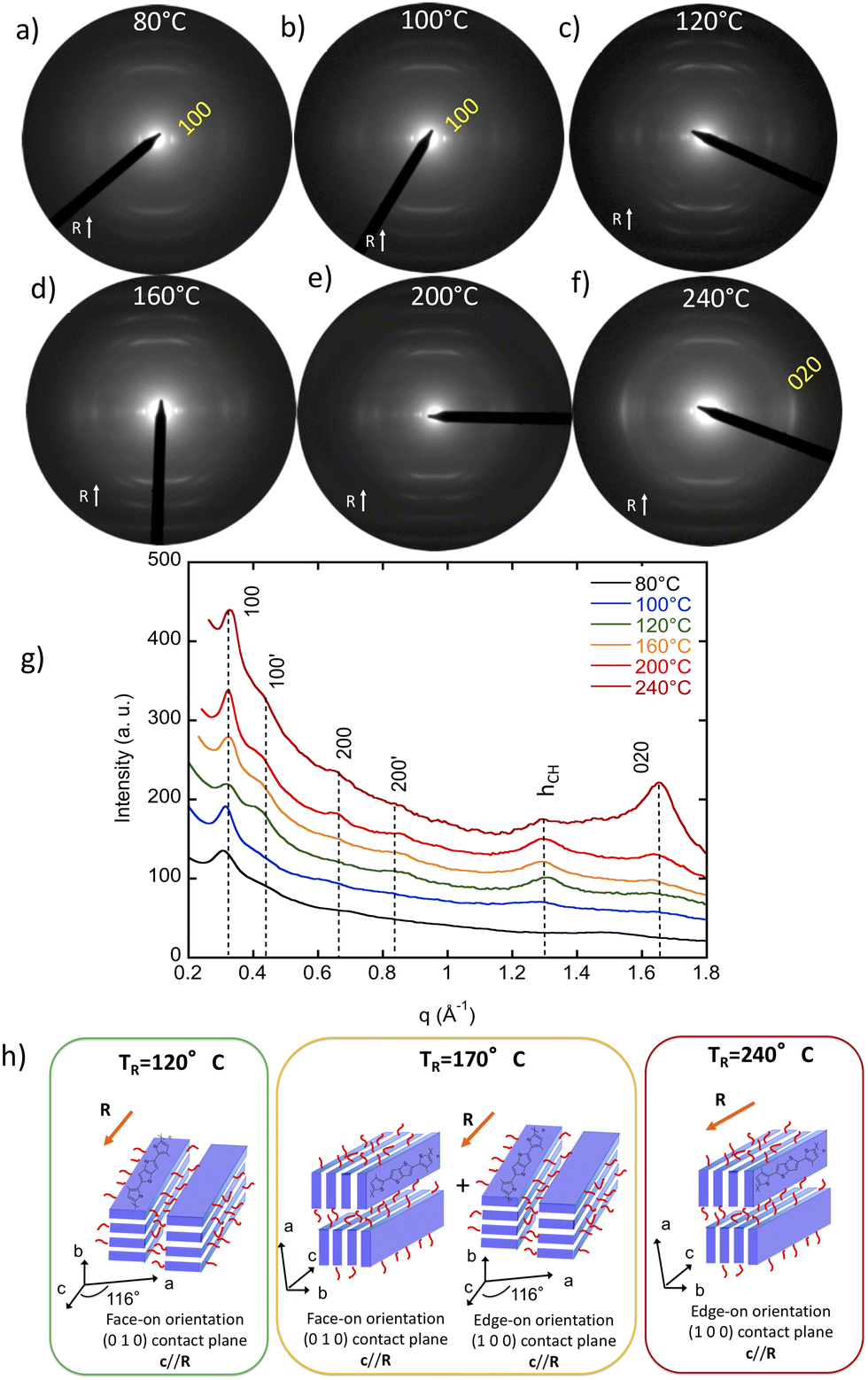 | ||
| Fig. 3 (a)–(f) Evolution of the electron diffraction pattern of PBTTT-8O thin films rubbed at different temperatures (rubbing direction is vertical). (g) Equatorial plot profile of the ED patterns as a function of TR. hCH originates from the scattering of C7OC4 side chains. (h) Schematic illustration of the dominant PBTTT-8O crystal contact plane in rubbed thin films as a function of TR. For all TR, the PBTTT-8O chains orient parallel to the rubbing direction R but the highest orientation is observed for TR = 170 °C. For TR = 170 °C, the film consists of a mixture of face-on and edge-on crystals. | ||
Further changes in the structure of PBTTT-8O can also be evidenced by polarized UV-vis-NIR spectroscopy. In particular, TR impacts the vibronic structure and the position of the absorption spectrum of PBTTT-8O.36 For POL⊥R, the absorption of the films corresponds essentially to amorphous and non-oriented PBTTT chains (see Fig. 2a and b showing the evolution of the UV-vis spectra with TR for POL‖R and POL⊥R). Interestingly, when TR increases between 80 °C and 180 °C, the absorption of amorphous PBTTT zones shifts to the blue from 517 nm to 485 nm whereas for TR approaching 240 °C, it shifts back to 510 nm. For POL‖R, the vibronic structure of the spectra changes slightly with TR. The 0–0 vibronic component is seen as a shoulder at 590 nm. It increases in intensity when increasing TR between 80 °C and 180 °C and decreases substantially for higher TR approaching 240 °C.
For polythiophenes such as P3HT, the intensity of the 0–0 vibronic contribution is related to the extension of planarized chain segments in the crystals: the 0–0 component shows enhanced intensity when the planarized chain segment increases.31,36 This result suggests chain segment planarization is enhanced around 180 °C in PBTTT-8O, in perfect agreement with the observed maximum of order parameter and charge conductivity in the doped films. Accordingly, the rubbed films of PBTTT-8O show a marked improvement of in-plane orientation and chain planarization around 180 °C which is of importance for the TE properties as shown hereafter.
The rubbing temperature TR is also a handle to modify the structure of the aligned thin films i.e. contact plane and crystal dimensions.31 As noted in our previous work, there is evidence for polymorphism in rubbed PBTTT-8O films. It is manifested by the coexistence of two lamellar reflections d100 and  at 19.7 Å and 14.6–15.0 Å, respectively. The polymorph with the larger d100 is dominant regardless of TR and d100 is close to the value found for PBTTT-C12. The section profile of the ED patterns in Fig. 3g suggests that the proportion of the 14.6–15.0 Å polymorph is most pronounced for TR = 120 °C and tends to decrease at larger TR.
at 19.7 Å and 14.6–15.0 Å, respectively. The polymorph with the larger d100 is dominant regardless of TR and d100 is close to the value found for PBTTT-C12. The section profile of the ED patterns in Fig. 3g suggests that the proportion of the 14.6–15.0 Å polymorph is most pronounced for TR = 120 °C and tends to decrease at larger TR.
This polymorph is possibly related to a structure with more tilted side chains and such polymorphism has been observed in the family of poly(alkylthiophene)s.37 It may correspond to a side chain conformation that is formed predominantly around TR = 120 °C because of the presence of the ether group that is expected to modify its conformation as compared to a linear C12 side chain. Indeed, due to the presence of the ether function, a significant increase of gauche effects, which lead to coil/random side chains, are expected for PBTTT-8O as regards to the usual PBTTT-C12.38 The majority of the reflections in the ED patterns for TR = 120 °C and 160 °C are attributed to the dominant polymorph with d100 = 19.7 Å.
As shown in our previous work, the reflections of this polymorph for TR = 170 °C can be indexed using a monoclinic unit cell with a = 21.9 Å, b = 7.6 Å, c = 13.7 Å and β = 116°. Modeling of the structure was performed to calculate ED patterns and compare them with the experimental ED pattern (see Fig. 4). To ease the structural refinement, we considered that side chains are ordered (DSC on powdered samples suggested that they are rather disordered, see ref. 12). The refined model involves a monoclinic unit cell with two chains per unit cell and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (see Fig. 4d and e). The off-meridional position of the most intense −3 0 3 reflection is the signature of this structure and it reflects the fact that side chains, although partially disordered, are tilted to the PBTTT backbone and located within the −3 0 3 planes. This is at variance with PBTTT-C12 for which the predominant meridional position of the 0 0 3 reflection (see Fig. S1, ESI†) indicates that the side chains are located in a plane perpendicular to the PBTTT backbone. The consequence of tilted C7–O–C4 side chains is that the successive PBTTT π-stacks are offset along the chain direction in the monoclinic unit cell of PBTTT-8O as compared to PBTTT-C12. Accordingly, the introduction of an ether function in the side chain modifies the packing of successive layers of π-stacked PBTTT backbones. Incidentally, the refined structure of PBTTT-8O suggests that oxygen atoms of the ether are located within a common plane inside the side chain layers, which may explain to some extent the higher side chain cohesion evidenced by DSC.16 This explains at least in part the better thermomechanical properties of PBTTT-8O that can be aligned up to very high temperatures close to the melting, contrary to PBTTT-C12.
space group (see Fig. 4d and e). The off-meridional position of the most intense −3 0 3 reflection is the signature of this structure and it reflects the fact that side chains, although partially disordered, are tilted to the PBTTT backbone and located within the −3 0 3 planes. This is at variance with PBTTT-C12 for which the predominant meridional position of the 0 0 3 reflection (see Fig. S1, ESI†) indicates that the side chains are located in a plane perpendicular to the PBTTT backbone. The consequence of tilted C7–O–C4 side chains is that the successive PBTTT π-stacks are offset along the chain direction in the monoclinic unit cell of PBTTT-8O as compared to PBTTT-C12. Accordingly, the introduction of an ether function in the side chain modifies the packing of successive layers of π-stacked PBTTT backbones. Incidentally, the refined structure of PBTTT-8O suggests that oxygen atoms of the ether are located within a common plane inside the side chain layers, which may explain to some extent the higher side chain cohesion evidenced by DSC.16 This explains at least in part the better thermomechanical properties of PBTTT-8O that can be aligned up to very high temperatures close to the melting, contrary to PBTTT-C12.
 | ||
| Fig. 4 (a) Electron diffraction pattern of an oriented PBTTT-8O film aligned by high-T rubbing at TR = 170 °C. (b) Schematic representation of the ED pattern, (c) calculated ED pattern in black for a monoclinic unit cell with [010] (black) and [0 −1 0] zone axes. b-Axis (d) and c-axis (e) projections of the refined structure of PBTTT-8O. | ||
Changes of the contact plane induced by the rubbing temperature are best visualized by plotting the equatorial section profiles of the ED patterns as a function of increasing rubbing temperature and comparing the relative intensities of the h 0 0 (h = 1–3) and 0 2 0 reflections (see Fig. 3). For TR ≤ 120 °C, the intensity of the equatorial 0 2 0 is very small and the intensities of the h 0 0 (h = 1, 2 and 4) are by far dominating. Thus, films formed at TR ≤ 120 °C are essentially made of face-on oriented PBTTT-8O crystals. For TR ≥ 200 °C, the situation changes progressively with a strong increase of the intensity of the 0 2 0 reflection that is dominant for TR = 240 °C. Accordingly, films rubbed at 240 °C are essentially made of edge-on oriented crystals. The films prepared at TR = 170 °C are composed of a mixture of edge-on and face-on crystals with a predominance of the latter. Thus, by changing TR it is possible to probe if the proportion of face-on/edge-on crystals impacts or not the TE properties of doped PBTTT-8O films.
2.2 Influence of the PBTTT-8O structure on the spectroscopic signatures of F6TCNNQ− anions in the oriented thin films
Having demonstrated that rubbing temperature determines in-plane orientation and structure of PBTTT-8O thin films, we have followed the doping of oriented thin films using polarized UV-vis-NIR spectroscopy. We have chosen three conditions: TR = 120 °C for mainly face-on oriented films, TR = 170 °C corresponding to a mixture of edge-on/face-on crystals with maximum in-plane orientation and TR = 240 °C for aligned films made of edge-on crystals (see Fig. S2, ESI†). For these three films, we have followed the evolution of the UV-vis-NIR spectra versus F6TCNNQ concentration in acetonitrile (Fig. 5). The advantage of using polarized light is that we can distinguish different contributions in the spectra such as oriented and ordered PBTTT-8O domains (POL‖R) or amorphous PBTTT-8O domains (POL⊥R). | ||
| Fig. 5 Evolution of the UV-vis-NIR spectrum of PBTTT-8O thin films as a function of the doping concentration with F6TCNNQ and for three films oriented by rubbing at 120 °C, 170 °C and 240 °C. The figures in the left column correspond to the spectra recorded for a light polarization POL‖R (R is the rubbing direction) whereas for the right column, POL⊥R. The broad absorption band near 800 nm that overlaps with the 0–2 band of the radical anion F6TCNNQ˙− is attributed to dimers of the dopant anion F6TCNNQ˙− and noted as D.39,40 | ||
Regarding the spectra at TR = 120 °C, the evolution as a function of increasing dopant concentration for POL⊥R is interesting. Indeed, for [F6TCNNQ−] ≤ 0.1 g l−1, the vibronic structure corresponding to the F6TCNNQ− anion is well observed with three components corresponding to 0–0, 0–1 and 0–2 contributions. For [F6TCNNQ−] ≥ 0.5 g l−1, the situation changes and beside the anion vibronic structure, a broad band around 800 nm that overlaps with the 0–2 of the F6TCNNQ− anion appears and tends to gain in intensity with increasing dopant concentration. In our previous work, this band was attributed to dimers of F6TCNNQ−, in agreement with earlier results on (TCNQ−)2 dimers as well as clustering of F6TCNNQ probed by electron paramagnetic resonance.39,40 The fact that the broad band at 800 nm appears only at larger dopant concentration is fully consistent with its attribution to dimers (F6TCNNQ−)2 since the probability to form them should increase with the dopant concentration in PBTTT-8O. Interestingly, the same trend i.e. a gradual increase of the dimer band at 800 nm with increasing doping concentration is also seen for the films prepared at 170 °C and 240 °C. It is worth to recall that for PBTTT-C12, no such dimer band was evidenced in the same range of [F6TCNNQ], indicating that the formation of such dimers is favored by the presence of C7–O–C4 side chains in PBTTT-8O.
Notably, it is possible to compare the films grown at various TR in terms of dimer concentration. As seen in Fig. 5, when considering the highest [F6TCNNQ] of 5 g l−1, there is a clear trend in the ratio of the dimer band versus 0–0 F6TCNNQ˙− band when the rubbing temperature is increasing. At 120 °C, the 0–0 contribution of F6TCNNQ˙− absorption is of lower intensity than the dimer band. For TR ≥ 170 °C, the trend is reversed, the 0–0 band becomes more intense. This observation indicates that the dimer/anion ratio in the F6TCNNQ-doped PBTTT-8O films changes with the structure of the films. As observed by TEM ED, the films grown at TR ≥ 170 °C show more mixed index reflections, suggesting that the films are more ordered and in particular the packing of C7–O–C4 chains is possibly more ordered than for TR = 120 °C. This is why we propose that the disorder in the C7–O–C4 side chains modulates the formation of F6TCNNQ˙− dimers: a higher level of disorder in the side chain packing favors the formation of dimers.
It is worth to note that similar spectroscopic signatures of dimer-like features were observed in F4TCNQ-doped P3HT films and attributed to the Charge Transfer Complex (CTC) between P3HT and F4TCNQ.41 The comparison with the present work and with ref. 41 suggests that the observed bands around 800 nm previously assigned to CTC correspond rather to dimers of the sole dopant radical anions F4TCNQ˙−.
For POL‖R, regarding the polaronic bands P1 and P2, a clear evolution with the dopant concentration and with the rubbing temperature is observed. In the PBTTT-8O films rubbed at 170 °C, the P2 band is redshifted from 790 nm at 0.05 g l−1 to 858 nm at 5 g l−1. This suggests that the polarons should be more delocalized with increasing dopant concentration and it illustrates the fact that the local environment of the polarons changes with dopant concentration. The same type of P2 band shift is also observed for the films prepared at 120 °C and 240 °C. However, For TR = 240 °C, the P2 band shifts to 797 nm at 5 g l−1. In other words, the P2 band is substantially more redshifted for TR = 170 °C than for TR = 240 °C, which suggests that the polarons are better delocalized as compared to the films prepared at 120 °C and 240 °C.9,10 A close look at the P1 band versus TR using FTIR spectroscopy confirms the previous observations for the P2 band. As seen in Fig. 6, the P1 band seems more red-shifted for TR = 170 °C than for TR = 120 °C and 240 °C. In addition, the FTIR spectra also show the typical C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching peak of the F6TCNNQ− anions at 2190 cm−1. Notably, no trace of the neutral F6TCNNQ dopant is observed at 2213 cm−1 for films doped at 1 g l−1 indicating that the majority of dopant molecules are ionized in the oriented PBTTT-8O films.34
N stretching peak of the F6TCNNQ− anions at 2190 cm−1. Notably, no trace of the neutral F6TCNNQ dopant is observed at 2213 cm−1 for films doped at 1 g l−1 indicating that the majority of dopant molecules are ionized in the oriented PBTTT-8O films.34
 | ||
| Fig. 6 Evolution of the normalized IR spectrum of PBTTT-8O thin films oriented at different rubbing temperatures and doped by ICD using F6TCNNQ in ACN (1 g l−1). Infrared-active vibrational (IRAV) modes are highlighted. The inset shows the expanded view of the FTIR spectrum around CN stretching peak of the dopant anion F6TCNNQ˙−. | ||
2.3 Structural evolution in oriented PBTTT-8O films versus doping
The evolution of the structure with increasing dopant concentration was followed for the three thin films aligned at 120 °C, 170 °C and 240 °C using electron diffraction. Fig. 7 exemplifies the variation of the lattice parameters corresponding to the lamellar periodicity along the side chains d100 and the π-stacking periodicity d020 as a function of increasing concentration of F6TCNNQ. For PBTTT with C12 alkyl side chains, doping with F6TCNNQ results in a small increase of d100 form 19.7 Å to 20.3 Å and a decrease of the π-stacking distance from 3.8 Å to 3.65 Å.24,31 For all rubbing temperatures, a different behavior is observed in the case of PBTTT-8O. For [F6TCNNQ] ≤ 0.5 g l−1, d100 tends to increase and d020 to decrease. This suggests that the PBTTT-8O lattice can accommodate a small amount of F6TCNNQ dopant in its lattice. However, beyond this concentration, a reverse trend is observed with a reduction of d100 and an increase of d020 for TR = 120 °C and 170 °C. Notably, for TR = 170 °C, the final values of d100 and d020 for [F6TCNNQ] = 5 g l−1 are almost identical to the values of the pristine PBTTT-8O. In our previous study, polarized UV-vis-NIR spectroscopy and EPR showed that the orientation of F6TCNNQ in PBTTT-8O grown at TR = 170 °C is random inside the side chain layers of PBTTT-8O, in strong contrast to PBTTT-C12 for which the dopants orient very well in the direction perpendicular to the alkyl side chains.16,33 The abnormal dependence of PBTTT-8O lattice parameters with increasing dopant concentration [F6TCNNQ] indicates some kind of re-organization of the lattice upon dopant incorporation. At a larger [F6TCNNQ], dopants are expected to interact more together inside the disordered side chain layers of C7–O–C4 and to cluster (see Fig. 7e). Our results suggest that the dopant's reorientation/reorganization in the side chain layers is a consequence of dopant anion's clustering and that this phenomenon depends on the order initially present in the side chain layers. Clustering seems more favorable when C7–O–C4 side chain layers are more disordered (TR ≤ 120 °C). It is also worth to mention that preferential location of dopant molecules in the only amorphous phase of PBTTT-8O, as observed for magic blue in semi-crystalline P3HT seems unlikely for PBTTT-8O. Indeed, for P3HT doped with MB, lattice parameters are unchanged whatever the MB dopant concentration, contrary to PBTTT-8O doped with F6TCNNQ.42 Moreover, polarized UV-vis-NIR spectroscopy measurements in Fig. 5 show that, similarly to PBTTT-C12, the amorphous phase of PBTTT-8O is not doped by F6TCNNQ (no bleaching of the neutral amorphous polymer band).42 This is in contrast to FeCl3 that can dope both crystalline and amorphous PBTTT-8O phases. It is therefore instructive to compare the structural variation upon doping observed for F6TCNNQ with that for FeCl3-doped films (see Fig. S3 and Fig. 7d). Indeed, contrary to F6TCNNQ-doping, a very different situation is observed when the PBTTT-8O films are doped with FeCl3 (see Fig. 7d). In that case, d100 increases steadily up to 24.0 Å at [FeCl3] = 5 g l−1 in the same way as observed previously for PBTTT-C12 doped with FeCl3. However, in strong contrast to doping with F6TCNNQ, doping PBTTT-8O with FeCl3 results in a total loss of reflections corresponding to order in the chain direction for [FeCl3] = 5 g l−1. In other words, stacking order within π-stacks is strongly altered in PBTTT-8O at high concentration of FeCl3 and more preserved in the case of F6TCNNQ doping. This explains, at least in part, the reduced TE properties measured for FeCl3-doped films as compared to F6TCNNQ (see next section).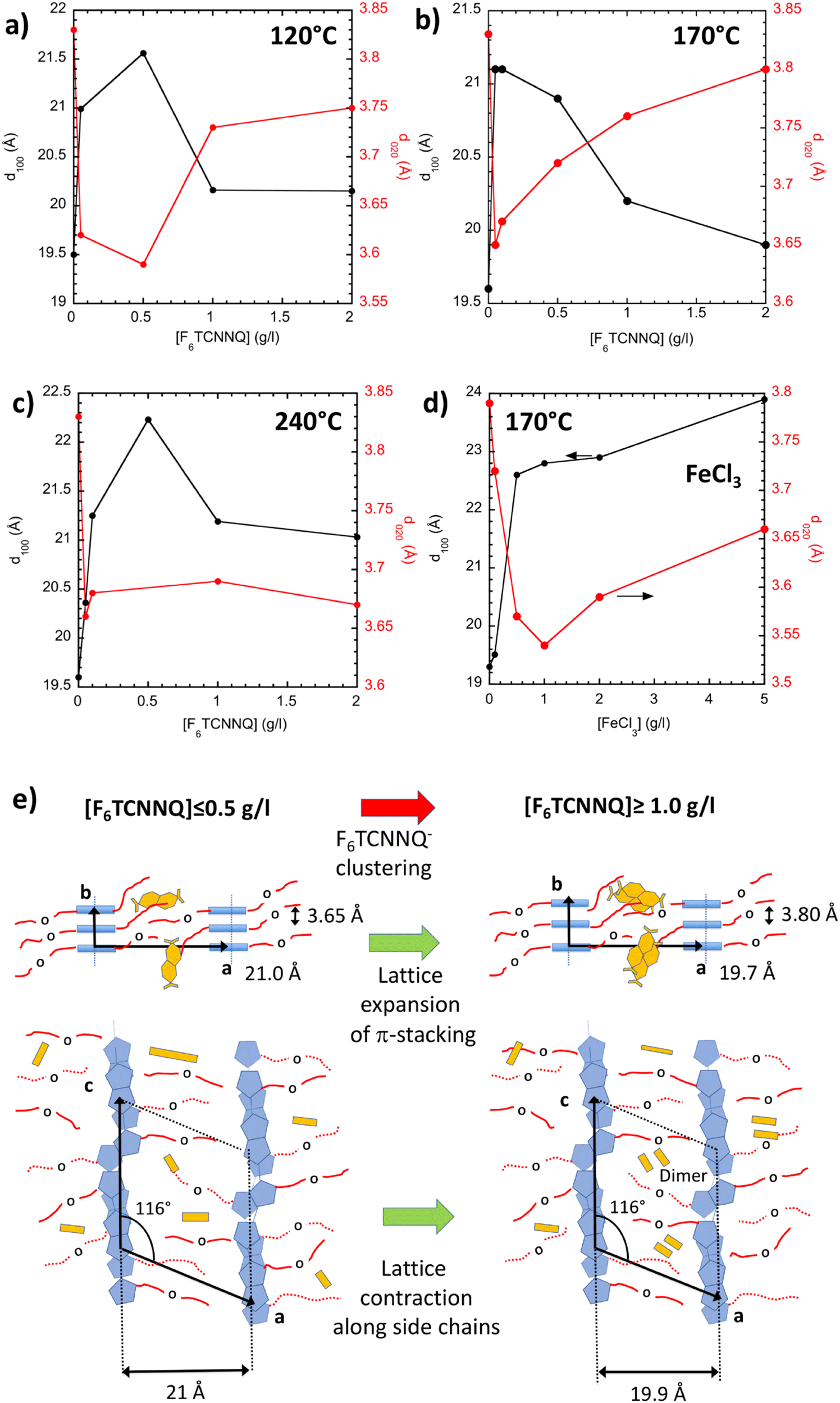 | ||
| Fig. 7 (a–c) Evolution of the reticular distance d100 and d020 corresponding the lamellar period and the π-stacking periodicity as a function of F6TCNNQ concentration in acetonitrile for PBTTT-8O thin films aligned by high-T rubbing at 120 °C (a), 170 °C (b) and 240 °C (c). For comparison, we show in (d) the evolution of the same lattice parameters of PBTTT-8O with increasing concentration of FeCl3 (TR = 170 °C). (e) Schematic illustration of the structural changes in PBTTT-8O induced by the clustering of F6TCNNQ˙− upon increasing doping concentration. | ||
2.4 Anisotropic TE properties versus rubbing temperature
Finally, we measured the anisotropic TE performances of the doped and oriented PBTTT-8O films for the three selected TR of 120 °C, 170 °C and 240 °C in the directions parallel and perpendicular to the rubbing (see Table 1 and Fig. 8). For all films, a marked anisotropy of TE properties is observed in the oriented films. The anisotropy of charge conductivity is in the range 3.4–29. It is most pronounced for the films prepared at TR = 170 °C, in agreement with the higher DR observed at this TR. As reported previously, the films grown at TR = 170 °C show a very high charge conductivity in the range 2–5 × 104 S cm−1 in the direction parallel to the rubbing. Even for a high dopant concentration of 5 g l−1, there is no true saturation of the conductivity but larger concentrations of F6TCNNQ are prohibited by the solubility limit of F6TCNNQ in ACN. As a means of comparison, FeCl3-doped PBTTT-8O films prepared at 170 °C yield charge conductivities σ‖ up to 9500 ± 3000 S cm−1 (for 5 g l−1, see Fig. S4, ESI†). This value is significantly below 2–5 × 104 S cm−1 obtained for F6TCNNQ-doped PBTTT-8O. This lower conductivity coincides with the strongly reduced order in the FeCl3-doped PBTTT-8O films as evidenced by TEM (see Fig. S3, ESI†) and supports the idea that introduction of dopants in the polymer host must preserve to the extent possible the original structure of the PBTTT-8O crystals.| T R | Dichroic ratio | 3D order parameter | Ratio P1/neutral | Maximum conductivity σ‖ (S cm−1) | Charge conductivity anisotropy σ‖/σ⊥ | Seebeck coefficient anisotropy S‖/S⊥ | Maximum power factor (μW m−1 K−2) |
|---|---|---|---|---|---|---|---|
| 120 °C | 13.5 | 0.8 | 0.38 | 3250 | 4.7 | 4.2 | 256 |
| 170 °C | 19.3 | 0.87 | 0.34 | 2–5 × 104 | 29 | 4.3 | 1000–2900 |
| 240 °C | 5.9 | 0.62 | 0.43 | 1100 | 3.4 | 3.0 | 16 |
| FeCl3 170 °C | 19.3 | 0.87 | — | 9500 | 21 | 4.7 | 100 |
 | ||
| Fig. 8 Evolution of the charge conductivity (a), the Seebeck coefficient (b) and the thermoelectric power factor (c) as a function of the dopant concentration of F6TCNNQ in the directions parallel and perpendicular to the rubbing for aligned PBTTT-8O thin films rubbed at different temperatures. Data for a non-oriented PBTTT-8O thin film are provided for comparison. | ||
Concerning the rubbing temperature, it has a drastic impact on the TE performances of the F6TCNNQ-doped PBTTT-8O. Both the PBTTT-8O films prepared at 120 °C and 240 °C show a much smaller charge conductivity σ‖ of 3250 S cm−1 and 1100 S cm−1, respectively, for [F6TCNNQ] = 5 g l−1. Notably, the conductivity at TR = 240 °C is similar to that of non-oriented thin films which is consistent with the low alignment level achieved at 240 °C i.e. OP = 0.62 (see Table 1). As a remark, these differences in charge conductivity are not related to strong differences in doping levels. The doping level can be tentatively approximated by the ratio of the P1 band absorbance at 2500 nm and the absorbance of the undoped polymer film (for parallel orientation).20 As seen in Table 1, the three films show similar values of this ratio in the 0.34–0.43 range, indicating that the observed charge transport differences must be due to differences in charge mobilities.
Regarding the Seebeck coefficient, it is larger when measured in the direction of chain alignment than perpendicular, but the observed anisotropy is substantially lower than for charge conductivity, S‖/S⊥ is in the range 3–4. Overall, the results are consistent with the differences in structure and alignment of the three films and they underline the beneficial role of orientation on the thermopower S.19 The highest conductivity is expectedly observed for the film that displays the best in-plane orientation and also the most extended planarized chain segments i.e. for TR = 170 °C and therefore a clear correlation between in-plane orientation and σ‖ is observed. The importance of in-plane orientation is predominant over other parameters and minor variations in the OP have a major impact on the charge conductivity in the chain direction. This is somehow expected from the theoretical work of Ihnatsenka et al. indicating that σ‖/σ⊥ increases exponentially with the order parameter.19 Regarding the anisotropy in Seebeck coefficients, it has been shown that it can be explained if anisotropic long-range repulsive Coulombic interactions between carriers are taken into account in the hopping between localized states. In particular, a stronger screening of Coulomb interactions in the direction parallel to the chains with respect to perpendicular direction can explain the strong anisotropy of S whereas the use of only on-site Coulomb interactions cannot capture this effect.19,43 For the range of very high charge conductivities (>104 S cm−1) for TR = 170 °C, the hopping regime might not be appropriate to describe transport properties. In that case, it might be the stronger delocalization of carriers due to enhanced chain segment planarization of PBTTT-8O that could explain a stronger screening of Coulomb interactions in the chain direction, hence the anisotropy of the Seebeck coefficient.
Notably, the best Power factors for the doped PBTTT-8O films are obtained for the films prepared at 170 °C and doped at 2 g l−1i.e. showing a structure close to that of the pristine undoped films prior to doping. PF reaches values in the range 1000–2900 μW m−1 K−2. The films of PBTTT-8O prepared at 120 °C and 240 °C the lattice of which is most altered upon doping show reduced TE performances with PF in the range 16–256 μW m−1 K−2i.e. comparable to 100 μW m−1 K−2 observed for FeCl3-doped films. This observation supports the idea that high TE performances can only be observed when (i) high in-plane orientation of the PBTTT chains is obtained by rubbing prior to doping and (ii) the structure of the doped material remains close to that of the undoped pristine polymer. Overall, these conclusions are in line with the recent work of Jacobs et al. on the influence of para-crystallinity measured in the π-stacking direction on the charge transport properties in doped polymer semiconductors.44 As a side remark, the polymorphism of PBTTT-8O evidenced in the pristine films seems to have little influence on the TE performances of doped films.
Finally, we investigated the S–σ correlations in the oriented PBTTT thin films that can help analyze transport phenomena in doped PSCs.45 For PBTTT-C12, we have demonstrated two different scaling laws in the directions parallel and perpendicular to the chain orientation, regardless of the type of dopant used (FeCl3, F4TCNQ and F6TCNNQ).22,42 In the chain direction, a power law of the type S‖ ∝ σ‖−1/4 was evidenced and a different relation of the type S⊥ ∝ −log(σ⊥) perpendicular to the chains.22 The power law dependence with an exponent s = −1/4 has been predicted by different models.46–49 Kemerink and coworkers proposed that variable range hopping transport in a system with a gaussain DOS and Coulomb trapping by ionized dopants can account for this type of correlation. However, such a model can only apply over a limited range of conductivities and can hardly be extrapolated to the range of conductivities >1000 S cm−1.43,46 Limelette and coworkers used a different approach considering rather delocalized carriers such as Dirac fermions with a parabolic DOS and a scattering mechanism by unscreened charged impurities.47 More recently, Gregory et al. proposed an improved version of the Kang and Snyder transport model i.e. a semi-localized transport (SLoT) model to bridge the gap between localized and delocalized transport in conducting polymers.48,49
In Fig. 9, we have merged the S, σ data points relative to the PBTTT-8O film oriented at 170 °C and doped with solutions of F6TCNNQ of different concentrations along with the data obtained for PBTTT-C12 doped with various dopants (FeCl3, F4TCNQ, F6TCNNQ). Most interestingly, the data points for the polymer with single ether C7OC4 side chains oriented at 170 °C fall onto the same master curves obtained previously for PBTTT-C12 for both directions ‖ and ⊥ to the rubbing. In strong contrast, the data points obtained for the films oriented at 120 °C and 240 °C do not comply with the trends of the master curves. In particular, the poor alignment of the PBTTT-8O films for TR = 120 °C and 240 °C results in totally different S–σ correlation curves closer to the non-oriented films and far away from the power law relation seen in the chain direction (see Fig. 9a). This result suggests that the PBTTT backbone, its in-plane orientation and π-stacking rather than the chemical natures of the side chains and dopant determines the type of S–σ scaling observed in the oriented PBTTT i.e. the anisotropic charge transport at play. It also suggests that neither the presence of polymorphism, nor the presence of dimers of F6TCNNQ˙− in PBTTT-8O thin films impacts negatively the TE performances. Conversely, poor in-plane orientation observed for TR = 120 °C and 240 °C and perturbation of π-stacking within individual PBTTT π-stacks modifies substantially the S–σ correlations. This result is important in the sense that anisotropic charge transport mechanism at play in oriented doped PSCs is primarily determined by structure and orientation control while the chemical nature of dopant and side chains seem more secondary factors in determining the upper limits of TE performances. These parameters become probably predominant for other aspects such as long-term stability of the doped systems or processing of thin films. Indeed, engineering of PSC side chains and of the dopant plays a primary role in the processing of polymer and dopant or long-term stability of the doped PSCs.50
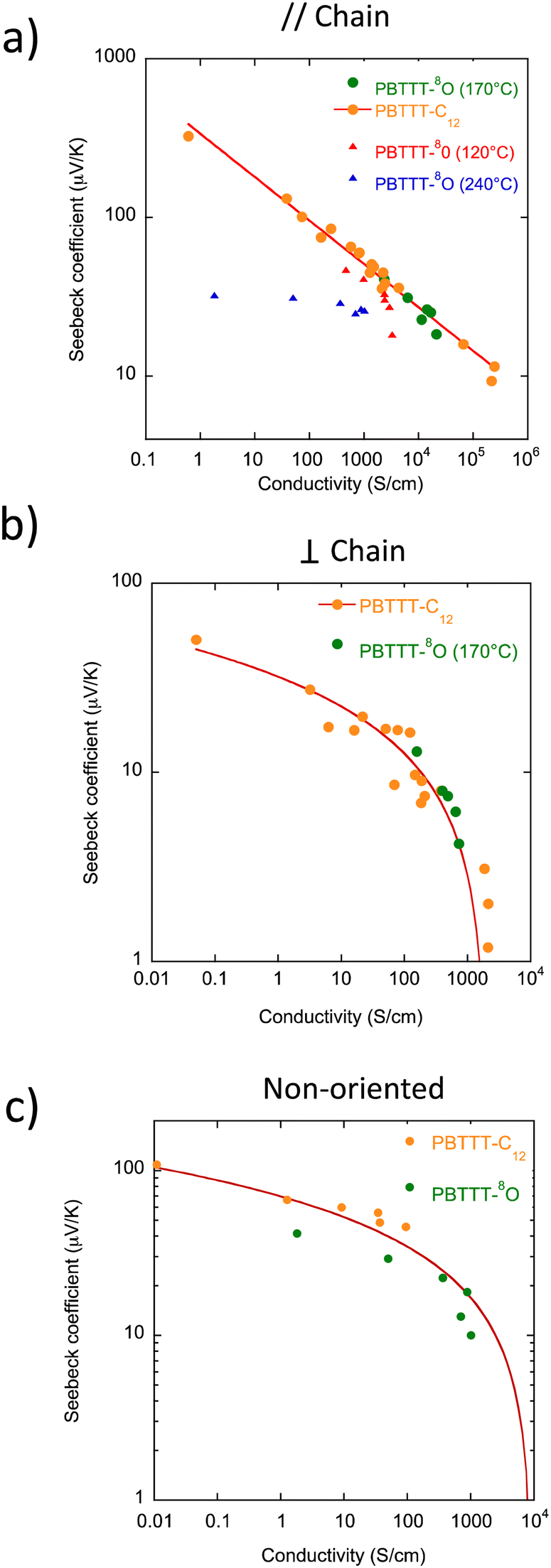 | ||
| Fig. 9 Correlations between the Seebeck coefficient and charge conductivity measured in the direction parallel (a) and perpendicular (b) to the rubbing direction for PBTTT-C12 and PBTTT-8O films rubbed at 125 °C and 170 °C, respectively. The continuous lines correspond to the fits with a power law (‖) and a logarithmic relation (⊥). (c) Correlations between the Seebeck coefficient and charge conductivity measured in non-oriented thin films of PBTTT-C12 and PBTTT-8O. | ||
The S–σ correlation in non-oriented films is very close to that for the films measured in the direction perpendicular to the chains with a logarithmic dependence. This result underlines the importance to measure the S–σ correlations in oriented films since the retained model (for instance the Kang–Snyder or the semi-localized transport model by Greogory et al.)48,49 to adjust the S–σ relationships in non-oriented samples is not representative of charge transport in the chain direction but may be dominated by transport in the direction perpendicular to the chains. Accordingly, applying transport models such as SLoT to non-oriented polymer films will inherently account for variable proportions of the intra-chain (delocalized) and inter-chain (localized) transport.
III. Conclusion
The anisotropic TE properties of PBTTT-8O thin films aligned by high-T rubbing and doped with F6TCNNQ have been studied. The rubbing temperature is a handle to control in-plane chain orientation of PBTTT backbones and the crystal's contact plane on the substrate. TR is a key parameter controlling the final TE performances of the aligned films. An optimum in TE performances corresponding to PF ≥ 2000 μW m−1 K−2 is identified for the films prepared at TR = 170 °C. It corresponds to (i) the best in-plane chain orientation, (ii) the highest extension of planarized chain segments and (iii) the best preservation of the crystal lattice of pristine PBTTT-8O after doping with F6TCNNQ. The contact plane of the PBTTT crystals or polymorphism determined by the rubbing temperature have apparently little influence on the final TE performances of the films. At low doping concentrations, F6TCNNQ˙− anions are incorporated into the lattice of PBTTT-8O with a contraction of the lattice along the π-stacking and expansion along the side-chain direction. However, at large dopant concentration, re-organization of the dopant radical anions in the form of dimers is observed and the lattice parameters of PBTTT-8O relaxed towards those of the pristine undoped structure. The formation of the (F6TCNNQ˙−)2 dimers seems to be influenced by the structure of the C7–O–C4 side chains. More generally, this study demonstrates that the identification of optimal TE performances requires a careful study of the pristine polymer film crystallization and orientation prior to doping. Precise control of structure and orientation are prerequisite to validate high performances of a given polymer semiconductor once doped. This study shows that orientation and packing of PBTTT backbones rather than chemical nature of side chains and dopants determine the physics of charge transport in doped PBTTT. Side chain layers play a key role in the hosting of dopants and can help preserve the packing of π-conjugated backbones. Side chain engineering of conjugated polymers impacts also the polymer film processing and may help reaching better chain alignment. Forthcoming studies will focus on the key question of long-term stability of such high performance TE polymer films.IV. Experimental section
(a) Materials and thin film preparation
PBTTT-8O was synthesized following the procedure described in our previous publication (Mn = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 Da and polydispersity of 1.79). The synthesis of F6TCNNQ is given in ref. 33 and references therein. Sodium poly(styrenesulphonate) (NaPSS), anhydrous solvents (99%) used for doping (acetonitrile) and film preparation (ortho-dichlorobenzene) were purchased from Sigma Aldrich. PBTTT-8O films are prepared by doctor blading at 165 °C from a solution in ODCB (10 g l−1) on substrates of glass covered with a thin NaPSS layer (spincoated from a 10 g l−1 aqueous solution at 3000 RPM).
200 Da and polydispersity of 1.79). The synthesis of F6TCNNQ is given in ref. 33 and references therein. Sodium poly(styrenesulphonate) (NaPSS), anhydrous solvents (99%) used for doping (acetonitrile) and film preparation (ortho-dichlorobenzene) were purchased from Sigma Aldrich. PBTTT-8O films are prepared by doctor blading at 165 °C from a solution in ODCB (10 g l−1) on substrates of glass covered with a thin NaPSS layer (spincoated from a 10 g l−1 aqueous solution at 3000 RPM).
(b) Orientation and doping of thin films
The orientation of the films by high-T rubbing followed the methodology described in previous publications.32,33,35 For high-T rubbing, a homemade set-up consisting of a rotating cylinder covered with a non-woven microfiber tissue and a translating hot plate was used. The film thickness was extracted from the UV-vis absorbance, assuming that the bulk densities of PBTTT-C12 and PBTTT-8O are similar (which is consistent with the similar crystal lattice volumes).24,51The doping of PBTTT-8O films with F6TCNNQ was performed by using the incremental concentration doping (ICD) procedure33 with full sample immersion for 40 s in the dopant solution of increasing concentration. No rinsing with the pure solvent was performed to avoid de-doping of the films. Both doping and rubbing were performed in a Jacomex glovebox (PN2 ≤ 1 ppm and PO2 ≤ 1 ppm).
(c) Structural analysis by TEM
Thin films of oriented PBTTT-8O are coated with a thin amorphous carbon film (<2 nm) using an Edwards Auto306 evaporator. The films were removed from the glass substrate by floating on distilled water and recovered on TEM copper grids. Doping was performed inside the glovebox on the TEM grids by dipping for 40 s in the solutions of F6TCNNQ in acetonitrile. Doped grids were rapidly transferred to the TEM for analysis. A CM12 Philips microscope (120 kV) equipped with a MVIII (Soft Imaging System) charge coupled device camera TEM was used for the structural analysis of the films (bright field and diffraction modes). The 0 0 3 reflection at 4.5 Å is not modified upon doping and was therefore used as an internal calibration to calculate the d100 and d020 reticular distances. Beam exposure was set to a minimum using the low dose system to avoid de-doping under the electron beam when the same zone is exposed for a prolonged period of time. Modeling of the structure of PBTTT-8O was performed using the appropriate modules of Cerius2 program and following the approach by Kayunkid et al. for P3HT and for other polymer semiconductors.52,53(d) Polarized UV-Vis-NIR absorption
A Varian Cary 5000 spectrometer with polarized incident light (spectral resolution of 1 nm) was used to probe the level of film orientation and the effect of film doping with F6TCNNQ (350–2500 nm) as a function of dopant concentration. The angle of light polarization is measured with respect to the rubbing direction (0° corresponding to the light polarization POL‖R and 90° corresponding to the light polarization POL⊥R).(e) Charge conductivity and Seebeck coefficient
The methodology for charge transport (four-point probe) and Seebeck coefficient measurements is described in ref. 21. Devices were fabricated on glass substrates cleaned by ultrasonication in acetone, ethanol, hellmanex and deionized water (×3 times). The cleaned substrates were dried under nitrogen and exposed to plasma prior to electrode deposition. Interdigitated gold electrodes (40 nm thick) in a four-points probe geometry (see ref. 21) were evaporated (0.4–0.6 nm s−1) through a shadow mask. The geometry of gold electrodes allows to measure both the charge transport and thermopower on a same substrate in both parallel and perpendicular directions to the rubbing. Oriented films of PBTTT-8O were floated on water and carefully recovered on the device with pre-deposited gold electrodes. They were subsequently dried under vacuum prior to doping in the glove box using the incremental concentration doping (ICD) method (see Fig. 1b).33DC conductivity and Seebeck coefficients were measured in a Jacomex glovebox under N2 atmosphere (<1 ppm H2O and <2 ppm O2). Four-point probe measurements of electrical conductivity were performed using a Keithley 2634B and a Lab Assistant Semiprobe station. The resistivity ρ was derived from the sheet resistance R following the relation ρ = 1.81·R·t where t is the film thickness (the geometrical correction factor was determined following the method in ref. 21).
For the thermopower, a differential temperature method was used whereby a temperature gradient ΔT was established across the sample along or perpendicular to the rubbing direction. ΔT was ramped between 0 and 12 K around room temperature and the Seebeck coefficient was extracted from the slope of the thermovoltage versus ΔT. A constantan wire was used to calibrate the Seebeck coefficient.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Bernard Lotz is gratefully acknowledged for fruitful discussions and careful reading of the manuscript. C. Blanck and M. Schmutz are acknowledged for technical support in TEM. P. Allgayer is acknowledged for technical support with the rubbing machine and N. Zimmermann is acknowledged for technical support in pre-patterned device preparation. Financial support from ANR grant Anisotherm ANR-17-CE05-0012 (ANISOTHERM) is acknowledged. P. Durand is grateful for financial support from Région Grand’Est. This work was financially supported by the European Commission through Marie Sklodowska-Curie project HORATES (GA-955837).References
- S. Ogawa, Organic Electronics, Springer, Japan, 2015, pp. 1–245 Search PubMed.
- M. Goel and M. Thelakkat, Macromolecules, 2020, 53, 3632 CrossRef CAS.
- O. Bubnova and X. Crispin, Energy Environ. Sci., 2012, 5, 9345 RSC.
- R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Müller, Chem. Soc. Rev., 2016, 45, 6147 RSC.
- M. Massetti, F. Jiao, A. J. Ferguson, D. Zhao, K. Wijeratne, A. Würger, J. L. Blackburn, X. Crispin and S. Fabiano, Chem. Rev., 2021, 121, 12465 CrossRef CAS.
- T. Degousée, V. Untilova, V. Vijayakumar, X. Xu, Y. Sun, M. Palma, M. Brinkmann, L. Biniek and O. Fenwick, J. Mater. Chem. A, 2021, 9, 16065 RSC.
- A. D. Scaccabarozzi, A. Basu, F. Aniés, J. Liu, O. Zapata-Arteaga, R. Warren, Y. Firdaus, M. I. Nugraha, Y. Lin, M. Campoy-Quiles, N. Koch, C. Müller, L. Tsetseris, M. Heeney and T. D. Anthopoulos, Chem. Rev., 2021, 122, 4420 CrossRef PubMed.
- R. Ghosh, A. R. Chew, J. Onorato, V. Pakhnyuk, C. K. Luscombe, A. Salleo and F. C. Spano, J. Phys. Chem. C, 2018, 122, 18048 CrossRef CAS.
- R. Ghosh, C. M. Pochas and F. C. Spano, J. Phys. Chem. C, 2016, 120, 11394 CrossRef CAS.
- T. J. Aubry, K. J. Winchell, C. Z. Salamat, V. M. Basile, J. R. Lindemuth, J. M. Stauber, J. C. Axtell, R. M. Kubena, M. D. Phan, M. J. Bird, A. M. Spokoyny, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2020, 30, 2001800 CrossRef CAS.
- D. T. Scholes, S. A. Hawks, P. Y. Yee, H. Wu, J. R. Lindemuth, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. Lett., 2015, 6, 4786 CrossRef CAS PubMed.
- S. D. Tyler, Y. Patrick Y., R. Lindemuth Jeffrey, O. Kang Hyeyeon, G. R. Jonathan, K. Luscombe Christine, C. Spano Frank, H. Tolbert Sarah and J. Schwartz Benjamin, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef.
- I. E. Jacobs, E. W. Aasen, J. L. Oliveira, T. N. Fonseca, J. D. Roehling, J. Li, G. Zhang, M. P. Augustine, M. Mascal and A. J. Moulé, J. Mater. Chem. C, 2016, 4, 3454–3466 RSC.
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140 CrossRef CAS PubMed.
- J. Hynynen, E. Järsvall, R. Kroon, Y. Zhang, S. Barlow, S. R. Marder, M. Kemerink, A. Lund and C. Müller, ACS Macro Lett., 2019, 8, 70 CrossRef CAS.
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef PubMed.
- P. Durand, H. Zeng, T. Biskup, V. Vijayakumar, V. Untilova, C. Kiefer, B. Heinrich, L. Herrmann, M. Brinkmann and N. Leclerc, Adv. Energy Mater., 2022, 12, 2103049 CrossRef CAS.
- S. Ihnatsenka, ACS Phys. Chem. Au, 2021, 2, 118 CrossRef.
- V. Untilova, T. Biskup, L. Biniek, V. Vijayakumar and M. Brinkmann, Macromolecules, 2020, 53, 2441 CrossRef CAS.
- A. Hamidi-sakr, L. Biniek, J.-L. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, Adv. Funct. Mater., 2017, 27, 1700173 CrossRef.
- V. Vijayakumar, Y. Zhong, V. Untilova, M. Bahri, L. Herrmann, L. Biniek, N. Leclerc and M. Brinkmann, Adv. Energy Mater., 2019, 9, 1900266 CrossRef.
- V. Untilova, H. Zeng, P. Durand, L. Herrmann, N. Leclerc and M. Brinkmann, Macromolecules, 2021, 54, 6073 CrossRef CAS.
- V. Vijayakumar, E. Zaborova, L. Biniek, H. Zeng, L. Herrmann, A. Carvalho, O. Boyron, N. Leclerc and M. Brinkmann, ACS Appl. Mater. Interfaces, 2019, 11, 4942 CrossRef CAS PubMed.
- Y. Huang, D. H. Lukito Tjhe, I. E. Jacobs, X. Jiao, Q. He, M. Statz, X. Ren, X. Huang, I. McCulloch, M. Heeney, C. McNeill and H. Sirringhaus, Appl. Phys. Lett., 2021, 119, 111903 CrossRef CAS.
- Z. Liang, Y. Zhang, M. Souri, X. Luo, A. M. Boehm, R. Li, Y. Zhang, T. Wang, D.-Y. Kim, J. Mei, S. R. Marder and K. R. Graham, J. Mater. Chem. A, 2018, 6, 16495 RSC.
- J. Min, D. Kim, S. G. Han, C. Park, H. Lim, W. Sung and K. Cho, Adv. Electron. Mater., 2022, 8, 2101142 CrossRef CAS.
- A. Salleo, R. J. Kline, D. M. DeLongchamp and M. L. Chabinyc, Adv. Mater., 2010, 22, 3812 CrossRef CAS PubMed.
- A. Gasperini and K. Sivula, Macromolecules, 2013, 46, 9349 CrossRef CAS.
- M. J. Lee, D. Gupta, N. Zhao, M. Heeney, I. McCulloch and H. Sirringhaus, Adv. Funct. Mater., 2011, 21, 932 CrossRef CAS.
- A. Hamidi-Sakr, L. Biniek, S. Fall and M. Brinkmann, Adv. Funct. Mater., 2016, 26, 408 CrossRef CAS.
- L. Biniek, N. Leclerc, T. Heiser, R. Bechara and M. Brinkmann, Macromolecules, 2013, 46, 4014 CrossRef CAS.
- V. Vijayakumar, P. Durand, H. Zeng, V. Untilova, L. Herrmann, P. Algayer, N. Leclerc and M. Brinkmann, J. Mater. Chem. C, 2020, 8, 16470 RSC.
- Y. Karpov, T. Erdmann, M. Stamm, U. Lappan, O. Guskova, M. Malanin, I. Raguzin, T. Beryozkina, V. Bakulev, F. Günther, S. Gemming, G. Seifert, M. Hambsch, S. Mannsfeld, B. Voit and A. Kiriy, Macromolecules, 2017, 50, 914 CrossRef CAS.
- L. Biniek, S. Pouget, D. Djurado, E. Gonthier, K. Tremel, N. Kayunkid, E. Zaborova, N. Crespo-Monteiro, O. Boyron, N. Leclerc, S. Ludwigs and M. Brinkmann, Macromolecules, 2014, 47, 3871 CrossRef CAS.
- J. Clark, J.-F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 163306 CrossRef.
- T. J. Prosa, M. J. Winokur and R. D. McCullough, Macromolecules, 1996, 29, 3654 CrossRef CAS.
- D. M. DeLongchamp, R. J. Kline, Y. Jung, E. K. Lin, D. A. Fischer, D. J. Gundlach, S. K. Cotts, A. J. Moad, L. J. Richter, M. F. Toney, M. Heeney and I. McCulloch, Macromolecules, 2008, 41, 5709 CrossRef CAS.
- R. H. Boyd and W. D. J. Phillips, Chem. Phys., 1965, 43, 2927 CAS.
- A. Privitera, R. Warren, G. Londi, P. Kaienburg, J. Liu, A. Sperlich, A. E. Lauritzen, O. Thimm, A. Ardavan, D. Beljonne and M. Riede, J. Mater. Chem. C, 2021, 9, 2944 RSC.
- O. Zapata-Arteaga, B. Dörling, A. Perevedentsev, J. Martín, J. S. Reparaz and M. Campoy-Quiles, Macromolecules, 2020, 53, 609 CrossRef CAS PubMed.
- Y. Zhong, V. Untilova, D. Muller, S. Guchait, C. Kiefer, L. Herrmann, N. Zimmermann, M. Brosset, T. Heiser and M. Brinkmann, Adv. Funct. Mater., 2022, 32, 2202075 CrossRef CAS.
- D. Scheunemann, V. Vijayakumar, H. Zeng, P. Durand, N. Leclerc, M. Brinkmann and M. Kemerink, Adv. Electron. Mater., 2020, 6, 2000218 CrossRef CAS.
- I. E. Jacobs, G. D’Avino, V. Lemaur, Y. Lin, Y. Huang, C. Chen, T. F. Harrelson, W. Wood, L. J. Spalek, T. Mustafa, C. A. O’Keefe, X. Ren, D. Simatos, D. Tjhe, M. Statz, J. W. Strzalka, J.-K. Lee, I. McCulloch, S. Fratini, D. Beljonne and H. Sirringhaus, J. Am. Chem. Soc., 2022, 144, 3005–3019 CrossRef CAS PubMed.
- A. M. Glaudell, J. E. Cochran, S. N. Patel and M. L. Chabinyc, Adv. Energy Mater., 2015, 5, 1401072 CrossRef.
- H. Abdalla, G. Zuo and M. Kemerink, Phys. Rev. B, 2017, 96, 241202(R) CrossRef.
- M. Lepinoy, P. Limelette, B. Schmaltz and F. T. Van, Sci. Rep., 2020, 10, 8086 CrossRef CAS PubMed.
- S. D. Kang and G. J. Snyder, Nat. Mater., 2016, 16, 252 CrossRef PubMed.
- S. A. Gregory, R. Hanus, A. Atassi, J. M. Rinehart, J. P. Wooding, A. K. Menon, M. D. Losego, G. J. Snyder and S. K. Yee, Nat. Mater., 2021, 20, 1414 CrossRef CAS PubMed.
- J. Liu, L. Qiu, G. Portale, M. Koopmans, G. ten Brink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2017, 29, 1701641 CrossRef.
- C. Y. Kao, B. Lee, L. S. Wielunski, M. Heeney, I. McCulloch, E. Garfunkel, L. C. Feldman and V. Podzorov, Adv. Funct. Mater., 2009, 19, 1906–1911 CrossRef CAS.
- N. Kayunkid, S. Uttiya and M. Brinkmann, Macromolecules, 2010, 43, 4961 CrossRef CAS.
- M. Brinkmann, Mater. Chem. Front., 2020, 4, 1916 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03600b |
| This journal is © The Royal Society of Chemistry 2022 |