
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of Yb3+ concentration on multiband upconversion in a single NaYF4:Yb/Er microcrystal determined via nanosecond time-resolved spectroscopy†
Hanchang
Huang‡
 a,
Maohui
Yuan‡
a,
Zhongyang
Xing‡
a,
Wenda
Cui
ab,
Tongcheng
Yu
ab,
Shuai
Hu
a,
Guomin
Zhao
ab,
Chuan
Guo
*ab and
Kai
Han
*ac
a,
Maohui
Yuan‡
a,
Zhongyang
Xing‡
a,
Wenda
Cui
ab,
Tongcheng
Yu
ab,
Shuai
Hu
a,
Guomin
Zhao
ab,
Chuan
Guo
*ab and
Kai
Han
*ac
aCollege of Advanced Interdisciplinary Studies, National University of Defense Technology, Changsha, 410073, China. E-mail: guochuan20@nudt.edu.cn; hankai0071@nudt.edu.cn
bHunan Provincial Key Laboratory of High Energy Laser Technology, Changsha, 410073, China
cState Key Laboratory of Pulsed Power Laser Technology, Changsha, 410073, China
First published on 23rd September 2022
Abstract
In lanthanide-sensitized upconversion (UC) nanomaterials, the typical sensitizer Yb3+ can significantly modulate the codoped activator (such as Er3+ ions) to generate multiband transitions. However, the kinetics of these multiband emissions affected by the Yb3+ concentration remains unclear. In this work, we employ nanosecond time-resolved spectroscopy to investigate a single optically trapped NaYF4:Yb/Er microcrystal (MC) doped with different Yb3+ concentrations. Interestingly, high doping Yb3+ ions shorten the UC response time by a factor of 10 (from ∼1 μs to ∼100 ns) compared to low doping Yb3+. Meanwhile, 430 nm light (4G7/2 → 4I11/2) emitted before 654 nm (4F9/2 → 4I15/2) with Yb3+ dramatically increasing (opposite to the normal low Yb3+ doping cases), representing a new four-photon pathway (4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 → 2H9/2 → 2D5/2 → 4G7/2). Furthermore, we observed a broad range of time-dependent emission color, changing from being initially green over near-white to red. Particularly, the red emission is dominated in the decay process, yielding insight into the fundamental multiband UC kinetics. The result shows that varying the Yb3+ doping concentration provides a way to control the energy flow between the energy levels of Er3+ ions, which can modulate the response time and color evolution of UC on Yb/Er doped materials. This can direct the prospective development of fast-response optoelectronic devices, photocatalysis, and UC displays in the future.
Introduction
Luminescence modulation of micron-sized materials has a wide range of applications in the field of optics.1,2 Lanthanide-based upconversion (UC) materials have the unique ability to convert two or more near-infrared photons to a higher energy photon,3 thus have been increasingly used in photocatalysis,4,5 photovoltaics,6 photodynamic therapy,7 and anticounterfeiting.8 One of the most studied UC converters is β-NaYF4:Yb/Er, mainly because of the low phonon energy of the host,9 large absorption cross-section of sensitizers (Yb3+) at near-infrared excitation (around 976 nm),10,11 and abundant energy levels of activators (Er3+).12,13 To improve the UC efficiency of β-NaYF4:Yb/Er, several strategies have been applied,14 such as changing substrates,15 using core–shell structures,16 and modifying the surface of particles.17 But all these works were based on a Yb/Er doping concentration at around 20/2 mol%,9,18 meaning that almost 80 mol% RE3+ contents (such as Y3+) remain optically inactive. Hence, there is still plenty of room to improve the UCL efficiency by further increasing the Yb3+ concentration, because a large number of sensitizers (Yb3+) can ensure more photons to be absorbed (by Er3+) in the system. However, when the Yb3+ doping concentration increases to a certain level, the UCL would decline or even quench due to the undesired competing processes such as back-energy transfer (BET) and nonradiative decay.19–22 For example, Li et al. demonstrated that the restriction for the doping ratio of Yb3+ is the Yb3+–Yb3+ energy migration resulting in surface quenching.22 However, the energy transfer between dense Yb3+ and relatively thin Er3+ has still not been explained yet. Therefore, investigating the kinetics of highly doped Yb3+ in the β-NaYF4:Yb/Er system can help us understand the UC mechanism and design UC crystal structures with higher UCL efficiency.To study the optical properties of high dopant concentration UC crystals, high excitation irradiance is required to overcome the decline or even quenching of UCL.23 Normally, when the near-infrared excitation power is not high enough, the β-NaYF4:Yb/Er materials mainly show green (545/525 nm) and red (650 nm) UC emissions, and sometimes relatively weak blue (410 nm) emission.9,18 But under high-irradiance excitation, the UCL is significantly enhanced and the multiband UC emission is effectively achieved as the silent energy levels of Er3+ are activated.24 To better understand the kinetics of β-NaYF4:Yb/Er systems with different Yb3+ doping concentrations (especially highly doped), high-power excitation laser is needed to enable multiband UC emissions. Previously, we had developed a sophisticated experimental setup called optical trapping time-resolved photoluminescence spectroscopy (OT-TRPLS), which combines a 1342 nm optical tweezer25,26 and an advanced time-resolved photoluminescence device with a 976 nm excitation nanosecond-pulsed (ns-pulsed) laser.27 This allows for measuring the transient spectra at a nanosecond timescale while trapping a single MC to avoid the anisotropy from the movement of the measured MC itself28–30 and interactions from the surrounding MCs.31
In this work, we systematically investigate the impact of Yb3+ concentration on multiband emissions in a single optically trapped NaYF4:Yb/Er MC through the OT-TRPLS platform. The kinetics of multiband emissions was determined based on the time-resolved spectra and the multiband color variation dependent on the decay time (from being green over near-white to red) was also quantitatively demonstrated. Interestingly, highly doped Yb3+ MC (over 40 mol%) populated the characteristic 4G7/2 energy level of Er3+ (the highest energy level that a four-photon absorption can achieve) as fast as ∼0.1 μs after excitation, whereas a timescale of ∼1 μs is obtained for the lowly doped Yb3+ MC (lower than 20 mol%). Besides, high doping Yb3+ concentration can generate a new populating pathway (4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 → 2H9/2 → 2D5/2 → 4G7/2) to achieve the four-photon UCL. By comparing highly doped Yb3+ systems with their low-Yb3+ doping counterpart, we find that massive Yb3+ ions can accelerate the four-photon population of high energy-level UC (populating 4G7/2). Meanwhile, we observed significant red-light emission (dropping from 4F9/2 to 4I15/2) in the decay process in highly Yb3+ doped systems and therefore prove that Yb3+ ions can trigger the BET process, making more electrons flow into 4F9/2.
Experimental
Materials
Ytterbium(III) chloride (YbCl3·6H2O, 99.99%), erbium(III) chloride (ErCl3·6H2O, 99.99%), and yttrium(III) chloride (YCl3·6H2O, 99.99%) were purchased from Aladdin Industrial Corporation. Sodium fluoride (NaF) and Na2-ethylenedia-metracetic acid (EDTA-2Na) were supplied by Sinopharm Chemical Reagent Co., Ltd.Synthesis of β-NaY(Yb)F4:Yb3+/Er3+ MCs
β-NaY(Yb)F4:Yb3+/Er3+ MCs were prepared by the hydrothermal method. According to the doping ratio, YbCl3·6H2O, ErCl3·6H2O, and YCl3·6H2O were dissolved in 22 mL of deionized water such that the total rare-earth-ion content was 1 mmol, and was stirred ultrasonically until the solution became transparent. 1 mmol EDTA-2Na was added to the mixed solution and stirred thoroughly for 30 minutes. Then 10 mmol NaF was added and stirred for 30 minutes until the solution resembled a jelly appearance. The mixtures above were annealed at 160 °C for 24 hours in a hydrothermal reactor and then cooled down to room temperature. The final products were washed three times with ethanol and deionized water sequentially.Sample characterization
The monocrystalline β-NaY(Yb)F4:Yb3+/Er3+ MCs are characterized by scanning electron microscopy (SEM). The result (see Fig. 1a–d) shows that the size of MCs slightly increases along with Yb doping concentration, but the overall crystals are about 7 μm in length as hexagonal prisms. The X-ray diffraction (XRD) patterns of the microcrystals were recorded using an X-ray diffractometer with Cu K radiation at 40 kV and 200 mA (Rigaku). The XRD data in the 2θ range from 10° to 70° were collected in the scanning mode at a scanning rate of 10° min−1. The results (see Fig. 1e) demonstrated that the prepared NaY(Yb)F4 MCs are β-phase crystalline according to the data of the Joint Committee on Powder Diffraction Standards (JCPDS). | ||
| Fig. 1 SEM micrographs of (a–d) β-NaY(Yb)F4:2% Er/x% Yb (x = 10, 40, 60, 90) MCs. (e) XRD patterns of the as-prepared β-NaY(Yb)F4:2% Er/x% Yb (x = 10, 40, 60, 90) MCs compared to the known peaks of the hexagonal phase of NaYF4. (f) Schematics of the OT-TRPLS setup for multiband UCL detection under laser excitation. The main components are as follows, M1: dichroic mirror for short wave pass, cutoff wavelength is 900 nm; M2: beamsplitter; M3: beam combining mirror for 976 nm laser and 1342 nm laser; M4: beamsplitter; 1342 CW laser and 976 ns-pulsed laser were coaxial before reaching the objective lens. (g–j) Real-time monitoring of the captured single β-NaY(Yb)F4:2% Er/x%Yb (x = 10, 40, 60, and 90) MCs by a CMOS camera under 976 nm ns-pulsed laser excitation at the integration time of 50 ms. | ||
Photoluminescence measurements
The time evolution spectra were recorded using the OT-TRPLS setup shown in Fig. 1f, which is equipped with a 976 nm ns-pulsed excitation source and a 1342 nm continuous-wave (CW) optical tweezer. Here the optical tweezer is used to keep the sample MC stable and isolated to avoid unfavorable interactions with the surrounding MCs. Particularly, the 1342 nm laser is chosen because both the Er3+ and Yb3+ ions have relatively low absorption coefficients at this wavelength.13,32 A color-complementary metal oxide semiconductor (CMOS) camera is used to monitor the in situ optical trapping process as shown in Fig. 1g–j. The excitation power density is ∼0.32 GW cm−2 with a 15 ns pulse width. A spectrometer equipped with an image intensified charge-coupled device (ICCD) camera is utilized to detect the time evolution spectra with the synchronous triggering of a 976 nm ns-pulsed laser at a repetition rate of 37 Hz. The power efficiency is shown in Fig. S1 and S2 (ESI†). Using the HAMAMATSU system C9920-02G, the quantum efficiencies of the prepared β-NaY(Yb)F4:Yb3+/Er3+ MCs (Yb 10/40/90 mol%) in aqueous solutions are found to be 0.04%, 0.016%, and 0.01%, respectively.Results and discussion
We obtained the time evolution spectra (Fig. 2a and b) of a single optically trapped NaYF4:Yb/Er (60/2 mol%) MC after the excitation of the 976 nm ns-pulsed laser and observed the rise and decay process of multiband UCL of Er3+ ions. The results show that the multiband UCL process has an obvious sequence of rising and decay at different wavelengths. Furthermore, based on the time evolution spectra, the CIE chromaticity coordinates are calculated and shown in Fig. 2c, which reveals the evolutionary color of the single NaYF4:Yb/Er (60/2 mol%) MC after 976 nm ns-pulsed excitation. The color of a single NaYF4:Yb/Er (60%/2%) MC changed from initially green over near-white to red, which demonstrates that we achieved the near-white color in a single NaYF4:Yb/Er doped with high Yb3+ concentration. The color evolution originates from the difference in the rise and decay of the UCL at different wavelengths, which requires analyzing populating processes of energy levels corresponding to these emissions. We subsequently focused on the main UCL peaks, which can be clearly distinguished from each other. According to the previous studies,12,24,33–37 transient spectra of multiband UCL are determined based on the emission wavelengths and their corresponding transitions, as shown in Fig. 2d.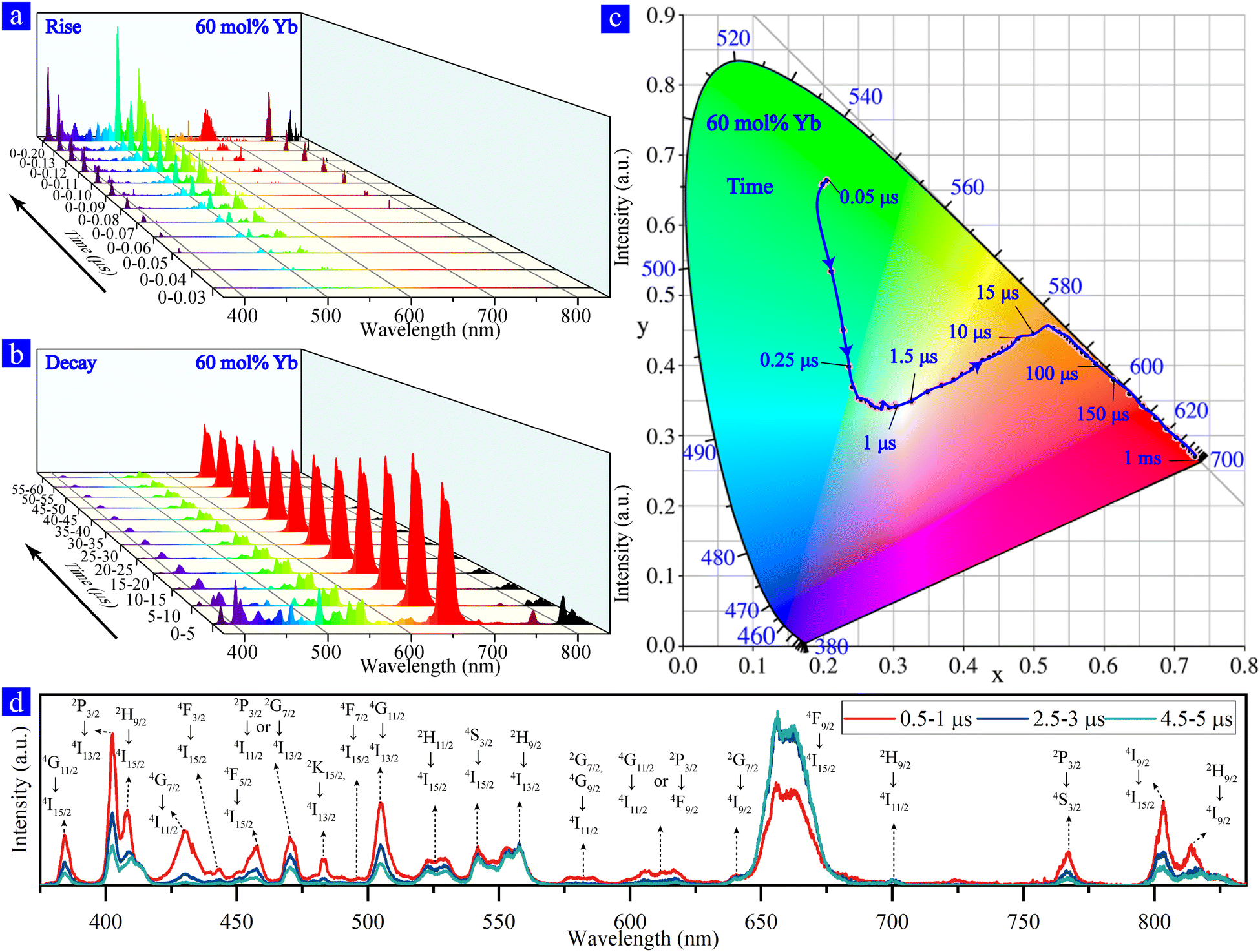 | ||
| Fig. 2 The 0 moment is the starting point of the 976 nm ns-pulsed excitation. Time evolution spectra of NaYF4:Yb/Er (60/2 mol%) determined using the superimposed transient spectrum at the same moment, 200 times. (a) The temporal evolution of spectra was acquired in a step of 0.01 μs from moment 0. (b) The temporal evolution of spectra was acquired with an integration time of 5 μs. (c) CIE chromaticity coordinates for the multiband UCL of a single β-NaYF4:Yb/Er (60/2 mol%) MC based on time evolution. (d) Transient spectra of the single β-NaYF4:Yb/Er (60%/2%) MC for different time periods with an integration time of 0.5 μs. | ||
To further investigate the UC kinetics of single NaYF4:Yb/Er (60%/2%) MC after excitation, we plot a diagram of the populating processes of Yb3+ and Er3+ ions based on the time evolution spectra. Fig. 3a presents the populating and multiband UCL emitting processes, as well as the decay times of different UC emissions (Fig. 3b). There are two main populating pathways in the Yb–Er systems, i.e., pathway A (4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 → 2H9/2 → 4F5/2 → 2K13/2) and pathway B (4I15/2 → 4I11/2 → 4I13/2 → 4F9/2 → 2H9/2 → 2D5/2).24,33,38,39 The UCL at 542 nm (4S3/2 → 4I15/2), 522 nm (2H11/2 → 4I15/2), 496 nm (4F7/2 → 4I15/2), 505 nm (4G11/2 → 4I13/2), and 384 nm (4G11/2 → 4I15/2) emits within 50 ns after excitation, which originated from the efficient populating channel of 4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 from pathway A and the processes of BET-1 (4S3/2 → 4I11/2) and BET-2 (4G11/2 → 4F9/2) owing to the high doping of Yb3+.19–22 In the timescale from 50 ns to 80 ns after excitation, the emergence of 402 nm (2P3/2 → 4I13/2) and 767 nm (2P3/2 → 4S3/2) emissions demonstrates the occurrence of the four-photon process. This reveals that the populating channel of 4F5/2 → 2K13/2 → 2P3/2 (pathway A) leads to the population of the 2P3/2 level. In addition, 482 nm (2K15/2 → 4I13/2), 575 nm (2G7/2 → 4I11/2), 585nm (4G9/2 → 4I11/2), 410nm (2H9/2 → 4I15/2) and 558 nm (2H9/2 → 4I13/2) emissions indicate that the 2K15/2, 2G7/2, 4G9/2 and 2H9/2 levels can accumulate electron population processes from pathway A and a nonradiative process (nRP). The radiative transition observed within 80 ns after excitation proves that the whole pathway A (including A-4 processes) has operated. However, the radiative transition on pathway B of the two-photon and four-photon processes has not been observed such as 654 nm (4F9/2 → 4I15/2) and 430 nm (4G7/2 → 4I11/2) emissions. Although the 2H9/2 energy level can accumulate electron population and produce radiative transition at 410 nm and 558 nm by A-3 and B-3 processes, no radiation transition was observed at 654 nm indicating that the B-3 and B-2 processes are ineffective. This demonstrates that pathway A is more efficient than pathway B in highly-Yb3+ doped NaYF4:Yb/Er MCs.
 | ||
| Fig. 3 (a) Schematic representation of the transition kinetics during the first 200 ns after the 976 nm ns-pulsed excitation. The purple part shows the process of establishing electron populations and the nRP. The blue part shows the all radiative transition of multiband UCL. The red part shows the main populating process of red emission enhancement. The green part shows the rise sequence of multiband UCL along with time. (b) The multiband UCL lifetimes of a single β-NaYF4:Yb/Er (60%/2%) MC. | ||
The existence of a new populating process (4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 → 2H9/2 → 2D5/2 → 4G7/2) is suggested according to the observation of 430 nm at 90 ns, which means the B-4 process has operated effectively. However, 654 nm emission was not observed at this time-scale indicating that the B-2 and B-3 processes are not efficient, which implies that pathway A in NaYF4:Yb/Er (60/2 mol%) can provide electrons to prompt the B-4 process. In NaYF4:Yb/Er (60/2 mol%) MCs, pathway A is completed first, then the B-4 process is followed, and finally the B-2 and B-3 processes are performed.
The observation of the 654 nm emission from 90 ns to 110 ns indicates that the B-2 and B-3 processes occur efficiently, which is associated with the 4I13/2 level.40 The electron population accumulation at the 4I13/2 level can prompt the B-2 (4I13/2 → 4F9/2) process and 654 nm emission (4F9/2 → 4I15/2), as well as the B-3 (4F9/2 → 2H9/2) process. Compared with pathway A, the reason for the slow operation of pathway B is the slow decay of 4I11/2 → 4I13/2 nRP, which was mentioned by Lee et al.41 The 803 nm emission (4I9/2 → 4I15/2), which occurs based on the 654 nm emission from 110 ns to 120 ns, implies that the nRP of 4F9/2 → 4I9/2 prompts the radiative transition of 4I9/2 energy level.
We have also observed distinct emissions at 456 nm (4F5/2 → 4I15/2), 443 nm (4F3/2 → 4I15/2), 617 nm (2P3/2 → 4F9/2 or 4G11/2 → 4I11/2), 640 nm (2G7/2 → 4I9/2), and 817 nm (2H9/2 → 4I9/2) from 90 ns to 200 ns. The UCL at these wavelengths appears relatively late, considering that the energy levels 4F5/2, 4F3/2, 2P3/2, 4G11/2, 2G7/2, and 2H9/2 have already accumulated the electron population before 90 ns as analyzed above. There might be two explanations for this; (1) these UC emissions are extremely weak that they cannot be detected before the moment of 90 ns; and (2) only the accumulation of a sufficient number of electron population at the corresponding emitting levels can generate the radiative transitions at these wavelengths.
We have noticed that the UCL at all the wavelengths declined significantly from 0–5 μs to 5–10 μs (except 654 nm), which showed an increasing trend in contrast (see Fig. 2b). The decline happened because the energy that the sensitizer Yb3+ can provide to the activator Er3+ decreases after excitation, and thus fewer electrons can be populated to the upper energy levels in the activator along with time. The lifetimes of multiband UCLs at different wavelengths are shown in Fig. 3b.42 In this figure, we divided the wavelengths into three groups according to the number of absorbed photons (two, three, or four), which were determined by the power of the incident laser originally.33 It shows that the lifetime of UCL decreased as the number of absorbed photons increased in lanthanide-doped systems, suggesting that the UCL lifetime relates not only to the energy level lifetime but also to the energy flow network of the whole UC system.43 On the other hand, the increase of the 654 nm emission (4F9/2 → 4I15/2) shows that more electrons were populated to 4F9/2, which can be caused by the paths relating to the processes of BET-1, BET-2, and B-2 (labeled as C-3, C-1, and C-2, respectively, in Fig. 3a). The first path is the BET-2 process, where the electrons drop from 4G11/2 to 4F9/2, while the second combines the BET-1 and B-2 processes, through which the electrons were first dropped from the energy level 4S3/2 to 4I13/2 (BET-1) and then pumped to 4F9/2 (B-2). Due to the BET-2 process, the lifetimes of the 505 nm emission and the 384 nm emission (both drops from 4G11/2) were much shorter compared to the other three-photon emissions (410 and 558 nm) as the electrons at 4G11/2 reduced (see Fig. 3b). In the two-photo group presented in Fig. 3b, the 542 nm emission (4S3/2 → 4I15/2) and 522 nm emission (2H11/2 → 4I15/2) decayed faster than the 654 nm emission, which might be due to the BET-1 process that reduced the electron population at the energy levels of 4S3/2 and 2H11/2.
To further investigate the effect of sensitizer Yb3+ on the kinetics of multiband UCL, we recorded the time evolution spectra of rising processes for single NaY(Yb)F4:Yb/Er MC doped with different concentrations of Yb3+ sensitizer, as shown in Fig. 4a–c. The spectra show that the multiband UCL appeared at ∼1 μs and ∼0.1 μs, respectively (schematic representation of the transition kinetics is shown in Fig. S3–S5, ESI†) when the Yb3+ doping concentration was low (10 mol%) and high (40, 60 and 90 mol%); and the 10% Yb3+ doping mainly presented two-photon UCL at 522 nm and 542 nm within 100 ns. After ns-pulsed excitation, the electrons in Yb3+ sensitizer ions were first populated to the energy level 2F5/2 and then dropped back to 2F7/2. Through this process, energy was transferred from the sensitizer ions to Er3+ activator ions, further populating the electrons in Er3+ to higher energy levels. Fig. 4d shows the schematics of how energy transfers within the β-NaY(Yb)F4:Yb/Er lattice for low Yb3+ doping and high Yb3+ doping. As the Yb3+ doping concentration grows, the surrounding Yb3+ ions per Er3+ became more and the energy migration is enhanced, resulting in the increase of the energy transferred from Yb3+ to Er3+. Particularly, for high Yb3+doping cases, the energy of Yb3+can be transferred to the surrounding Er3+ ions as well as the neighboring Yb3+ ions.44–46 Therefore, Er3+ ions can acquire more instantaneous energy, and thus the multiband UCL process can be speeded up when the Yb3+ doping concentration is high enough.
 | ||
| Fig. 4 The 0 moment is the starting point of the 976 nm ns-pulsed excitation. Time evolution spectrum of NaY(Yb)F4:Yb/Er (X/2 mol%) determined using the superimposed transient spectrum at the same moment. X = (a) 10, (b) 40, and (c) 90. (d) Schematic representation showing the distributing difference of ions between high-doping and low-doping in a single NaY(Yb)F4:Yb/Er MC. (e) Schematic representation showing the difference of the populating processes between high-doping and low-doping in a single NaY(Yb)F4:Yb/Er MC. | ||
Apart from the acceleration of the UC process, there are also changes in the populating process between low and high doping (Fig. 4e). Commonly, in Yb/Er systems the populating process operates through pathway A (A-1, A-2, A-3, A-4, and nRP) and pathway B (B-1, B-2, B-3, B-4 and nRP), with B-3 happening after A-4.27 But when Yb doping concentration is high, B-4 (2H9/2 → 2D5/2) happens superior to B-3 (4F9/2 → 2H9/2) because 2H9/2 accumulates abundant electrons due to the enhancement of pathway A-3 and the following nRP. This shows that pathway A can promote the operation of pathway B.
Furthermore, we have investigated the decay processes at different Yb3+ doping concentrations (as shown in Fig. 5) and found a strong red emission (at 654 nm) for high doping (Fig. 5b and c). This confirms our previous studies on 60% Yb3+ that Yb3+ ions can enhance the 654 nm emission through the processes of C-1 C-2 and C-3. As shown in Fig. 5d, for highly Yb3+ doped MCs (40 and 90 mol%), the CIE chromaticity diagram shows the almost same color variation trend. For the 90% Yb3+ doped MC, the near-white color appeared at around 500 ns and the red color appeared at around 100 μs. However, the low Yb3+ doped MC (10 mol%) initially emits green color (dominated by the two-photon UCL), and gradually includes blue-violet color (originated from the three- or four-photon UCL). As the Yb3+ concentration increases, the lifetime of multiband UCL decreases as shown in Fig. 5e, possibly owing to the energy loss of Yb3+ sensitizers at considerably high concentrations.43 The phenomena of measuring β-NaY(Yb)F4: Yb/Er (90/2 mol%) and β-NaYbF4: Er (2 mol%) MCs are similar, and relevant results are detailed in Fig. S6–S9 (ESI†).
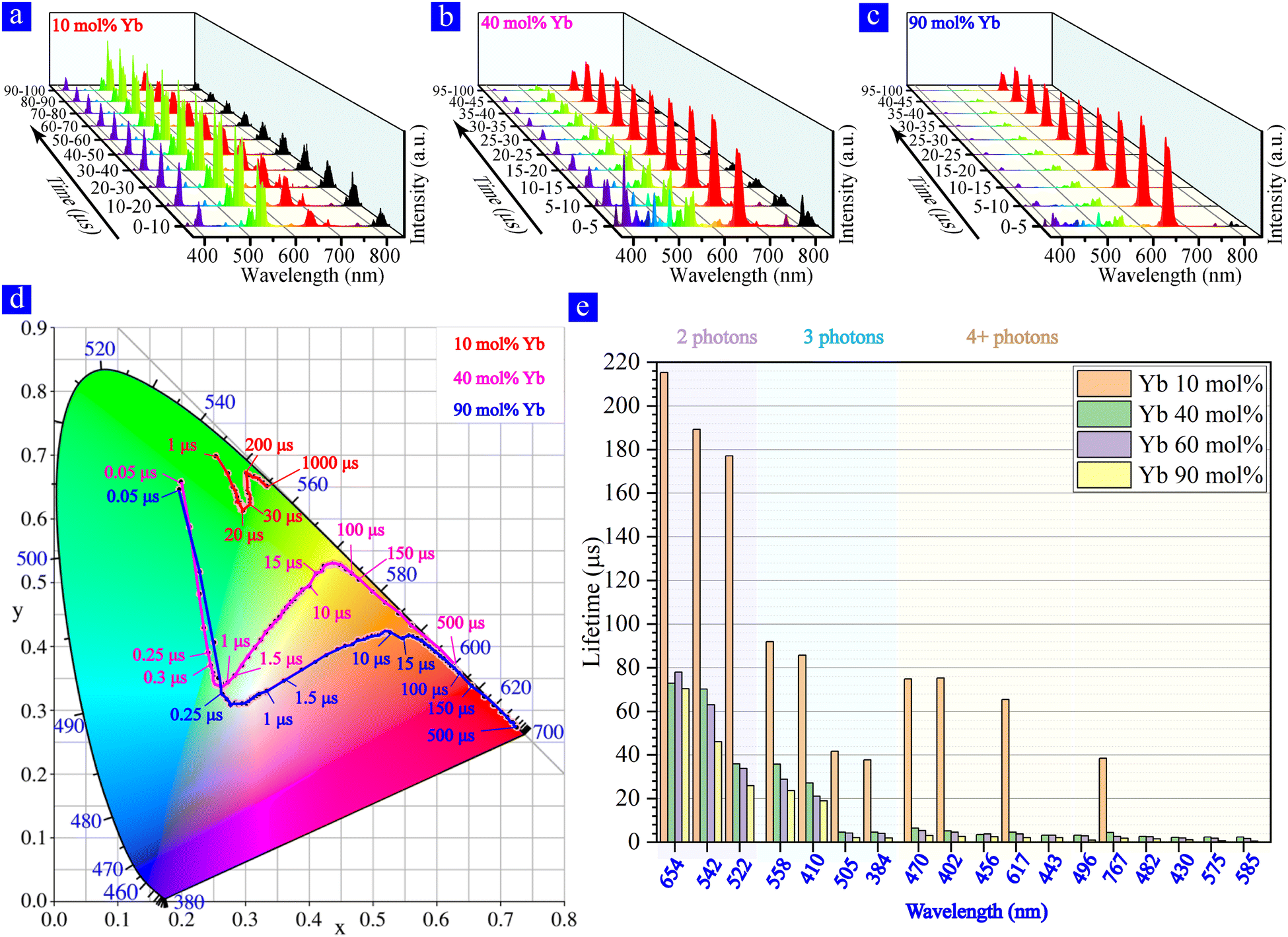 | ||
| Fig. 5 The 0 moment is the starting point of the 976 nm ns-pulsed excitation. Time evolution spectrum of NaY(Yb)F4:Yb/Er (X/2 mol%) determined using the superimposed transient spectrum at the same moment. X = (a) 10, (b) 40, and (c) 90. (d) CIE chromaticity coordinates for the multiband UCL of the single β-NaY(Yb)F4:Yb/Er MC with different Yb3+ concentrations based on the time evolution spectrum. (e) The multiband UCL lifetimes of a single β-NaY(Yb)F4:Yb/Er MC with different doping concentrations. | ||
Conclusions
In summary, we applied nanosecond time-resolved spectroscopy to an optically trapped single β-NaY(Yb)F4:Yb/Er MC varying the doping concentration of the Yb3+ sensitizer. First, we recorded the time evolution spectra of a β-NaYF4:Yb/Er (60/2 mol%) MC after ns-pulsed high-irradiance excitation, from which we calculated CIE chromaticity to show color evolution (from initially green over near-white to red). The sequences of UCL rising processes at different wavelengths are determined based on the time evolution spectra, which reveals a new 4-photon populating process (4I15/2 → 4I11/2 → 4F7/2 → 2H11/2 →4S3/2 → 2G7/2 → 4G11/2 → 2H9/2 → 2D5/2 → 4G7/2). Furthermore, we show the difference in the populating pathway between high and low doping concentrations. We found that the timescale to achieve pathway A and pathway B is one order of magnitude smaller for high doping (∼0.1 μs) compared to low doping (∼1 μs). However, limited by the BET process and the energy decay originating from the Yb3+ sensitizer, the multiband UCL lifetime has shortened, and the proportion of red-light emission keeps increasing, as the Yb3+ doping concentration increases. The result indicates that a reasonably large number of Yb3+ ions can effectively modulate the response speed of the multiband UC process, change photophysical pathways, and control energy flow to the 4F9/2 energy level leading to a huge percentage of red emission. Our findings provides a way to understand the impact of Yb3+ concentration on UC, which has potential applications in fast-response optoelectronic devices and UC displays in the future.Author contributions
H. H., M. Y. and K. H. conceived the work. M. Y., Z. X., C. G., K. H. and G. Z. provided guidance and supervision. T. Y., S. H. and W. C. characterized the upconversion microcrystals by SEM and XRD. H. H. and C. G. built the photoluminescence platform. H. H. performed the spectroscopy experiment. H. H., M. Y. and Z. X. analyzed the data and wrote the manuscript. All authors participated in the discussion and finalization of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported financially by the Science Fund for Excellent Young Scholars (2017X016), Innovation Fund for Young Scholars (19QNCXJ) and the State Key Laboratory of Laser Interaction with Matter Foundation (SKLLIM1708). We are thankful to Xi’an ruixi Biological Technology Co., Ltd for providing us with the β-NaYF4 microcrystals.Notes and references
- Z. Dong, N. Zhang, Y. Wang, J. Wu, Q. Gan and W. Li, Adv. Funct. Mater., 2019, 29, 1904453 CrossRef CAS.
- Z. Y. Yang, J. Y. Xu, P. Wang, X. J. Zhuang, A. L. Pan and L. M. Tong, Nano Lett., 2011, 11, 5085–5089 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS.
- S. Liu, J. Huang, L. Yan, N. Song, P. Zhang, J. He and B. Zhou, J. Mater. Chem. A, 2021, 9, 4007 RSC.
- Q. Tian, W. Yao, Z. Wu, J. Liu, J. Liu, W. Wu and C. Jiang, J. Mater. Chem. A, 2017, 5, 23566 RSC.
- F. Xu, Y. Sun, H. Gao, S. Jin, Z. Zhang, H. Zhang, G. Pan, M. Kang, X. Ma and Y. Mao, ACS Appl. Mater. Interfaces, 2021, 13, 2674–2684 CrossRef CAS PubMed.
- X. Wu, P. Yan, Z. Ren, Y. Wang, X. Cai, X. Li, R. Deng and G. Han, ACS Appl. Mater. Interfaces, 2019, 11, 385–393 CrossRef CAS PubMed.
- X. Liu, Y. Wang, X. Li, Z. Yi, R. Deng, L. Liang, X. Xie, D. T. B. Loong, S. Song, D. Fan, A. H. All, H. Zhang, L. Huang and X. Liu, Nat. Commun., 2017, 8, 899 CrossRef PubMed.
- J. F. Suyver, J. Grimm, M. K. van Veen, D. Biner, K. W. Krämer and H. U. Güdel, J. Lumin., 2006, 117, 1–12 CrossRef CAS.
- F. Vetrone, J.-C. Boyer, J. A. Capobianco, A. Speghini and M. Bettinelli, J. Appl. Phys., 2004, 96, 661–667 CrossRef CAS.
- B. S. Cao, Y. Y. He, L. Zhang and B. Dong, J. Lumin., 2013, 135, 128–132 CrossRef CAS.
- D. G. O'Shea, J. M. Ward, B. J. Shortt, M. Mortier, P. Féron and S. N. Chormaic, Eur. Phys. J.: Appl. Phys., 2007, 40, 181–188 CrossRef.
- G. Yao, C. Lin, Q. Meng, P. Stanley May and M. T. Berry, J. Lumin., 2015, 160, 276–281 CrossRef CAS.
- S. Han, R. Deng, X. Xie and X. Liu, Angew. Chem., Int. Ed., 2014, 53, 11702–11715 CrossRef CAS PubMed.
- G. Y. Lee, K. Jung, H. S. Jang, J. Kyhm, I. K. Han, B. Park, H. Ju, S. J. Kwon and H. Ko, Nanoscale, 2016, 8, 2071–2080 RSC.
- F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924–2929 CrossRef CAS.
- P. Yuan, Y. H. Lee, M. K. Gnanasammandhan, Z. Guan, Y. Zhang and Q. H. Xu, Nanoscale, 2012, 4, 5132–5137 RSC.
- M. Haase and H. Schafer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- J. Wang, R. Deng, M. A. MacDonald, B. Chen, J. Yuan, F. Wang, D. Chi, T. S. Hor, P. Zhang, G. Liu, Y. Han and X. Liu, Nat. Mater., 2014, 13, 157–162 CrossRef CAS PubMed.
- D. Xu, C. Liu, J. Yan, S. Yang and Y. Zhang, J. Phys. Chem., 2015, 119, 6852–6860 CrossRef CAS PubMed.
- D. Gao, X. Zhang, B. Chong, G. Xiao and D. Tian, Phys. Chem. Chem. Phys., 2017, 19, 4288–4296 RSC.
- B. Shen, S. Cheng, Y. Gu, D. Ni, Y. Gao, Q. Su, W. Feng and F. Li, Nanoscale, 2017, 9, 1964–1971 RSC.
- J. Zhao, D. Jin, E. P. Schartner, Y. Lu, Y. Liu, A. V. Zvyagin, L. Zhang, J. M. Dawes, P. Xi, J. A. Piper, E. M. Goldys and T. M. Monro, Nat. Nanotechnol., 2013, 8, 729–734 CrossRef CAS PubMed.
- M. Yuan, R. Wang, C. Zhang, Z. Yang, W. Cui, X. Yang, N. Xiao, H. Wang and X. Xu, J. Mater. Chem. C, 2018, 6, 10226–10232 RSC.
- I. D. Stoev, A. Caciagli, Z. Xing and E. Eiser, Conference on Optical Trapping and Optical Micromanipulation XV, San Diego, CA, 2018, vol. 10723 DOI:10.1117/12.2318019.
- R. W. Bowman and M. J. Padgett, Rep. Prog. Phys., 2013, 76, 026401 CrossRef PubMed.
- H. Huang, M. Yuan, S. Hu, Y. Zhong, W. Cui, C. Guo, C. Song, G. Zhao and K. Han, J. Mater. Chem. C, 2022, 10, 9208–9215 RSC.
- P. Rodriguez-Sevilla, Y. Zhang, N. de Sousa, M. I. Marques, F. Sanz-Rodriguez, D. Jaque, X. Liu and P. Haro-Gonzalez, Nano Lett., 2016, 16, 8005–8014 CrossRef CAS PubMed.
- P. Rodriguez-Sevilla, L. Labrador-Paez, D. Wawrzynczyk, M. Nyk, M. Samoc, A. K. Kar, M. D. Mackenzie, L. Paterson, D. Jaque and P. Haro-Gonzalez, Nanoscale, 2016, 8, 300–308 RSC.
- J. Kim, R. Chacon, Z. Wang, E. Larquet, K. Lahlil, A. Leray, G. Colas-des-Francs, J. Kim and T. Gacoin, Nat. Commun., 2021, 12, 1943 CrossRef CAS.
- P. Rodriguez-Sevilla, H. Rodriguez-Rodriguez, M. Pedroni, A. Speghini, M. Bettinelli, J. G. Sole, D. Jaque and P. Haro-Gonzalez, Nano Lett., 2015, 15, 5068–5074 CrossRef CAS.
- C. Renero-Lecuna, R. Martín-Rodríguez and R. Valiente, Chem. Mater., 2011, 23, 3442–3448 CrossRef CAS.
- F. Frenzel, C. Würth, O. Dukhno, F. Przybilla, L. M. Wiesholler, V. Muhr, T. Hirsch, Y. Mély and U. Resch-Genger, Nano Res., 2021, 14, 4107–4115 CrossRef CAS.
- Z. X. Cheng, S. J. Zhang, F. Song, H. C. Guo, J. R. Han and H. C. Chen, J. Phys. Chem. Solids, 2002, 63, 2011–2017 CrossRef CAS.
- D. K. Sardar, J. B. Gruber, B. Zandi, J. A. Hutchinson and C. W. Trussell, J. Appl. Phys., 2003, 93, 2041–2046 CrossRef CAS.
- X. Y. Chen, E. Ma and G. K. Liu, J. Phys. Chem. C, 2007, 111, 10404–10411 CrossRef CAS.
- R. T. Wegh, E. V. D. Van Loef, G. W. Burdick and A. Meijerink, Mol. Phys., 2003, 101, 1047–1056 CrossRef CAS.
- T. Jung, H. L. Jo, S. H. Nam, B. Yoo, Y. Cho, J. Kim, H. M. Kim, T. Hyeon, Y. D. Suh, H. Lee and K. T. Lee, Phys. Chem. Chem. Phys., 2015, 17, 13201–13205 RSC.
- R. B. Anderson, S. J. Smith, P. S. May and M. T. Berry, J. Phys. Chem. Lett., 2014, 5, 36–42 CrossRef CAS.
- K. Shin, T. Jung, E. Lee, G. Lee, Y. Goh, J. Heo, M. Jung, E. J. Jo, H. Lee, M. G. Kim and K. T. Lee, Phys. Chem. Chem. Phys., 2017, 19, 9739–9744 RSC.
- E. Lee, M. Jung, Y. Han, G. Lee, K. Shin, H. Lee and K. T. Lee, J. Phys. Chem. C, 2017, 121, 21073–21079 CrossRef CAS.
- M. Inokuti and F. Hirayama, J. Chem. Phys., 1965, 43, 1978 CrossRef CAS.
- A. Teitelboim, B. Tian, D. J. Garfield, A. Fernandez-Bravo, A. C. Gotlin, P. J. Schuck, B. E. Cohen and E. M. Chan, J. Phys. Chem. C, 2019, 123, 2678–2689 CrossRef CAS.
- Y. Liang, Z. Zhu, S. Qiao, X. Guo, R. Pu, H. Tang, H. Liu, H. Dong, T. Peng, L. D. Sun, J. Widengren and Q. Zhan, Nat. Nanotechnol., 2022, 17, 524–530 CrossRef CAS PubMed.
- B. Zhou, L. Yan, J. Huang, X. Liu, L. Tao and Q. Zhang, Nat. Photonics, 2020, 14, 760–766 CrossRef CAS.
- J. Zuo, D. Sun, L. Tu, Y. Wu, Y. Cao, B. Xue, Y. Zhang, Y. Chang, X. Liu, X. Kong, W. J. Buma, E. J. Meijer and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3054–3058 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03013f |
| ‡ These authors contributed equally to this work and should be considered co-first authors. |
| This journal is © The Royal Society of Chemistry 2022 |