
Sequential oxidation of sulfur annulated perylenediimide: an efficient strategy to generate ultra-stable radical anions and dianions†
Aasif
Khan‡
a,
Ashutosh
Agrahari‡
 a,
Supriya
Saha
b and
Apurba Lal
Koner
*a
a,
Supriya
Saha
b and
Apurba Lal
Koner
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-pass Road, Bhauri, Bhopal 462066, Madhya Pradesh, India. E-mail: akoner@iiserb.ac.in
bAdvanced Computation and Data Sciences Division, CSIR-North East Institute of Science and Technology, Jorhat 785006, Assam, India
First published on 7th September 2022
Abstract
Sulfur annulated PDI (SAN-PDI) has been oxidized to three sequential levels (from sulfide/disulfide to sulfoxide/sulfone) using meta-chloroperoxybenzoic acid (mCPBA) to achieve electron-deficient PDI derivatives (1, 2, and 3). Each oxidation level leads to a shift towards a more positive reduction potential and thus increases the electron affinity. These PDI derivatives 1, 2, and 3 have been employed as precursors for the efficient generation of radical anions and dianions. The radical anion formation has been investigated using UV-Vis-NIR absorption and validated with EPR spectroscopy. The generated reduced species were found to be exceptionally stable (t1/2 > 5 years) under ambient conditions. The compound 3 shows radical anion generation in a solid-state thin-film upon exposure to amine vapors. Furthermore, the oxidized products have been optimized using DFT calculations to obtain their physiochemical properties. Hence, the extraordinary stability of radical anions and dianions of oxidized SAN-PDI under ambient conditions will make them a potential candidate for molecular magnetism, catalysis, and photovoltaics applications.
Introduction
Organic radicals and their anions have attracted outstanding attention in recent years for their applications in molecular magnetism,1 spintronics,2 photovoltaics,3 catalysis,4,5etc.6 One of the major challenges in dealing with such species is their high intrinsic reactivity making them prone to dimerization and reoxidation upon exposure to air. Hence, radical anion/dianion species are known to have poor ambient stability. Molecules with large π-surface with low lying energy of the lowest unoccupied molecular orbital (LUMO) are one of the ways to suppress undesired reactivity. In this regard, perylene diimides (PDIs) have been shown as one of the potential candidates to be explored due to their inherent electron-deficient character.7–10 PDI is best known for its near unity emission quantum yield, photostability, and chemical robustness, which make it suitable for various applications.11–17 Thanks to the synthetic versatility, the properties of PDI can be tuned by functionalizing the perylene core at the imide, ortho, and bay positions (Scheme 1). Alterations at the imide position by incorporating sterically demanding alkyl or aryl groups affect the solubility of PDIs, whereas modifications at the bay- and ortho-positions have an impact on the electronic and optical properties due to direct attachment to the perylene core.18 Delocalized π-electrons along with the imide groups lower down the LUMO energy of PDIs. Therefore, these molecules have been explored as precursors to generate stable radical anions.19 However, the ease of synthetic functionalization in PDIs makes it possible to introduce other electron-withdrawing groups to the perylene core to achieve more electron-deficient PDIs. With low-lying LUMOs, such PDIs can efficiently capture electrons and generate stable radical anions spontaneously.20 Introduction of conventional electron-withdrawing groups such as nitrile (–CN), nitro (–NO2), fluoride (–F), bromide (–Br), etc. to π-conjugated scaffolds has been shown to fulfil this goal.4,7,10,21–25 Earlier reports have shown the aforementioned strategy to be successful. In fact, a visible-light-triggered generation of stable radical anions was shown in the case of nitro substituted PDIs.10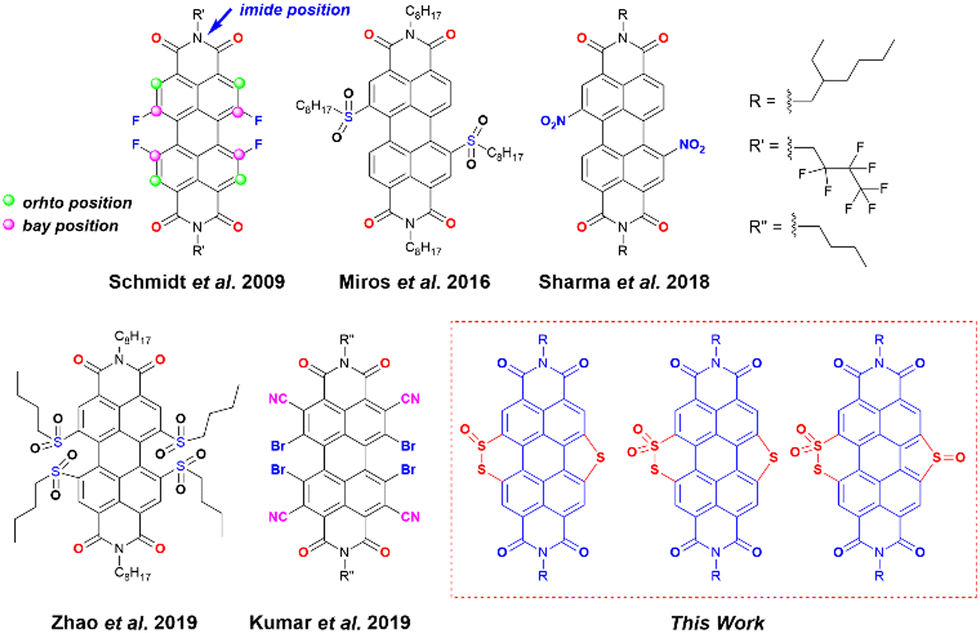 | ||
| Scheme 1 The structure of previously reported electron-deficient PDIs used for the generation of a stable radical anion and designed PDI molecules in this work. | ||
Kumar et al. showed the synthesis of an ortho-nitrile substituted PDI as a precursor of the stable radical anion.7 Apart from introducing the traditional electron-withdrawing groups, heteroatom (such as N, S, and Se) functionalization at the bay-position of PDI is a strategic step toward fine-tuning their electronic structures and energy levels.26–30 Matile and co-workers have shown that the LUMO energy of the naphthalene diimides (NDIs) and PDIs with sulfur bridges attached to the core can be lowered by simple sulfur oxidation.5,31,32 Zhou et al. showed efficient tuning of the LUMO energy of sulfur-containing PDIs via sulfur oxidation having low lying LUMOs (lowest −4.75 eV) with sulfones and sulfoxides attached at the bay positions.33 Chen et al. have reported sulfur annulation of PDI with the LUMO energy −3.46 eV and twisting in the perylene core, which made it possible to apply it in an organic field-effect transistor (OFET).18 Li et al. have applied similar annulated molecules for Organic Thin-Film Transistor (OTFT) applications.34 Nonetheless, the generation and characterization of radical anions from these sulfur annulated PDIs and their corresponding oxidized products remain unexplored.
Motivated by the findings mentioned earlier, we hypothesize the oxidation of sulfur atoms present in sulfur annulated PDI (SAN-PDI) towards modulation of the LUMO energy. In this work, we have demonstrated an elegant approach for the generation of super-stable radical anions and dianions. A simple as well as efficient oxidation of sulfur atoms of sulfide/disulfide of SAN-PDI using mCPBA results in sulfoxide/sulfone annulated PDI derivatives 1O-SAN-PDI (1), 2O-SAN-PDI (2), and 3O-SAN-PDI (3). To understand the effect of sulfur oxidation on the HOMO-LUMO energy levels, the structural optimization and density functional theory (DFT) calculations were performed. Photophysical and electrochemical properties were investigated for all three derivatives 1, 2, and 3. Furthermore, we investigated the efficacy of these electron-deficient precursors to generate radical anions via UV-Vis-NIR absorption spectroscopy. These electron-poor oxidized molecules were able to accept electrons from traces of amine present in N,N-dimethylformamide (DMF) solvent in an efficient and reversible manner. Moreover, the radical anions and dianions can also be generated in other solvents such as chloroform and THF in the presence of externally added amines like triethylamine (TEA) and diethylamine (DEA). The formation of the radical anions was characterized and validated with EPR spectroscopy. Compound 3 was also employed as a radical anion precursor for an efficient generation of radical anions in a solid-state thin-film upon exposure to amine vapors.
Results and discussion
The synthetic route for the designed compounds 1, 2, and 3 is shown in Scheme 2. The parent PDI (N,N-bis(2-ethylhexyl)perylenediimide) was nitrated to get dinitro-PDI (1,7-dinitro PDI and 1,6-dinitro PDI) using concentrated nitric acid and cerium(IV) ammonium nitrate in dichloromethane (DCM) at room temperature.10 Without further purification of 1,7-dinitro PDI and 1,6-dinitro PDI, we progressed to synthesize the SAN-PDI by treating dinitro-PDI with sulfur powder in N-methylpyrrolidone (NMP) at 190 °C for 4 h (as shown in Scheme S1, ESI†).18 Afterward, to oxidize the sulfur atoms of SAN-PDI to sulfoxide/sulfone, it was treated with mCPBA in DCM to get the final compounds 1, 2, and 3 (Scheme 2). The reaction conditions, yields for compounds 1, 2, and 3, and details of the synthetic procedures are given in the ESI† (see Table S1 in ESI†). Oxidation started at the most nucleophilic sulfur i.e., one of the sulfurs in the disulfide region (confirmed by DFT calculation, vide infra), and extended subsequently. Compounds 1–3 were characterized by both NMR spectroscopy (1H NMR, 13C[1H] NMR), and atmospheric-pressure chemical ionization high resolution mass spectrometry (APCI-HRMS). The compounds 4, 5, and 6 (shown in faded color in Scheme 2) were characterized only by mass spectrometry from the reaction mixture. We were not able to isolate them in pure form even after rigorous efforts. In addition to that, significant efforts were made to synthesize 4, 5, and 6 using other strong oxidizing agents such as H5IO6/CrO3, and hydrogen peroxide (H2O2) which were proved to be challenging for this oxidation step due to their poor solubility in DCM.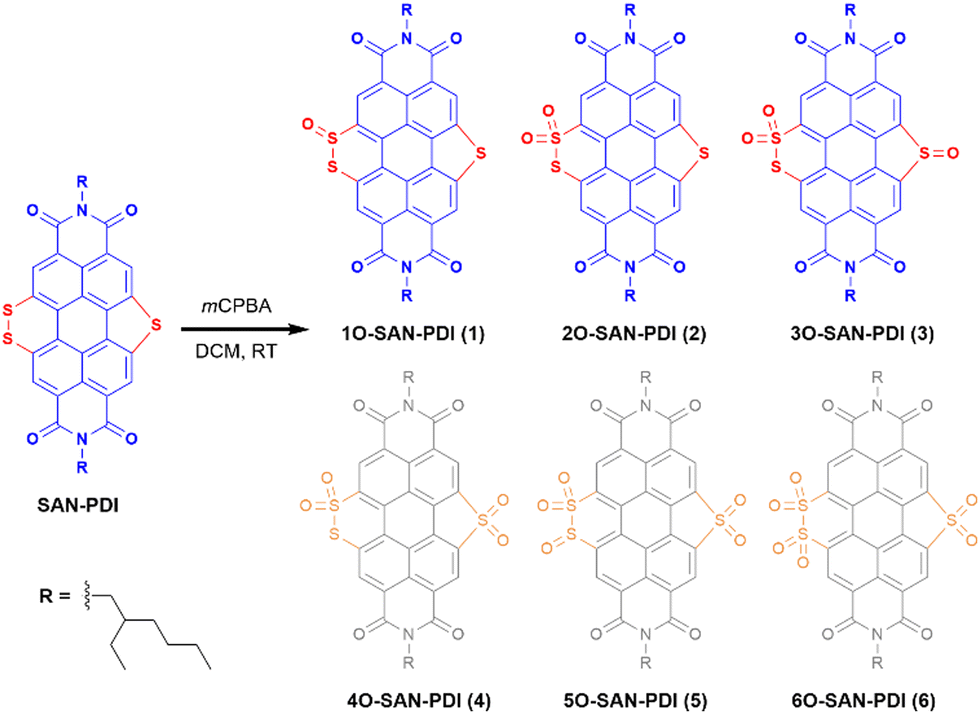 | ||
| Scheme 2 Synthesis of 1O-SAN-PDI (1), 2O-SAN-PDI (2), and 3O-SAN-PDI (3) [the molecules 1, 2, and 3 shown in blue color were isolated and fully characterized spectroscopically while molecules 4, 5, and 6 shown in faded color were not isolated but characterized by mass spectrometry in the reaction mixture]. | ||
To gain an insight into the electronic states and geometric structures of all the oxidized products of SAN-PDI, density functional theory (DFT) calculations were performed with Becke's three-parameter hybrid exchange functional35 and the Lee–Yang–Parr correlation functional (B3LYP).36,37 The 6-31G(d) basis sets were used for all involved atoms. All the optimization calculations were performed with the Gaussian 16 (G16)38 suite of programs. The optimization of all the molecules was performed in DCM solvent. The frontier orbital distributions of the optimized structures, as well as calculated values of the HOMO and LUMO energy levels of SAN-PDI, 1, 2, and 3, have been shown in Fig. 1. The HOMO and LUMO energy of SAN-PDI was found to be −5.90 eV and −3.56 eV respectively. As evident from the DFT calculated LUMO energy levels, each sequential oxidation of SAN-PDI has lowered the LUMO level in an increasing order while the bandgap does not change significantly. Structural twisting in the perylene core in SAN-PDI due to sulfur annulation has been observed to be 5.18°. Oxidation at sulfur doesn’t seem to affect this twisting much suggesting that it only assists in lowering the LUMO energy level (Fig. S1, ESI†).
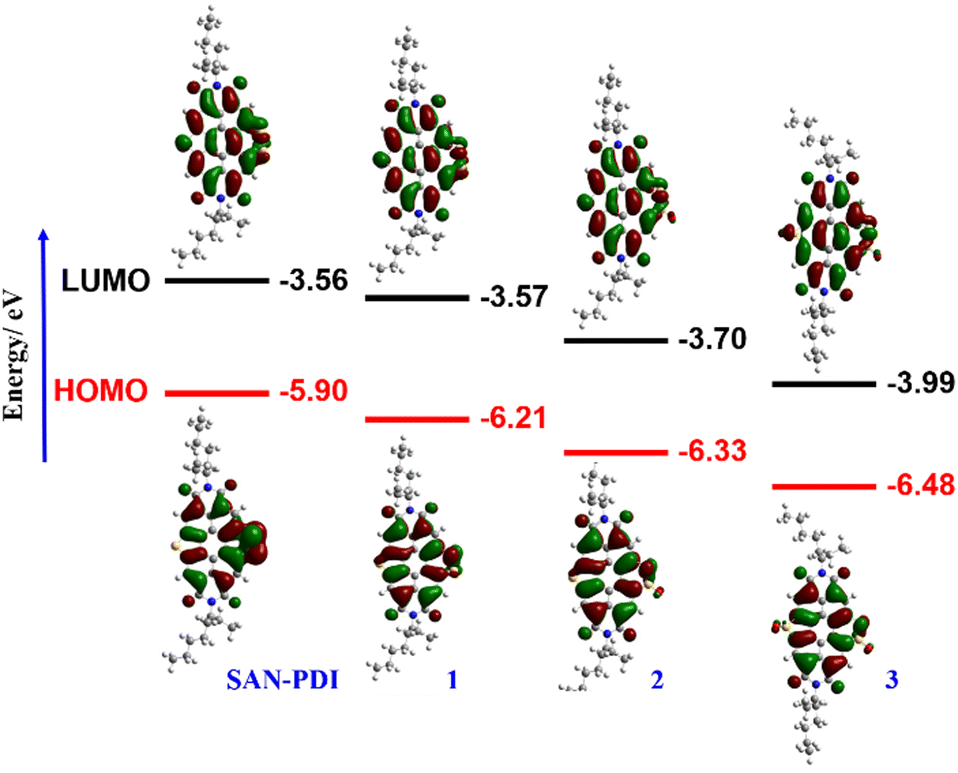 | ||
| Fig. 1 Sequential oxidation of SAN-PDI leads to a lowering of the energy levels. FMO pictures of SAN-PDI, 1, 2, and 3 obtained from density functional theory (DFT) calculations [basis set: B3LYP/6-31G(d)]. | ||
Since oxidation of sulfur atoms of SAN-PDI could afford different isomers corresponding to isolated compounds 1, 2, and 3 which are quite challenging to distinguish using 1H NMR spectroscopy only, to elucidate the structure of the isolated isomers, we have considered energy optimization of different isomers using DFT calculations to find the energetically most stable structure. Furthermore, 1H NMR spectroscopy, mass spectrometry, and the possible reactivity of sulfur atoms of SAN-PDI were also considered for structure elucidation. A detailed analysis of isolated isomers has been shown in the ESI† (see Scheme S2 and Table S2).
After synthesis and characterization of the designed compounds by NMR and mass spectrometry (see ESI† for details), the optical purity of compounds 2 and 3 was also verified from superimposable excitation and absorption spectra in DCM (Fig. S2 and S3, respectively, ESI†). Later, we performed concentration-dependent UV-Vis measurements. The absorption spectra in DCM solvent for SAN-PDI, 1, 2, and 3 show a linear relationship with the absorbance up to 25 μM suggesting that there is no aggregation up to this concentration (see Fig. S4–S7, ESI†). Hence, we decided on 10 μM as a working concentration for further spectroscopic experiments. First, we recorded the absorption and emission spectra of the compounds SAN-PDI, 1, 2, and 3 (10 μM) in DCM, as shown in Fig. 2. The absorption spectra of SAN-PDI exhibit broad absorption bands covering most of the visible region with characteristic peaks for the perylene core in the 400 to 500 nm region, while the broad peak nearby 600 nm is attributed to intramolecular charge transfer (ICT) from the sulfur to the perylene core.18,39 On the other hand, the UV-Vis spectra of 1, 2, and 3 show a narrower but bathochromic shift of the absorption maxima with respect to SAN-PDI. The disappearance of the ICT band in the absorption spectra of 1, 2, and 3 is evident (Fig. 2a), indicating the diminished ICT property from sulfur atoms to the perylene core because of their oxidation. The fluorescence emission intensity of SAN-PDI was found to be much less (Fig. 2c), which made it difficult to measure the emission quantum yield and excited-state lifetime. Single level oxidation in compound 1 did not show an observable change in the fluorescence emission intensity, but interestingly, 2 and 3 show a significant increase in emission intensity and this was reflected in their determined absolute fluorescence quantum yield too (Table 1). Such observation in the cases of 2 and 3 is due to the decrease in the photoinduced intramolecular electron transfer (PIET) from the sulfur to the perylene core.18 The fluorescence lifetime measurements of 2 and 3 in DCM solvent reveal an average lifetime of 1.6 ns and 8.0 ns, respectively (see Fig. S8, ESI† and Table 1).
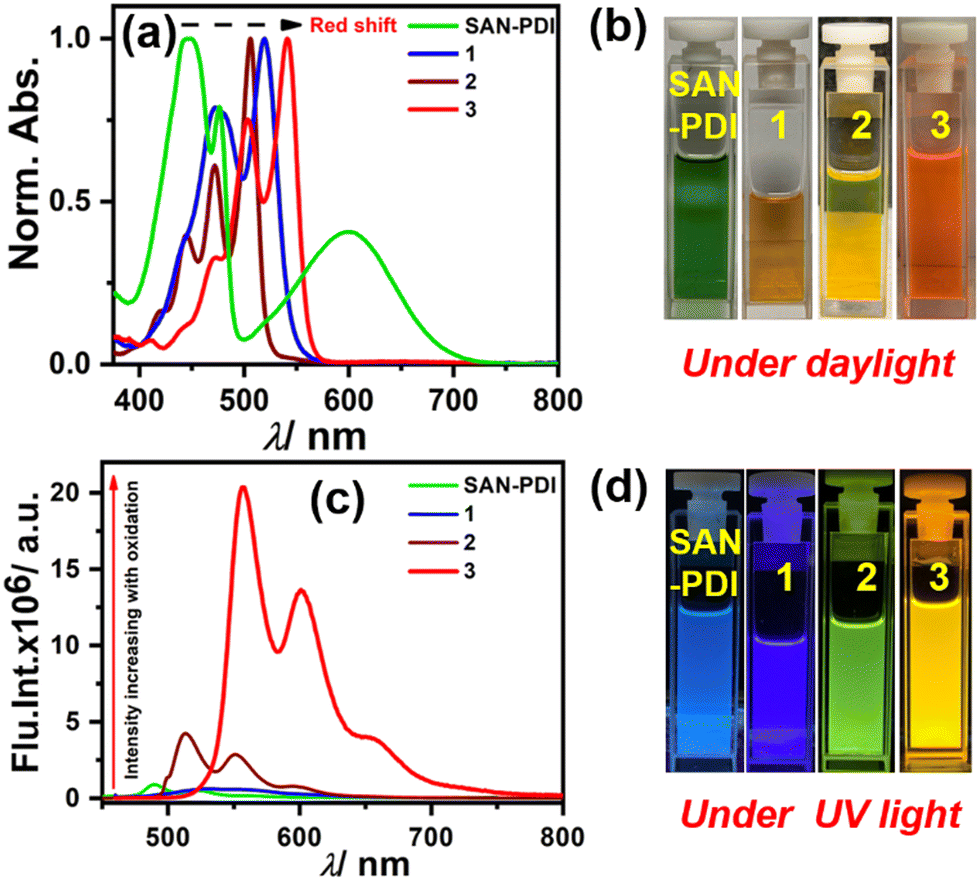 | ||
| Fig. 2 UV-Vis absorption (a) and emission (c) of compounds 1–3 (10 μM) in DCM solvent; (b) and (d) cuvette pictures of the molecules under daylight and UV light (365 nm), respectively. | ||
| λ maxabs/nm (ε/M−1 cm−1)a |
λ
maxem![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/nm a/nm |
SS/nm | Φ F | τ avg /ns |
E
Red.11/2b [V] |
E
Red.21/2b [V] |
|
|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2, absolute quantum yield determined by an integrating sphere system within ±0.1% error. b Measured in chloroform, nd not measured due to very low fluorescence emission intensity, SS: Stokes Shift. | |||||||
| SAN-PDI | 446 (13,125) | 490 | 44 | nd | nd | −0.57 | −0.80 |
| 1 | 521 (10,619) | 531 | 10 | 0.12 | nd | −0.47 | −0.72 |
| 2 | 507 (26,572) | 514 | 7 | 0.25 | 1.6 | −0.38 | −0.67 |
| 3 | 542 (11,925) | 558 | 16 | 0.83 | 8.0 | −0.01 | −0.33 |
After the primary spectroscopic studies of the synthesized molecules, their efficiency to generate the respective radical anions was investigated. In this context, single-electron transfer from an electron-rich species to the electron-deficient precursor molecule is required. We started with DMF as a solvent for the reduction of each molecule in the absence of light at room temperature.10 A trace amount of dimethylamine present in DMF acts as an electron-rich species, which can reduce 1, 2, and 3 into the respective radical anions. We have also tried some other amines such as diethylamine (DEA) and triethylamine (TEA) as a reducing agents for the generation of the radical anions.
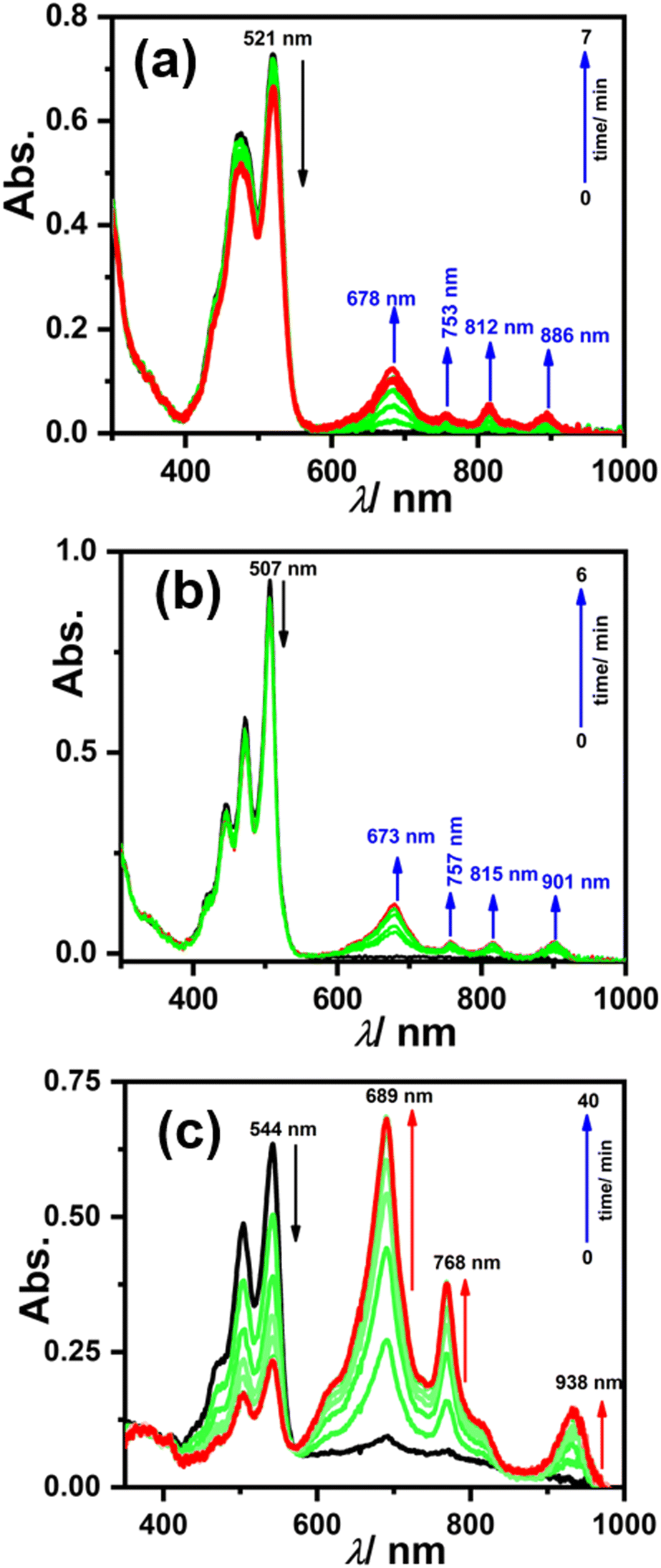
To generate the radical anion, we started with compound 3 since it is the most oxidized and thus most electron-deficient, among all (1, 2, and 3). When 10 μM of compound 3 was added to DMF solvent under the dark condition at room temperature (298 K), an immediate visible color change from yellowish to purple was observed which finally turned almost colorless and became non-fluorescent (shown in inset Fig. 3b). This change was monitored using UV-Vis-NIR absorption spectroscopy at different time intervals (Fig. 3a). New vibrational bands in the Vis-NIR region at 689 nm, 768 nm, and 938 nm were observed in the absorption spectra. The peaks corresponding to the perylene core between 400 and 600 nm disappeared completely. These absorption bands associated with the newly-formed species were increased exponentially up to 30 minutes and saturated thereafter (see Fig. 3b). This spectral change took place with a very clear isosbestic point at 570 nm indicating the transformation of 3 to a new species. Electron paramagnetic resonance (EPR) spectroscopy confirmed the identity of the newly generated species as a radical anion with a g-value of 2.002 (Fig. 3d) corresponding to a free electron.40 Similar spectral features were also observed when 3 was treated with 100 μM TEA in dry CHCl3 (Fig. 3c) and DEA in dry THF (both in the dark) (Fig. S9, ESI†). This confirms that radical anions can be generated using amines like TEA and DEA in solvents other than DMF as well. Similarly, compounds 1 and 2 were also taken in DMF solvent and monitored using UV-Vis-NIR absorption spectroscopy (Fig. 4 and Fig. S10, respectively, ESI†). A significant generation of radical anions was not observed in identical conditions; this is likely because of the comparatively higher LUMO energy, associated with the low level of oxidation making it less electron deficient than 3. Hence, the amount of trace dimethylamine present in DMF is not sufficient to generate radical anions (Fig. 4a and Fig. S10a, ESI† for 1 and 2, respectively). However, when the same solutions of 1 and 2 in DMF were exposed to visible light, the bands at 698, 812, and 886 nm in the case of 1 (Fig. 4b) and 683, 815, and 901 nm for 2 were observed while peaks corresponding to the perylene core between 400–600 nm disappeared Fig. S10b, ESI†). The addition of external amine (e.g., TEA) in DMF with visible light exposure for just 15 seconds proved to be even more efficient for radical anion generation for 1 (see Fig. 4c) and 2 (see Fig. S10c, ESI†). The presence of a radical anion for 1 (see Fig. 4d) and 2 (see Fig. S10d, ESI†) was confirmed with EPR spectroscopy with a g-value of 2.001 and 2.002, respectively. Longer exposure (10–15 min) of visible light resulted in the gradual disappearance of the absorption bands centered at 886 nm and 812 nm for 1˙− while 901 nm and 815 nm for 2˙−. At the same time, a new prominent peak appears at 698 nm and 683 nm for 1 and 2, respectively. This change appears to be due to further reduction of radical anions to their respective dianion (see Fig. S11 and S12, ESI†).41 This observation is consistent with the cyclic voltammetry (CV) results (vide infra), which show two reduction potentials corresponding to the radical anion and dianion.
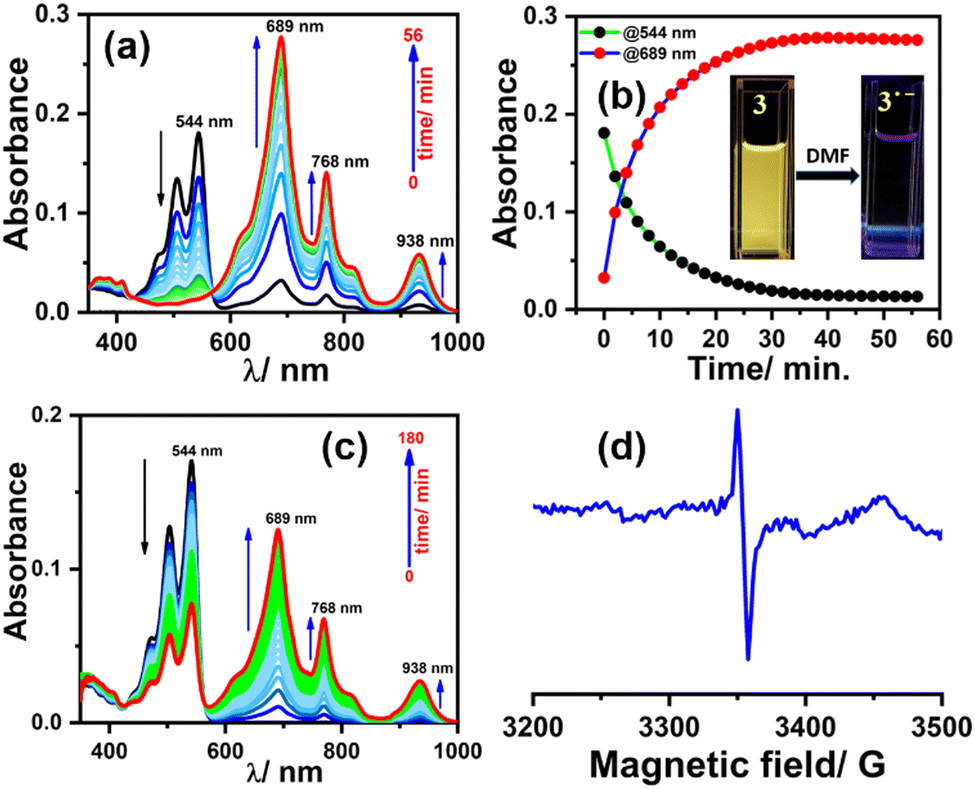 | ||
| Fig. 3 (a) Radical anion generation monitored using UV-Vis-NIR absorption spectroscopy with the electronic absorption spectra for 3 (10 μM) in DMF only in the dark, (b) kinetics plot of change in absorbance at 544 nm and 689 nm with time, and inset showing the fluorescence color change under UV illumination from yellow to colorless due to radical anion formation, (c) in CHCl3 solvent with 100 μM triethylamine (TEA) in the dark, and (d) confirmation of the radical anion by the EPR spectrum. | ||
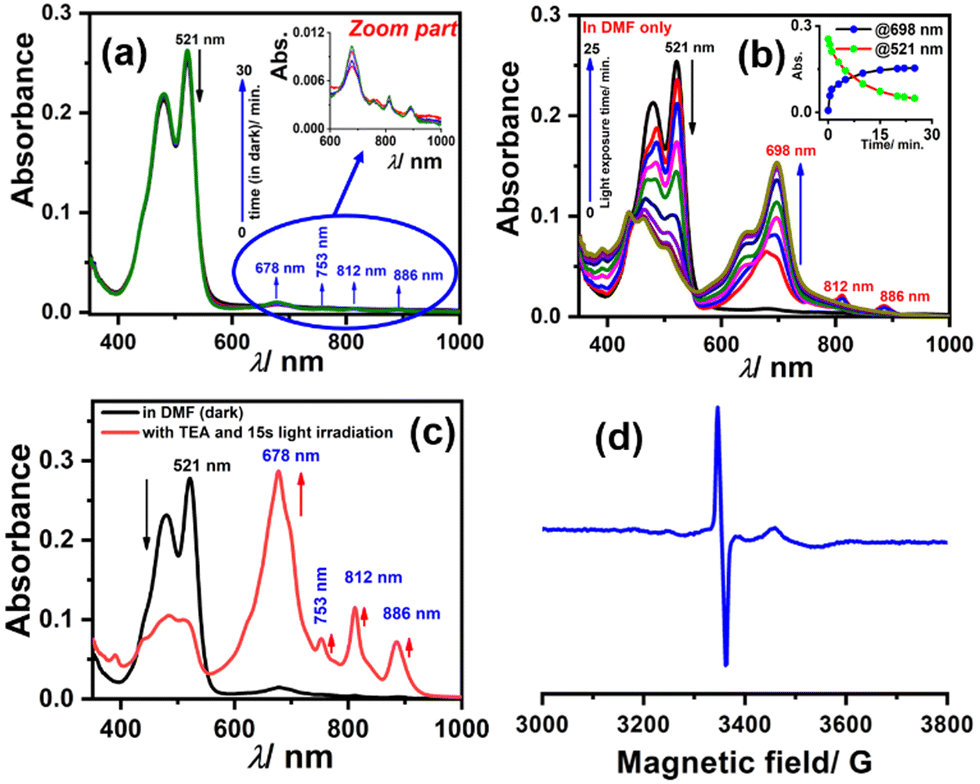 | ||
| Fig. 4 Radical anion generation monitored using UV-Vis-NIR absorption spectroscopy; electronic absorption spectra for 1 (10 μM) in DMF only (a) in the dark, (b) upon visible light irradiation and (c) with the addition of external TEA and visible light irradiation using a photoreactor (lamp intensity ∼960 l× measured by lux meter and wavelength range 400–700 nm) for just 15 seconds, and (d) confirmation of the radical anion by the EPR spectrum. | ||
To investigate the redox properties of the synthesized molecules and elucidate the effect of each sequential oxidation of SAN-PDI, we performed the CV experiment. The CV measurements were carried out in dry CHCl3 solvent with respect to the saturated calomel electrode (SCE) and tetrabutylammonium hexafluorophosphate as a supporting electrolyte. The cyclic voltammograms of SAN-PDI (parent molecule), 1, 2, and 3 are similar, which show two reversible reductions and no oxidation (see Fig. 5). The first reduction potential of SAN-PDI was found at −0.57 V vs. SCE while in the case of molecules 1, 2, and 3 it was observed at −0.47 V, −0.38 V, and −0.01 V, respectively. These reduction potential values indicate that upon each level of oxidation of SAN-PDI the reduction potential shifted from more negative potential to less negative potential. This reveals that the electron-accepting capability of the oxidized molecules (1, 2, and 3) increases in comparison to SAN-PDI upon the increasing level of oxidation. Thus, the order of electron affinity is 3 ≫ 2 > 1 > SAN-PDI.
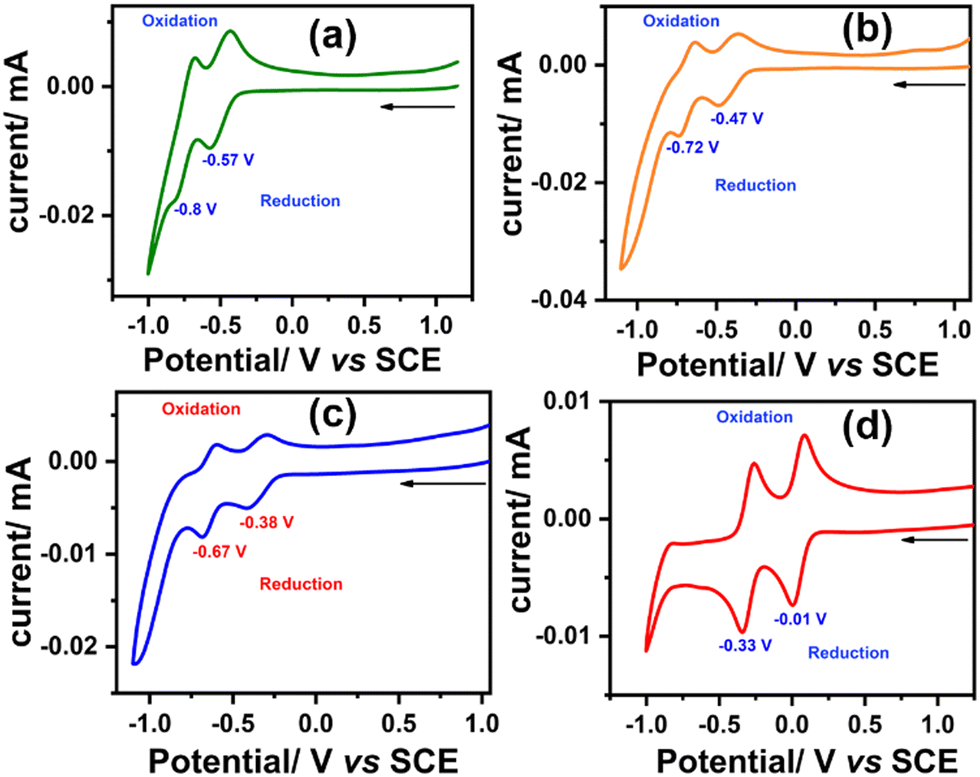 | ||
| Fig. 5 Cyclic voltammograms of SAN-PDI (a), 1 (b), 2 (c), and 3 (d), respectively, in dry CHCl3 solution at 100 mV s−1 scan rate with respect to saturated calomel electrode (SCE) with tetrabutylammonium hexafluorophosphate as the supporting electrolyte. | ||
To generate the radical anion electrochemically, we carried out a spectro-electrochemistry experiment (see Fig. 6). Upon applying the potential of −0.47 V, −0.38 V, and −0.01 V associated with the first reduction potential for molecules 1, 2, and 3, respectively, the new absorption bands in the NIR region (600–1000 nm) were found to be increasing over time while the absorption bands between 400 and 600 nm corresponding to neutral molecules 1, 2, and 3 decreased. These spectral changes were exactly similar to those observed earlier with (photo)reduction in the presence of amine (vide supra) indicating the formation of the radical anion.
 | ||
| Fig. 6 Spectro-electrochemistry data of the molecule 1 (a), 2 (b), and 3 (c), respectively, in dry CHCl3 solution with tetrabutylammonium hexafluorophosphate as a supporting electrolyte. (Potential applied: −0.47 V for 1, −0.38 V for 2 and −0.01 V for 3). | ||
To check the reversibility of reduced species as reflected from the CV experiment showing two reversible reductions, we dissolved 3 in DMF, which immediately started to generate a radical anion and shows their characteristic absorption bands (as shown in Fig. 7a red spectrum). But upon the addition of 100 equivalents of strong oxidant i.e., nitrosonium tetrafluoroborate (NOBF4), the generated radical anion reverts to neutral species 3, which can be seen in the spectral changes (see Fig. 7a blue spectrum). Similarly, the solution of 3 in DMF upon exposure to visible light for just 10 s results in an almost complete generation of the radical anion, and when NOBF4 was added to that the generated radical anion again reverted to its neutral state (as shown in Fig. 7b). These results validate that the reduction of 3 is completely reversible in nature.
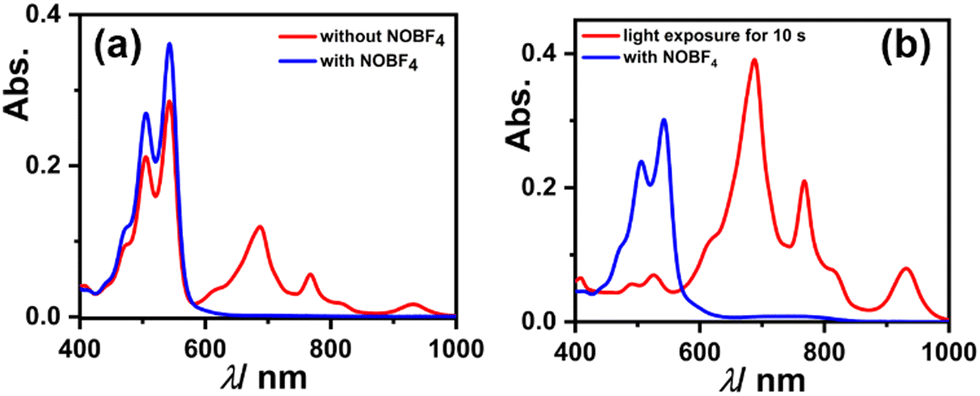 | ||
| Fig. 7 UV-Vis-NIR absorption spectra showing reversibility of the reduced species of 3 using NOBF4 (a) in the dark and (b) with visible light exposure. | ||
We monitored the stability of radical anions/dianions using UV-Vis-NIR absorption spectroscopy. Initially, the absorption bands for radical anions 1, 2, and 3 were observed, which subsequently converted to the characteristic peaks corresponding to two electron reduced dianion species, which is plausible considering the cyclic voltammetry experiment (Fig. 8).41,42 To our surprise, the spectral features remained unchanged for dianion 1, 2, and 3 up to several months to years under ambient conditions (air and moisture). Interestingly, even after several months, the same DMF solution of 1, 2, and 3 also showed an EPR signal, which indicates the equilibrium between the radical anion and dianion (Fig. S13, ESI†). This appreciable stability is attributed to the electron-deficient nature along with the large π-conjugated surface of the perylene core providing delocalization throughout the surface. The half-life time (t1/2) was also calculated from the change in absorbance of the dianion over a number of days (see the calculation of t1/2 in the ESI† and inset of Fig. 8) which was found to be t1/2 > 5 years in the case of each dianion. PDI-based dianions are shown to be emissive in nature as shown recently.12 Predictably, all three dianions of 1, 2, and 3 were found to be far-red emissive with an emission maxima centered around 700 nm (see Fig. S14, ESI†). The lifetime decay shows that the dianions have ca. 5 ns excited state lifetime (see Fig. S15 and Table S3, ESI†).
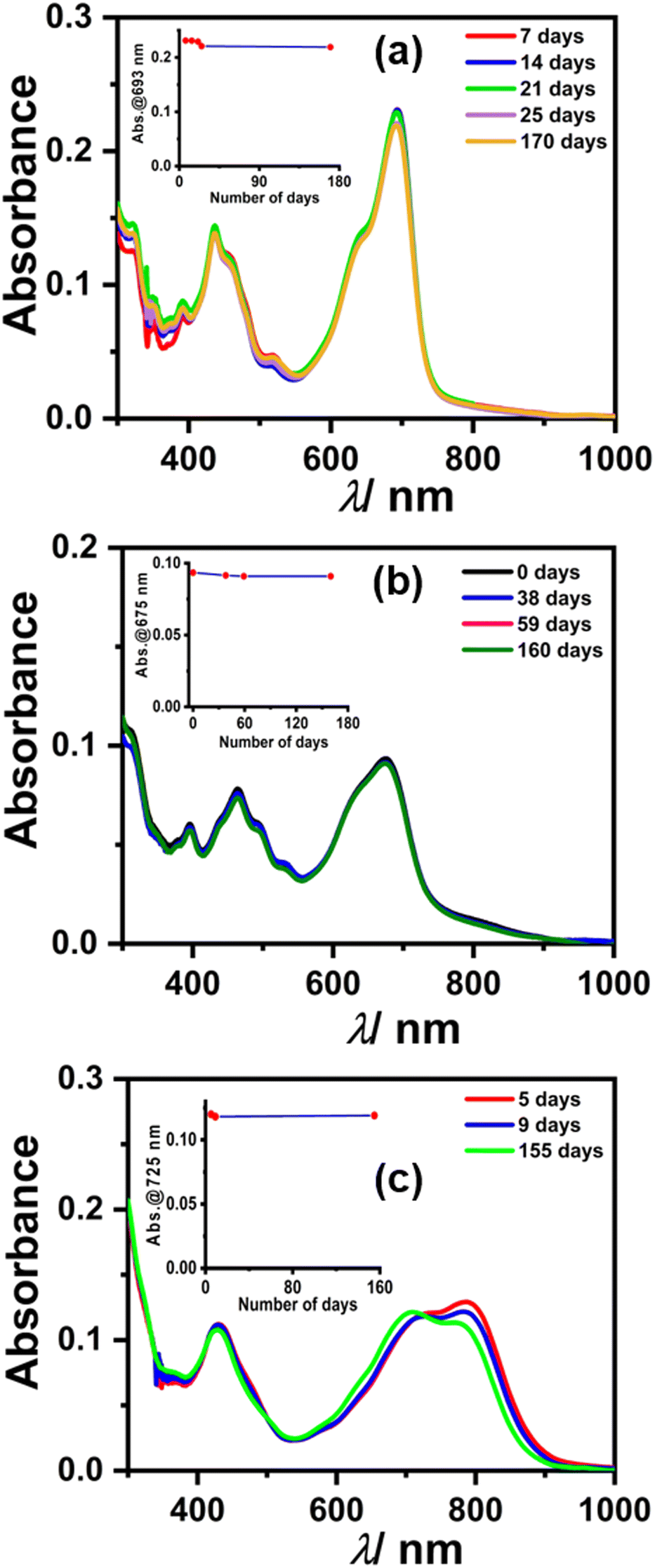 | ||
| Fig. 8 Stability plots under ambient conditions (air and moisture) of generated dianions in the presence of a trace amount of dimethylamine present in DMF for 1 (a), 2 (b), and 3 (c), respectively, monitored by using UV-Vis-NIR absorption spectroscopy; inset plots showing the change in absorbance vs. number of days. | ||
Among the compounds 1, 2, and 3, compound 3 shows solid-state fluorescence. Moreover, due to its highly electron-deficient nature, we envisioned generating a radical anion in the solid-state thin film with vapors of Lewis bases (amines) like DEA & TEA. In this regard, we drop cast compound 3 on a quartz plate, dried it, and first recorded the UV-Vis absorption and emission spectra of this solid-state thin film (as shown in Fig. 9a and b, respectively). The UV-Vis absorption spectra show structural absorption bands centered at 555 nm, 512 nm, and 475 nm while the emission spectrum shows a broad emission with the maximum centered at 637 nm. Later, this solid-state thin film was exposed to a vapor of DEA in the dark and the UV-Vis-NIR absorption spectrum was recorded (as shown in Fig. 9c). The observed spectrum shows the emergence of new vibrational bands in the NIR region between 600 and 1000 nm, while the structural absorption bands corresponding to the perylene core between 400 and 600 nm completely diminished. The new species was confirmed by recording the EPR spectrum, which shows a clear signal with a g value of 1.9907 (as shown in Fig. 9d) and validates the formation of the radical anion. This solid-state radical anion was also found to be stable under ambient conditions for up to one month (see Fig. S16, ESI†).
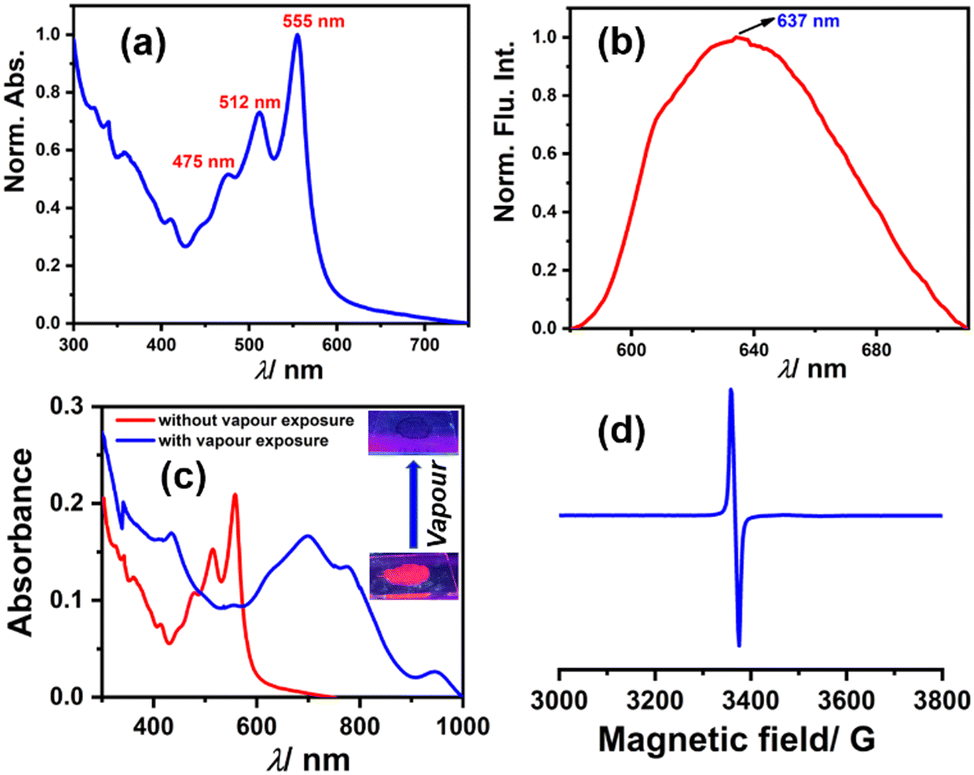 | ||
| Fig. 9 Solid-state properties of 3. (a) Normalized absorption spectrum, (b) emission spectrum, (c) radical anion generation using DEA vapors in a solid-state thin-film in the dark monitored by UV-Vis-NIR absorption spectroscopy; the inset shows the color change of the thin-film under UV light illumination, and (d) the EPR spectrum in the solid-state confirming the formation of the radical anion. | ||
Conclusions
In summary, a series of electron-deficient PDI derivatives (1, 2, and 3) have been synthesized from sulfur annulated PDI via simple oxidation of sulfur atoms of SAN-PDI using mCPBA as an oxidizing agent. All three synthesized molecules were found to be suitable precursors for the generation of super-stable radical anions and dianions. All of them showed spontaneous generation of radical anions/dianions in the presence of a trace amount of amines present in DMF and free amines like TEA and DEA. Compound 3, however, remains the most interesting among all due to its efficient generation of radical anions in DMF and upon the treatment of amines like TEA and DEA in non-polar solvents. On the other hand, 1 and 2 show radical anion conversion by accepting an electron from amines in polar and electron-rich solvents. The efficacy of radical anion generation can be accelerated with exposure to visible light. The generated radical anions 1˙−, 2˙−, and 3˙− have also been validated with EPR spectroscopy. Cyclic voltammetry results reveal the effect of sequential oxidation on reduction potential and thus increase the electron-accepting capability of the precursor molecules. Compound 3 is the most electron-deficient and exhibited radical anion generation in the solid-state thin film. To our surprise, the extraordinary stability of radical anions and dianions for months to years under ambient conditions (air and moisture) in the presence of amines as a reducing agent, proves that this strategy is successful in achieving electron-deficient PDI derivatives for radical anion and dianion generation. To the best of our knowledge, this is the first report of such extraordinary stability of radical anions/dianions of annulated PDI derivatives. Hence, the structural and electronic change made on SAN-PDI upon a sequential oxidation strategy leverages these molecules with a potential candidacy for further applications in photocatalysis and optoelectronic devices.Author contributions
The synthesis of all the compounds, and spectroscopic and electrochemical measurements were performed by A. K. and A. A. DFT calculation was done by S. S. The manuscript was prepared from the contribution of all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ALK acknowledges the financial and infrastructural support from the Indian Institute of Science Education and Research Bhopal (IISERB). AK thanks the University Grant Commission (UGC) for his doctoral fellowship and AA wishes to acknowledge Department of Science and Technology (DST), for INSPIRE fellowship for his BS-MS program. SS thanks CSIR-NEIST for the infrastructure and funding. Central Instrumentation Facility (CIF) at IISERB is also highly acknowledged.Notes and references
- H. Akamatu, H. Inokuchi and Y. Matsunaga, Nature, 1954, 173, 168–169 CrossRef.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- X. Zhang, X. Hao, L. Liu, A.-T. Pham, J. López-Andarias, A. Frontera, N. Sakai and S. Matile, J. Am. Chem. Soc., 2018, 140, 17867–17871 CrossRef CAS PubMed.
- Y. Zhao, C. Beuchat, Y. Domoto, J. Gajewy, A. Wilson, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101–2111 CrossRef CAS PubMed.
- M. R. Ajayakumar, D. Asthana and P. Mukhopadhyay, Org. Lett., 2012, 14, 4822–4825 CrossRef CAS.
- Y. Kumar, S. Kumar, D. Bansal and P. Mukhopadhyay, Org. Lett., 2019, 21, 2185–2188 CrossRef CAS PubMed.
- B. Lü, Y. Chen, P. Li, B. Wang, K. Müllen and M. Yin, Nat. Commun., 2019, 10, 767 CrossRef PubMed.
- D. Schmidt, D. Bialas and F. Wurthner, Angew. Chem., Int. Ed., 2015, 54, 3611–3614 CrossRef CAS.
- V. Sharma, U. Puthumana, P. Karak and A. L. Koner, J. Org. Chem., 2018, 83, 11458–11462 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- H. Li and O. S. Wenger, Angew. Chem., Int. Ed., 2022, 61, e202110491 CAS.
- N. Kapuria, V. Sharma, P. Kumar and A. L. Koner, J. Mater. Chem. C, 2018, 6, 11328–11335 RSC.
- M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513–1528 RSC.
- S. Betzold, S. Herbst, A. A. P. Trichet, J. M. Smith, F. Würthner, S. Höfling and C. P. Dietrich, ACS Photonics, 2018, 5, 90–94 CrossRef CAS.
- R. Roy, A. Khan, O. Chaterjee, S. Bhunia and A. L. Koner, Org. Mater., 2021, 3, 417–454 CrossRef CAS.
- R. Roy, A. Khan, T. Dutta and A. L. Koner, J. Mater. Chem. B, 2022, 10, 5352–5363 RSC.
- L. Chen, P. Xia, T. Du, Y. Deng and Y. Xiao, Org. Lett., 2019, 21, 5529–5532 CrossRef CAS.
- F. S. Goodson, D. K. Panda, S. Ray, A. Mitra, S. Guha and S. Saha, Org. Biomol. Chem., 2013, 11, 4797–4803 RSC.
- A. Zhang, W. Jiang and Z. Wang, Angew. Chem., Int. Ed., 2020, 59, 752–757 CrossRef CAS PubMed.
- G. Battagliarin, Y. Zhao, C. Li and K. Müllen, Org. Lett., 2011, 13, 3399–3401 CrossRef CAS PubMed.
- Y. Kumar, S. Kumar, S. Kumar Keshri, J. Shukla, S. S. Singh, T. S. Thakur, M. Denti, A. Facchetti and P. Mukhopadhyay, Org. Lett., 2016, 18, 472–475 CrossRef CAS PubMed.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS.
- F. Würthner, P. Osswald, R. Schmidt, T. E. Kaiser, H. Mansikkamäki and M. Könemann, Org. Lett., 2006, 8, 3765–3768 CrossRef PubMed.
- O. Chatterjee, R. Roy, A. Pramanik, T. Dutta, V. Sharma, P. Sarkar and A. L. Koner, Adv. Opt. Mater., 2022, 10 DOI:10.1002/adom.202201187.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375–380 CrossRef CAS PubMed.
- J. Cann, B. S. Gelfand and G. C. Welch, Mol. Syst. Des. Eng., 2020, 5, 1181–1185 RSC.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- X. Li, H. Wang, H. Nakayama, Z. Wei, J. A. Schneider, K. Clark, W.-Y. Lai, W. Huang, J. G. Labram, J. R. de Alaniz, M. L. Chabinyc, F. Wudl and Y. Zheng, ACS Appl. Energy Mater., 2019, 2, 3805–3814 CrossRef CAS.
- H. Zhong, C.-H. Wu, C.-Z. Li, J. Carpenter, C.-C. Chueh, J.-Y. Chen, H. Ade and A. K.-Y. Jen, Adv. Mater., 2016, 28, 951–958 CrossRef CAS PubMed.
- N.-T. Lin, A. Vargas Jentzsch, L. Guénée, J.-M. Neudörfl, S. Aziz, A. Berkessel, E. Orentas, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1121–1127 RSC.
- F. N. Miros and S. Matile, ChemistryOpen, 2016, 5, 219–226 CrossRef CAS PubMed.
- Y. Zhou, B. Xue, C. Wu, S. Chen, H. Liu, T. Jiu, Z. Li and Y. Zhao, Chem. Commun., 2019, 55, 13570–13573 RSC.
- G. Li, D. Li, X. Liu, H. Xu, J. Zhang, S. Wang, Z. Liu and B. Tang, Chem. Commun., 2019, 55, 9661–9664 RSC.
- R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, 1st edn, 2020 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Wallingford (CT), 2016 Search PubMed.
- D. Sahoo, V. Sharma, R. Roy, N. Varghese, K. Mohanta and A. L. Koner, Chem. Commun., 2019, 55, 103–106 RSC.
- Y. Che, A. Datar, X. Yang, T. Naddo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2007, 129, 6354–6355 CrossRef CAS PubMed.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663–1667 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and materials, synthesis, NMR, mass, optical and electrochemical properties, and DFT analysis. See DOI: https://doi.org/10.1039/d2tc02938c |
| ‡ These authors contributed to the work equally. |
| This journal is © The Royal Society of Chemistry 2022 |