
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of nitrophenols with a fluorescent Zr(IV) metal–organic framework functionalized with benzylamino groups†
Amina
Chatz-Giachia
 a,
Athanasia E.
Psalti
a,
Anastasia D.
Pournara
b,
Manolis J.
Manos
bc,
Christina
Pappa
a,
Konstantinos
Triantafyllidis
a and
Theodore
Lazarides
*a
a,
Athanasia E.
Psalti
a,
Anastasia D.
Pournara
b,
Manolis J.
Manos
bc,
Christina
Pappa
a,
Konstantinos
Triantafyllidis
a and
Theodore
Lazarides
*a
aDepartment of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: tlazarides@chem.auth.gr
bDepartment of Chemistry, University of Ioannina, 45110 Ioannina, Greece
cInstitute of Materials Science and Computing, University Research Center of Ioannina, 45110 Ioannina, Greece
First published on 1st August 2022
Abstract
Nitroaromatic compounds (NACs) are known explosives and environmental pollutants posing a risk for public health and national security. Thus, the development of efficient sensors for their rapid and efficient in-field detection is of high importance. Analytical methods based on fluorescence are gaining interest as current light detection technology allows the fabrication of miniaturized portable devices suitable for in-field use. Herein, we report the rational design and synthesis of Zr-1, a Zr(IV) based metal–organic framework (MOF) which is structurally analogous to UiO-66 with the assigned formula {Zr6O4(OH)8(H2O)4(L-1)4−x(NH2bdc)x}, (L-1 = 2-((benzyl)amino)-terephthalate; NH2bdc2− = 2-aminoterephthalate). Zr-1 incorporates a fluorescent ligand (L-1) with a pendant π-electron rich aromatic group and a basic secondary amine functionality, thereby targeting the selective detection of electron deficient and acidic NACs, 2,4,6-trinitrophenol (TNP) and 2,4-dinitrophenol (DNP). The stability of Zr-1 in an aqueous environment was confirmed by powder X-ray diffraction analysis on water treated samples. Fluorescence titration experiments on aqueous suspensions of acid activated Zr-1 (pZr-1) demonstrate that the material responds to small concentrations of TNP and DNP by displaying strong emission quenching, even in the presence of potentially competing compounds. The estimated limits of detection were found to be as low as 0.011 μM (2.5 ppb) for TNP and 0.026 μM (4.8 ppb) for DNP.
Introduction
Nitroaromatic compounds (NACs) are among the most important groups of industrial chemicals as they are widely used precursors and/or intermediates in the preparation of a variety of chemical products including pesticides, explosives, and pharmaceuticals. They are potently toxic and/or carcinogenic compounds with a highly explosive nature and their extensive industrial use renders them widely distributed environmental pollutants presenting a considerable danger to human population. Furthermore, NACs constitute one of the main ingredients for the preparation of explosive devices, thereby posing a major human safety and national security risk. NACs are xenobiotics resistant to chemical and biological oxidation and hydrolysis due to the electron-withdrawing character of nitro groups. Consequently, they are persistent contaminants of soil, groundwater and wastewater, and their constant monitoring is indispensable for the protection and safeguarding of the environment.1 Presently, determination of NACs is based mainly on traditional separation (i.e. GC, HPLC) and spectrometric (i.e. MS, IMS, IR) techniques, while their in-field detection is dominated by the use of trained canines. Despite their effectiveness, traditional analytical methods suffer from high cost, the requirement for highly trained personnel and lack of portability.2In search of more convenient and cost-effective alternatives, great attention is directed to luminescence-based techniques that employ fluorescent materials as sensing elements. These sensors respond to their interaction with targeted analytes by producing detectable changes in their emission characteristics (i.e. emission intensity, wavelength of emission maxima, quantum yield). The advances in light generation and detection technologies allow detection limits at the single-molecule level to be easily attainable. Furthermore, device miniaturization is feasible, thereby allowing the potential development of small, portable, and user-friendly systems suitable for in-field use.3
Organic Conjugated Polymers (CPs) have been successfully employed in building optical sensors for nitro-explosives. Swager and Yang first reported the synthesis of luminescent polymeric films displaying high sensitivity towards TNT and DNT.4 Conjugated polymers provide a key advantage for optical sensing applications: the polymer chain, featuring multiple interconnected chromophores, provides a platform with high exciton mobility. The energy migration processes that occur in such systems amplify the material's response, as a single analyte binding event may affect a large number of chromophores thereby resulting in greatly enhanced detection signals.5 This is in contrast to sensors which are based on small molecules, where each analyte binding event essentially only affects one chromophore.6
Luminescent Metal Organic Frameworks (LMOFs) is another promising class of luminescent materials that produce signal gain upon interaction with analytes.7 MOFs are crystalline porous organic/inorganic hybrid materials that can be self-assembled from their corresponding metal ions/clusters and organic bridging ligands.8 The introduction of luminescent properties in MOFs can be achieved by incorporation of fluorescent bridging ligands and/or photoactive metal anions in their framework, that leads to infinite arrays of chromophores, analogous to what is observed for CPs.9 Moreover, MOFs exhibit well defined crystalline structures with permanent porosity, structural diversity and tunability, and surface functionality that distinguishes them from luminescent organic CP sensors. Luminescent MOFs can respond to the encapsulation of various guest species within their structure's pores and therefore, display great potential for use in next generation sensing devices for in-field detection.10 In recent years, they have been extensively studied as optical sensors, however, relatively few research works focus on the rational design of MOF's pores structural design with the employment of tailored functional ligands.11
Herein, we study the rational design and synthesis of a hydrolytically stable Zr(IV) UiO-type12 MOF with an average connectivity of 8 (Zr-1), based on a strongly fluorescent dicarboxylic ligand with a pendant π-electron rich benzylamino aromatic group (L-1). The incorporation of L-1 into the MOF structure yields a material with pores lined with π-electron rich units, which, in combination with the basic amino groups, form a favourable environment for electron-deficient and acidic nitroaromatic guests. Fluorescence titrations with suspensions of protonated Zr-1 (pZr-1) and 2,4-dinitrophenol (DNP) and 2,4,6-trinitrophenol (TNP),‡ have shown that our material can act as an efficient and selective optical sensor for the determination of these analytes in water displaying emission quenching with exceptionally low detection limits.
Results and discussion
Synthesis and characterization
We synthesized the porous Zr4+ MOF Zr-1 using the functionalized dicarboxylic ligand 2-benzylaminoterephthalic acid, L-1. The syntheses of L-1 and Zr-1 are summarized in Scheme 1.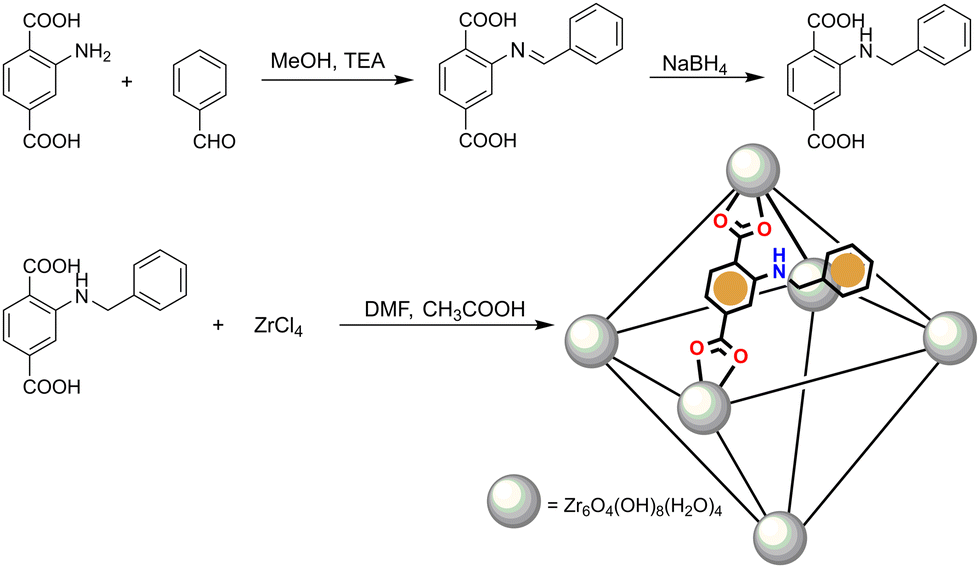 | ||
| Scheme 1 Schematic representation of the synthesis of ligand L-1 and MOF Zr-1. | ||
L-1 was prepared in a two-step process previously reported by our group13 using the approach of reductive alkylation whereby the reaction of 2-aminorerephthalic acid with benzaldehyde affords an intermediate imine which is reduced in situ with sodium borohydride to the desired product. Zr-1 was prepared following well-established solvothermal synthetic methods12a,14 by the reaction of L-1 with ZrCl4 in dimethylformamide (DMF) in a closed vial at 120 °C using acetic acid as the reaction modulator. As will be discussed below, the incorporation of the pendant benzylamino groups of L-1 within the structure of Zr-1 ensures that the material's pores are lined with functional groups which possess both a π-electron-rich system and Brønsted basicity. To use this material as a fluorescent sensor for nitrophenols, we treated Zr-1 with 4.0 M HCl for 8 h to protonate the material's secondary amino groups to ArRNH2+ Cl−, to produce a cationic and π-electron rich framework with exchangeable Cl− anions (pZr-1) thus forming a favourable environment for hosting π-electron deficient and anionic nitroaromatic guests such as nitrophenol anions.
It has been observed by many researchers that ligands which are based on N-alkyl substituted 2-aminoterephthalic acid, often show various degrees of elimination of the alkyl substituent to form the parent ligand during MOF synthesis.15 Therefore, in order to examine the stability of L-1 once it is incorporated into the framework of Zr-1, we measured the 1H-NMR spectra of digested MOF samples in D2O/NaOD both before and after acid activation. As seen in Fig. S5 (ESI†), the spectrum of the digested as-synthesized MOF clearly shows the 1H-NMR signals corresponding to L-1 along with weak signals due to 2-aminoterephthalate (NH2bdc2−) indicating less than 10% elimination of the benzyl groups. However, 1H-NMR spectra of digested samples of pZr-1, showed increased signals due to 2-aminoterephthalate. After conducting protonations of Zr-1 with different HCl concentrations and reaction times, we found that the use of 4.0 M HCl for 8 h produces highly active pZr-1 in terms of nitrophenol sensing, with ca. 30% benzyl group elimination (Fig. S7, ESI†). Thus, pZr-1 henceforth refers to the activated material produced under these conditions. In accordance with the 1H-NMR and thermogravimetric (see next section) analysis of Zr-1 and pZr-1, the MOF formula is proposed to be {Zr6O4(OH)8(H2O)4(L-1)4−x(NH2bdc)x} (x = 0.4–1.2).
Powder X-ray diffraction (PXRD) patterns of as synthesized Zr-1, pZr-1 and the parent material UiO-66 can be seen in Fig. 1. All 2θ peaks of the diffraction pattern of Zr-1 are consistent with UiO-66, which demonstrates that the two materials are structurally equivalent. However, as will be discussed below, the average connectivity of Zr-1 is reduced to 8 compared to the 12-connectivity12a of UiO-66.
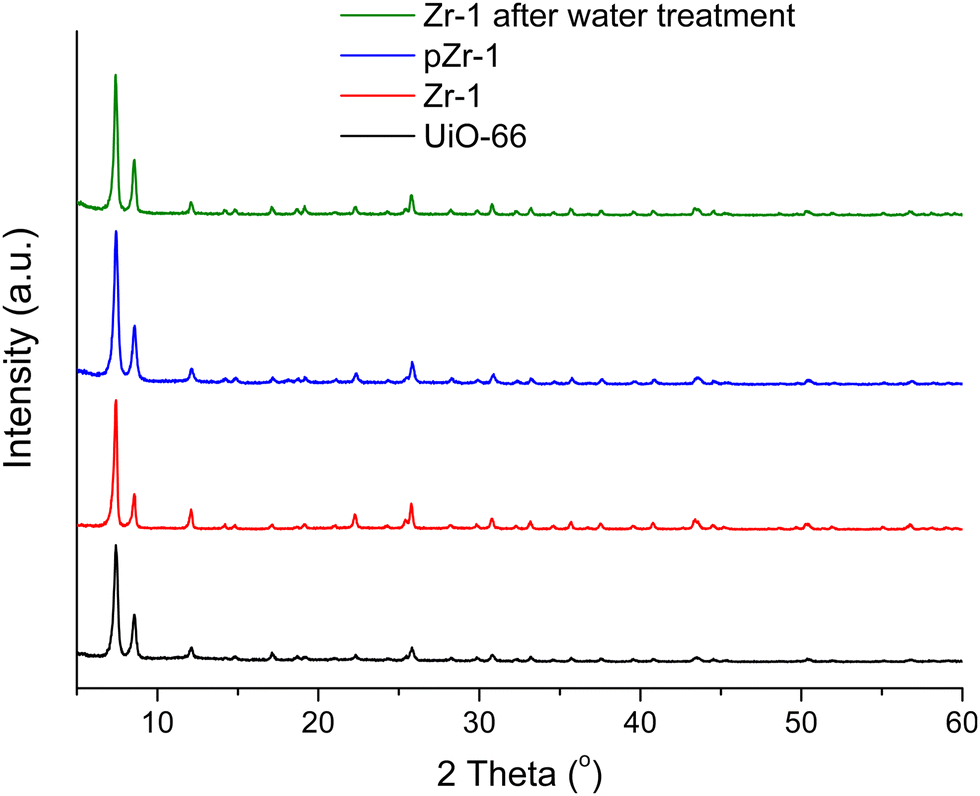 | ||
| Fig. 1 PXRD patterns of UiO-66 (black line), Zr-1 (red line), pZr-1 (blue line) and Zr-1 after water treatment (green line). | ||
Chemical and thermal stability
Stability in water is a prerequisite for the potential application of a material in pollutant detection processes in aqueous environments. The stability of Zr-1 towards H2O was studied by treating the material with H2O for 24 h at room temperature.After treatment, the solid was isolated via centrifugation and dried at 50 °C for 24 h before PXRD analysis was performed. The PXRD pattern of Zr-1 after water treatment is identical to the PXRD pattern of the as-synthesized material (Fig. 1). Evidently, the overall framework remains unaltered confirming that Zr-1 displays high stability in aqueous environment.
The thermogravimetric analysis (TGA) curve for Zr-1 after H2O exchange is seen in Fig. 2. TGA analysis of the water treated sample under air reveals that Zr-1 loses weight in two distinct steps: the first is completed at ca. 200 °C, and is attributed to the loss of lattice and coordinated water molecules, while the second, which is completed at ca. 550 °C, corresponds to complete ligand loss and formation of ZrO212b,16 (see ESI† for details). It is interesting to mention that Zr-1 exhibits a much lower weight loss due to the removal of water in comparison to what is observed for UiO-66-NH2 (ca. 10% vs. 30%, Fig. S10, ESI†). This is attributed to the presence of benzyl side groups in Zr-1, which (i) decrease the free volume within the material's pores and (ii) render the material less hydrophilic. In contrast to Zr-1, HCl treated pZr-1 shows a more extensive initial weight loss due to (i) an increased water content because of the partial elimination of benzyl side groups (vide supra) and (ii) the possible additional elimination of HCl (Fig. S10, ESI†).
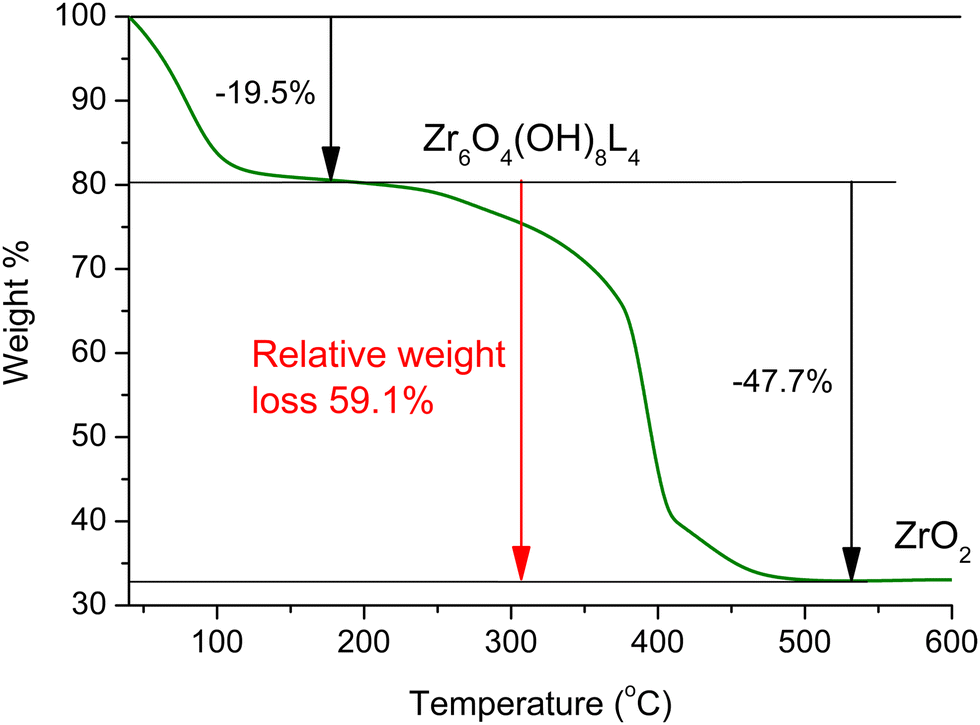 | ||
| Fig. 2 TGA curve for Zr-1 after water treatment, under air. | ||
Assuming 10% cleavage of the benzyl side group (as estimated by 1H-NMR analysis on digested Zr-1), we would expect a 12-connected material to have the formula {Zr6O4(OH)8(H2O)4(L-1)5.4(NH2-bdc)0.6}, while an 8-connected material would have the formula {Zr6O4(OH)8(H2O)4(L-1)3.6(NH2-bdc)0.4}. Thus, the expected relative weight loss from 200 to 600 °C is expected to be 68.1% and 58.7% in the case of 12- and 8-connectivity, respectively. Based on the above, the experimentally determined 59.1% relative weight loss (Fig. 2) agrees with Zr-1 having an average connectivity of 8. The missing linker defects of our material can be attributed to the steric hindrance induced by the bulky side group of ligand L-1. The lower connectivity of Zr-1 possibly accounts for its lower decomposition temperature (∼390 °C) compared to that observed for UiO-66 (>500 °C).12a The presence of missing-linker defects is a common feature of UiO-66(Zr)-type MOFs and defective materials reported in literature retain high crystallinity and thermal stability,17 as is also observed in our work.
BET surface area and porosity
To evaluate the porosity and specific surface area, we performed N2 adsorption–desorption measurements (77 K) on both Zr-1 and pZr-1 (Fig. 3). Both isotherms can be classified as Type I physisorption isotherms, typical of microporous materials.18 The surface area was calculated by the Brunauer–Emmett–Teller (BET) model to 893 m2 g−1 for Zr-1 and 932 m2 g−1 after activation with 4.0 M HCl (pZr-1). These values fall within the range of surface area values calculated for functionalized UiO-66-type materials prepared with solvothermal reactions.19 The partial benzyl group cleavage that occurs during MOF activation possibly accounts for the slightly higher surface area determined for pZr-1. For comparison, we performed the same measurement on a sample of UiO-66-NH2,20 the 12-connected amino derivative of UiO-66, (Fig. S11, ESI†) and the BET surface area was determined to be 1274 m2 g−1, in good agreement with literature results20b,21 The more open framework due to the 8-connectivity of Zr-1 partially compensates for the presence of the bulky benzyl side group thereby resulting in a BET surface area which is not dramatically lower than that of UiO-66-NH2.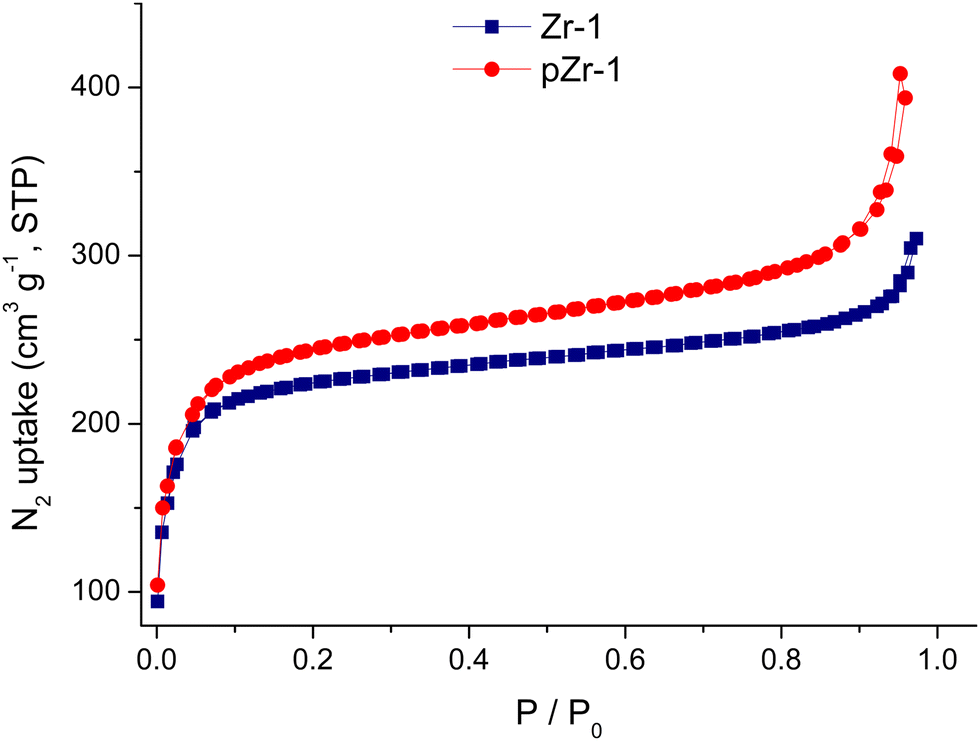 | ||
| Fig. 3 Nitrogen sorption isotherms at 77 K of Zr-1 and pZr-1 after treatment with acetone and overnight evacuation (120 °C). | ||
Photophysical properties
As described in the Experimental Part, Zr-1 was treated with 4.0 M HCl prior to its use in sensing studies. Acid activation is an essential step to achieve detection of nitrophenols. The pKa values for TNP and DNP are 0.3 and 4.0, respectively, indicating that the analyte molecules are found in their deprotonated form in the aqueous environment of the MOF suspension (pH = 4.5). Treatment with HCl ensures that the MOF's secondary amine units are protonated and introduces exchangeable Cl− counter-ions. Combined, these features yield a favourable environment inside the MOF's pores so that the material can function as an ion exchange sensor for the anionic nitrophenols.The activated material pZr-1 readily forms a fine suspension in water and exhibits turquoise fluorescence upon selective ligand excitation at 400 nm. To examine the stability of the suspension we repeated multiple fluorescence measurements on a suspension of pZr-1 in H2O (0.1 mg mL−1) in the span of 1.5 h and as can be seen in Fig. S12 (ESI†), the emission spectrum remains unaltered. Our group has previously reported the capability of a similar protonated Zr4+-terephthalate MOF (MOR-2) to form fine and stable suspensions.13
The emission spectrum of pZr-1 consists of a broad band with maximum at 470 nm (Fig. 4), similar to the emission spectrum of ligand L-1 (Fig. S13, ESI†). The excitation spectrum of pZr-1 (Fig. 4) consists of a broad band with a maximum at 370 nm that shows good agreement with the UV-vis absorption band of L-1 (Fig. S13, ESI†) and with what is observed in UiO-66-NH2(Zr/Hf)-type MOFs.3b The emission quantum yields of aqueous suspensions of Zr-1 and pZr-1 were found to be ca. 1.2% (Fig. S14 and S15, ESI†), double compared to that of a suspension of UiO-66-NH2 (ca. 0.56%, Fig. S16, ESI†). The emission quantum yields of MOFs are much lower than those of the free ligands in methanol solution (ΦL-1 = 39%; ΦNH2bdc = 62%, Fig. S17 and S18, ESI†). This can be attributed to self-quenching phenomena due to the relative proximity of the chromophores within the MOF matrix.7a The latter assumption is further supported by the much higher emission intensity displayed by the 8-connected pZr-1 compared to the 12-connected UiO-66-NH2, despite the free ligand NH2bdc having a higher quantum yield than L-1. Despite its modest fluorescence quantum yield, pZr-1 (aqueous suspension 0.1 mg mL−1) displays a clear fluorescence signal which can be recorded easily and accurately. This phenomenon has also been observed in MOFs that emit in the near IR region and has been attributed to the large number of luminophores per unit volume enabling the material to emit a large number of photons thereby leading to a clear and strong emission sigmal which is suitable for sensing and imaging studies.22
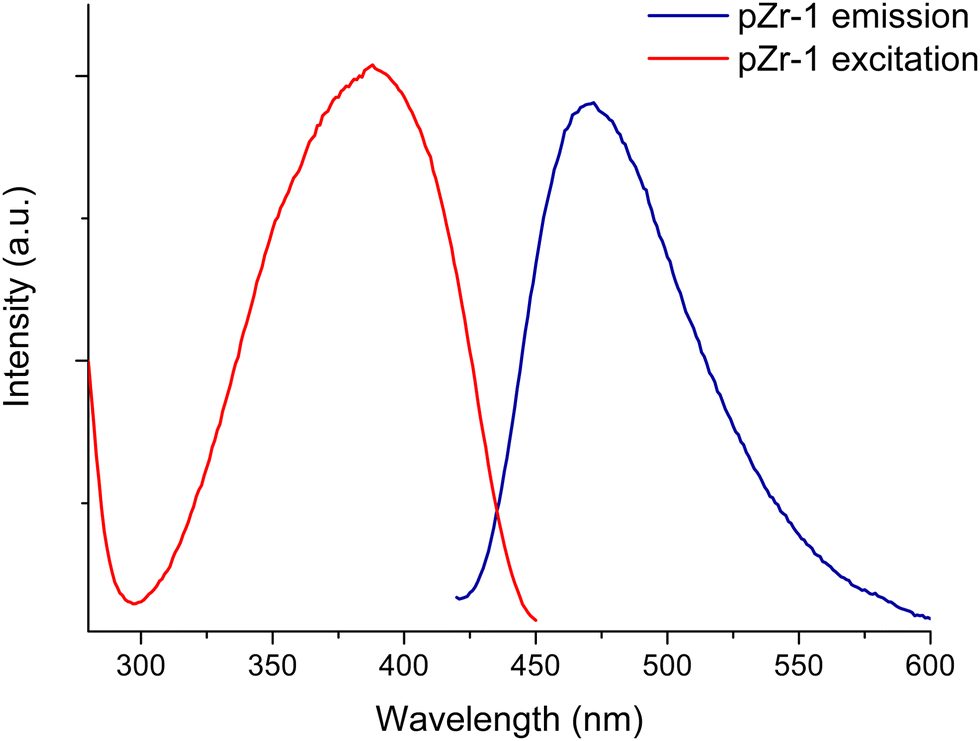 | ||
| Fig. 4 Excitation and emission spectra (λexc = 400 nm) of pZr-1 suspended in H2O (0.1 mg mL−1). | ||
It has been demonstrated, through electron paramagnetic resonance, time-resolved absorption spectroscopy and theoretical calculations, that the fluorescent excited state in Zr4+ MOFs with 2-aminoterephtahlate as bridging ligand predominantly involves shift of electron density from an orbital which is largely localized on the amino group to an unoccupied antibonding orbital of the aromatic ring without an appreciable contribution from a ligand-to-metal-charge-transfer (LMCT) component.23 This type of intraligand charge transfer (ILCT) transitions show sensitivity to the local environment around the chromophore, such as the presence of hydrogen bond donors and/or electron accepting/donating moieties, and are thus well suited for optical sensing applications.3b,9a,24
Fluorescence titrations
Benefitting from the stability in water and its strong emission signal, we investigated the ability of pZr-1 to sense trace amounts of nitrophenols in aqueous media. To evaluate the sensing ability of the material, we performed fluorescence titration experiments with gradual addition of aqueous solutions (5.0 × 10−5 M) of TNP and DNP to a suspension of pZr-1 in H2O (0.1 mg mL−1) at pH 4.5. It should be noted that we applied Inner Filter Effect (IFE) correction to all our experimental data (see ESI† for details). As can be seen in Fig. 5, fluorescence measurements clearly demonstrate that TNP and DNP have a strong quenching effect on the initial emission intensity of pZr-1, up to 92% and 86% at the end of the titration, respectively. For both titration experiments, the calibration curves show good linearity with respect to analyte concentration up to ca. 250 ppb. Through analysis of the linear fitting, we determined the Limits of Detection (LOD) and Quantification (LOQ) (see ESI† for details). LOD and LOQ values were calculated to be 0.011 and 0.037 μM (2.5 and 8.5 ppb) for TNP and 0.026 and 0.086 μM (4.8 and 16.0 ppb) for DNP detection, respectively. These values demonstrate that pZr-1 exhibits effective sensing behaviour towards TNP and DNP in aqueous solutions.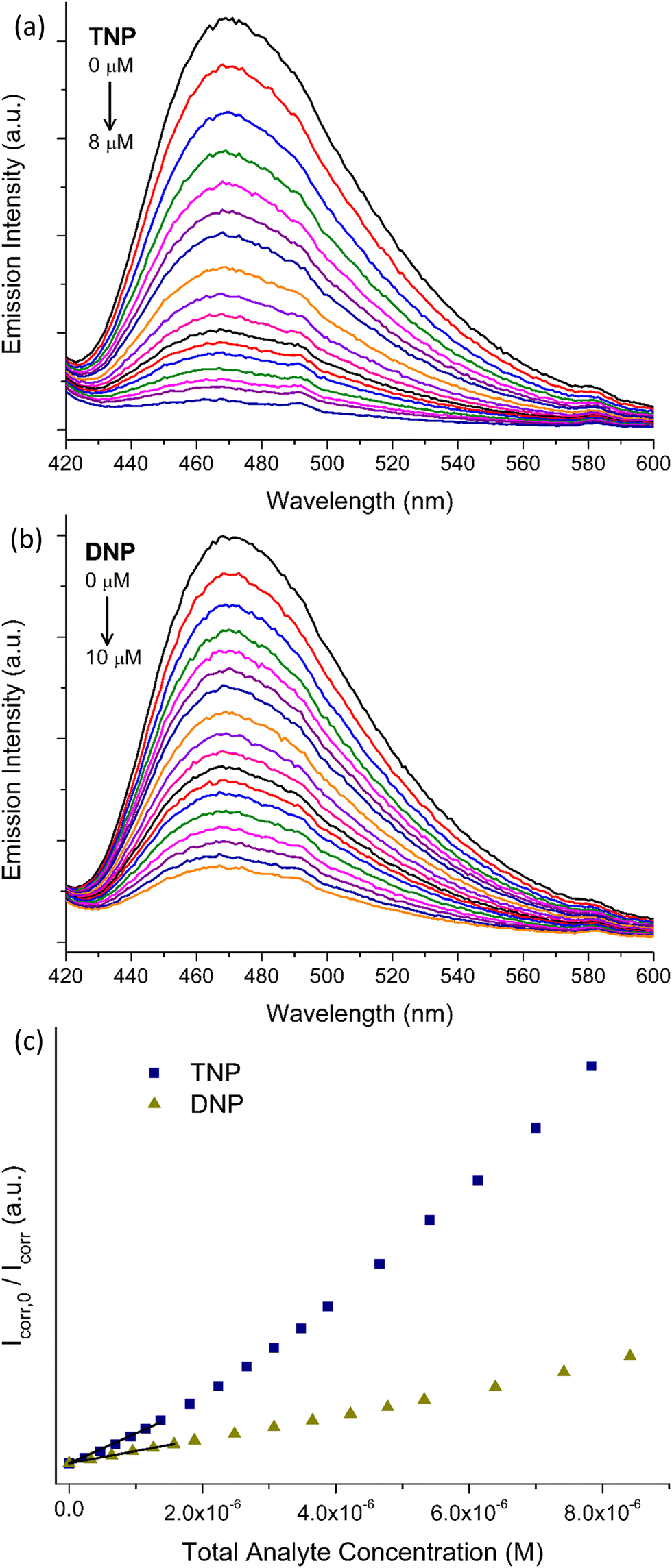 | ||
| Fig. 5 Fluorescence titrations (λexc = 400 nm) of pZr-1 suspended in H2O (2 mL, 0.1 mg mL−1) at pH 4.5 upon gradual addition of aqueous solution (5 × 10−5 M) of (a) TNP (b) DNP (c) Corresponding Stern–Volmer plots for both titrations, showing linearity at low analyte concentrations. | ||
The pores of pZr-1 are lined with π-electron rich units which are potentially suitable for donor–acceptor association with the electron-deficient nitroaromatic compounds. Furthermore, we theorize that entry of analytes in the pores could be further assisted by H bonding interactions and electrostatic interactions between the positively charged protonated secondary amine units and the anionic nitroaromatic guests. An identical titration experiment with TNP carried out using Zr-1, the as synthesized material that is not fully protonated, produced non consistent results with higher LOD and LOQ values (0.030 μM and 0.139 μM, respectively). Additionally, the same experiment with a deprotonated MOF sample, treated with triethylamine/MeOH solution, resulted in much weaker fluorescence quenching. (Fig. S23, ESI†) These observations support our proposition that the MOF-analyte interaction has a strong electrostatic component.
To further examine the quenching efficiency, we analyzed the data with the Stern–Volmer equation, eqn (1)
| (I0/I) = 1 + KSV × [A] | (1) |
To further test our MOF's sensing performance, we performed detection experiments using a sample of acid activated UiO-66-NH2, (Fig. S24 and S25, ESI†) and the results are shown in Table 1. Comparison of the fluorescence measurements for the two functionalized UiO-66-type MOFs demonstrates that pZr-1 shows superior quenching response and sensitivity. The increased sensing ability of our MOF is the result of the strategical design that incorporates a π-electron-rich system inside the MOF's pores, thus enhancing the interaction with electron deficient nitrophenolic analytes.
| Analyte | LOD (μM) | LOQ (μM) | K SV (M−1) | Maximum quenching % | |
|---|---|---|---|---|---|
| UiO-66-NH2 | TNP | 0.040 | 0.136 | 1.6 × 105 | 69 |
| DNP | 0.042 | 0.140 | 1.7 × 105 | 64 | |
| pZr-1 | TNP | 0.011 | 0.037 | 7.2 × 105 | 92 |
| DNP | 0.026 | 0.086 | 2.9 × 105 | 86 | |
Detection of nitrophenols by luminescent MOF sensors has been reported in numerous studies over the last years.10e In Table 2 we show LOD and LOQ values and Ksv values of some of the most efficient sensors reported. (A more extensive collection of representative recent examples can be found in Table S1, ESI†). Comparison of the data confirms that our material is among the best performing sensors and shows extremely high sensitivity. The LOD value for TNP (0.011 μM) is comparable with the lowest detection limit so far reported for TNP detection in H2O, by Joarder et al. (12.9 nM).27 The standout feature of pZr-1 is that the material is hydrolytically stable and forms fine and stable suspension in water and, thus, can be used in analysis of aqueous samples. Furthermore, the λexc (400 nm) used in titration experiments lies in lower energy than the absorption maximum region of nitrophenols and the emission maximum of pZr-1 does not overlap with the analytes’ absorption bands, ensuring that the inner filter effect on the emission measurements is kept at a minimum.
| Luminescent MOF | Analyte | Solvent | LOD (μM) | K SV (M−1) | λ exc (nm) | λ em,max (nm) | Ref. |
|---|---|---|---|---|---|---|---|
| [Zn2(TCPE)(tta)2]·2DMF·4H2O·2Me2NH2+ | TNP | DMF | 0.036 | 3.06 × 104 | 392 | 461 | 26 |
| DNP | 0.029 | 3.75 × 104 | |||||
| [Zn8(ad)4(BPDC)6O·2Me2NH2]·G | TNP | H2O | 0.012 | 4.6 × 104 | 340 | 405 | 27 |
| Zr6O4(OH)8(H2O)4(TTNA)8/3 | TNP | H2O | 0.043 | 5.1 × 105 | 324 | 399 | 28 |
| {Zr6O4(OH)8(H2O)4(L-1)4−x(NH2bdc)x} | TNP | H2O | 0.011 | 7.2 × 105 | 400 | 470 | This work |
| DNP | H2O | 0.026 | 2.9 × 105 |
Encouraged by these results we investigated the sensing selectivity of pZr-1 towards DNP and TNP. We performed fluorescence measurements to examine the quenching efficiency towards several competing non-acidic compounds, including nitrobenzene (NB) 1,3-dinitrobenzene (1,3-DNB), 2-nitrotoluene (2-NT), 4-nitrotoluene (4-NT), 2,4-dinitrotoluene (DNT), 2,6-dinitrotoluene (2,6-DNT), 2-nitrophenol (2-NP), 3-nitrophenol (3-NP), 4-nitrophenol (4-NP), aniline (NA) and 3-nitroaniline (3-NA). The bar graphs in Fig. 6, illustrate the fluorescence response suspensions of activated pZr-1 in H2O (pH = 4.5) in the presence of competing compounds. These non-acidic compounds have a negligible quenching effect compared to the strong quenching observed upon subsequent addition of equal amounts of TNP or DNP (IFE correction was applied to all experimental data).
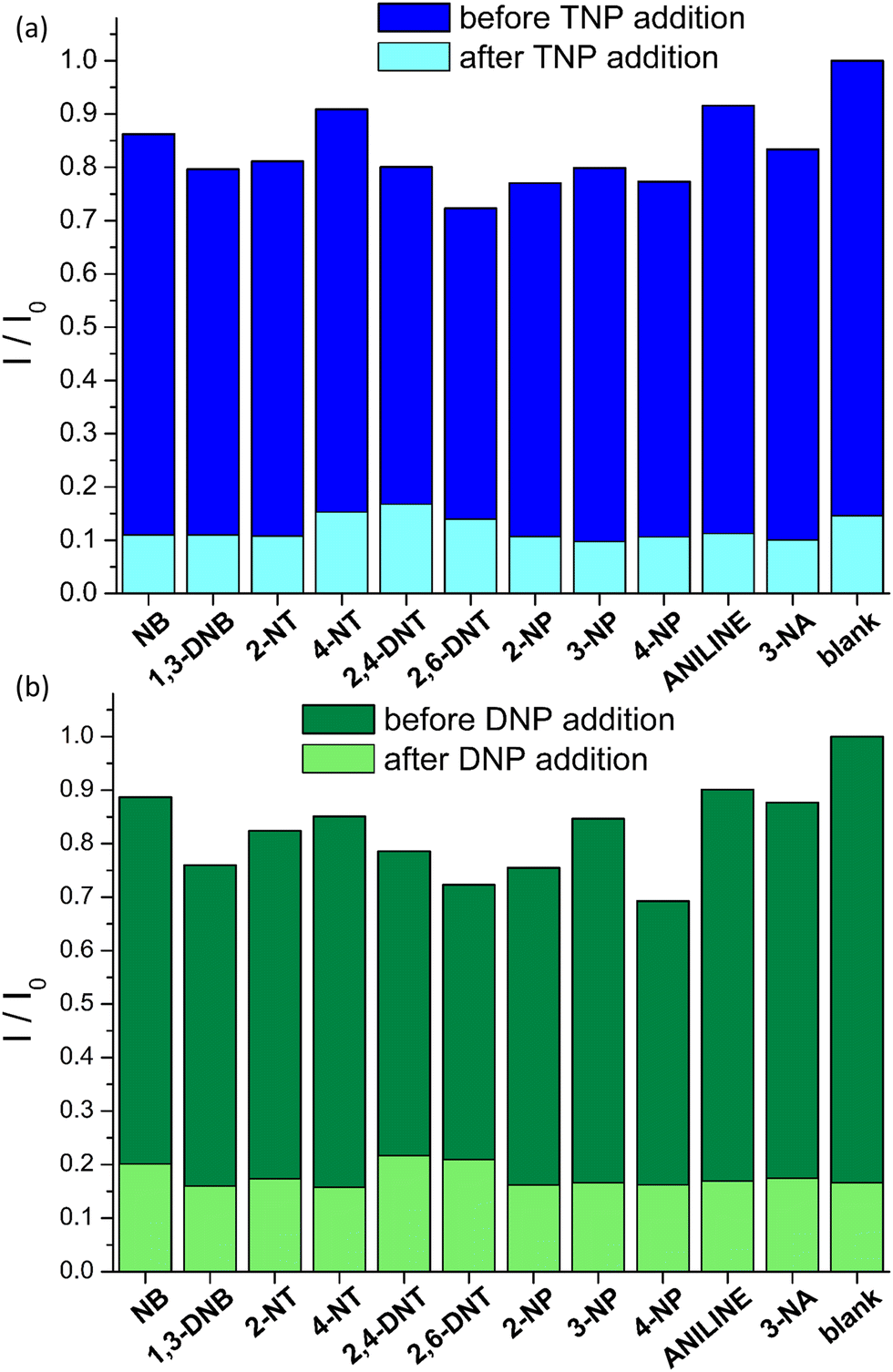 | ||
| Fig. 6 Normalized fluorescence response I/I0 (λexc = 400 nm) of Zr-1 suspended in H2O (2 mL, 0.1 mg mL−1) at pH 4.5 in the presence of various analytes (12 μM) The dark bars represent the response in the presence of competing analytes and the light bars represent the response upon addition of (a) TNP (10 μM) and (b) DNP (12 μM). I0 corresponds to emission intensity before the addition of analytes. | ||
To further investigate our MOF's selectivity, we carried out fluorescence titration experiments with the above compounds (Fig. S26, ESI†). The S–V plots of these titrations are shown in Fig. 7 along with those for TNP and DNP. In addition to the expected low quenching effect, the competing compounds produce strictly linear S–V plots within the studied concentration range. This series of fluorescence measurements with a range of different analytes clearly demonstrates that pZr-1 offers a remarkable combination of sensitivity and selectivity towards TNP and DNP. The lack of quenching response in the presence of mono-nitrophenols can be explained by considering that the pKa values of these molecules are in the range 7.0–8.3, meaning that the hydroxylic groups are not deprotonated in the aqueous MOF suspension.
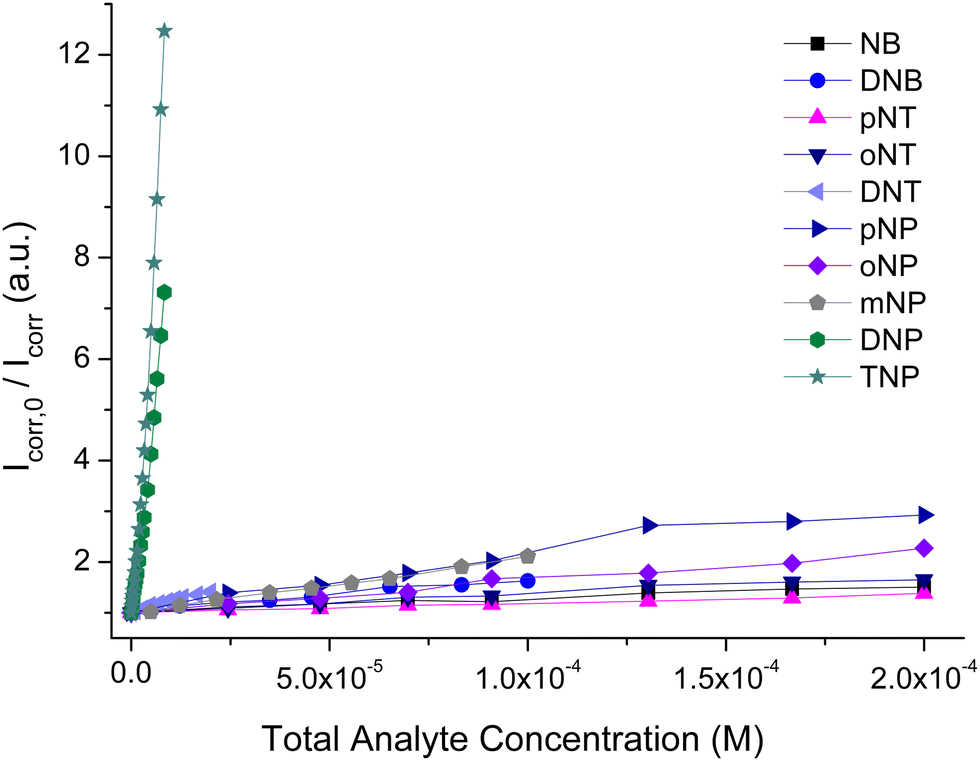 | ||
| Fig. 7 Comparison of Stern–Volmer plots of fluorescence titrations (λexc = 400 nm) of pZr-1 suspended in H2O (2 mL, 0.1 mg mL−1) at pH 4.5 with different nitroaromatic analytes. | ||
The treatment of pZr-1 with saturated aqueous solutions of TNP and DNP (ca. 10−3 M) results in an instant color change from yellow to orange. After washing with MeOH to remove any loosely attached DNP or TNP, we digested the samples in 40% NaOD in D2O, to perform 1H-NMR analysis. The 1H-NMR spectra confirmed the presence of the analytes within Zr-1 (Fig. S28 and S30, ESI†) with peak area analysis showing guest-to-bridging ligand molar ratios of ca. 0.14 for TNP and ca. 0.22 for DNP. The higher molar ratio that we observed for the DNP guest could be attributed to a size effect. When we performed an analogous experiment in the concurrent presence of both analytes (ca. 10−3 M), subsequent 1H-NMR analysis (Fig. S32, ESI†) showed that the material was loaded with equal amounts of TNP and DNP, thus reflecting their solution ratio. (The low solubility of DNP prevented us from performing further experiments at higher concentrations.)
Comparison of the PXRD pattens and IR spectra of pZr-1 before and after being loaded with DNP and TNP confirm the material's structural stability upon its interaction with the analytes (Fig. S27, S29, S31 and S34, ESI†). It is worth noting that the IR spectrum of pZr-1 shows the characteristic broad signal in the 3000–3800 cm−1 region due to absorbed water. In line with water molecules being displaced by the guest analyte, the water signal is greatly decreased in DNP-loaded pZr-1 revealing the characteristic peak at 3382 cm−1 due to the NH stretch of the secondary amino groups (Fig. S33, ESI†).
To quantify the kinetic response of pZr-1 towards TNP we performed a kinetic fluorescence scan (Fig. 8) where TNP was added to a suspension of pZr-1. The data could be satisfactorily fitted to two pseudo-first order processes29 with time constants of 5.17(2) and 48.2(3) s, with the faster process having a far greater contribution (Fig. S36, ESI†). Thus, the material's fluorescence response to the presence of TNP is dominated by fast kinetics, as is desirable for an effective chemosensor.30
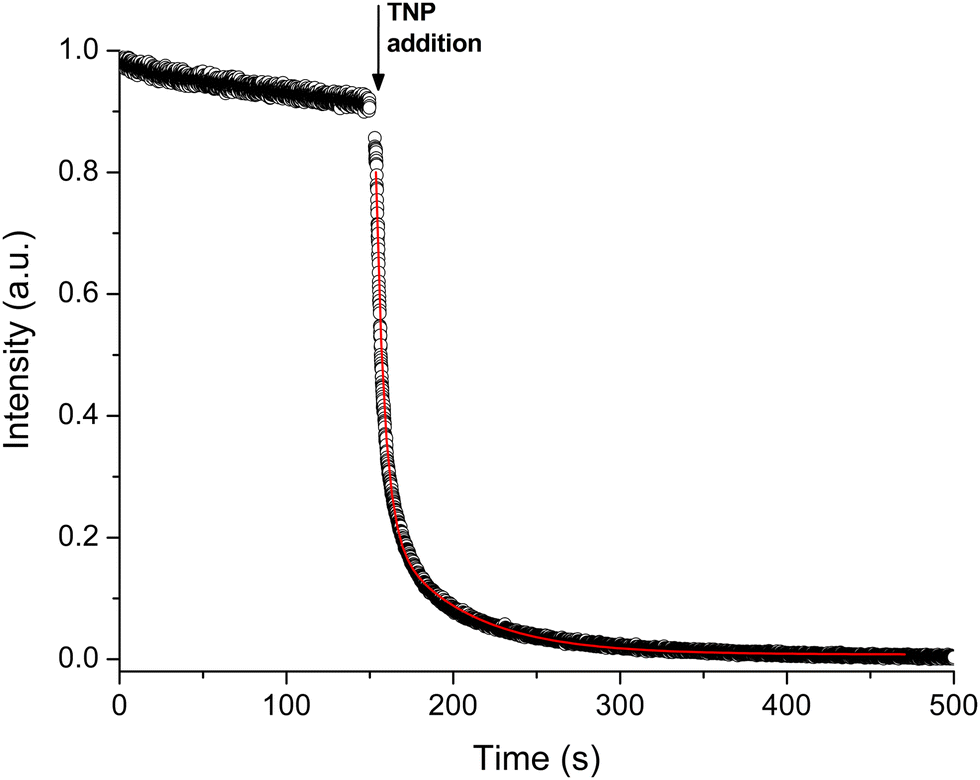 | ||
| Fig. 8 Time-dependent fluorescence response (λexc = 400 nm, λem = 470 nm) of pZr-1 suspended in H2O (2 mL, 0.1 mg mL−1) at pH 4.5 upon addition of 25 μL an aqueous solution of TNP (10−3 M). The concentration of TNP corresponds to that at the end of a typical titration experiment (8 μM). Black circles represent experimental data, the red curve represents the best fit based on a biexponential decay. | ||
To evaluate the structural integrity and reusability of Zr-1, we examined the sensing ability of an activated MOF sample that was treated with TNP aqueous solution and re-activated. Fig. S34 (ESI†) displays the PXRD patterns of pZr-1 after TNP sorption and reactivation confirming that the material retains good crystallinity apart from some minor. As shown in Fig. S35 (ESI†), fluorescence titration with gradual addition of TNP (5.0 × 10−5 M) on a suspension of the recycled sample in H2O (2 mL, 0.1 mg mL−1) resulted in quenching efficiency up to 89% with a quenching constant of 4.5 × 10−5 M−1. Thus, we observe that after one cycle of analyte sorption and re-activation, the quenching constant value of pZr-1 is slightly reduced, albeit the regenerated material still exhibits strong quenching in the presence of TNP.
Conclusions
We have presented the synthesis and study of a porous Zr(IV) MOF (Zr-1) incorporating a bridging ligand (L-1) with a pendant benzylamino group, thereby creating a π-electron rich and basic environment within the material's pores, which is favourable for the hosting of acidic nitroaromatic guest species. Powder X-ray diffraction and thermogravimetric analysis showed that Zr-1 possesses a UiO-66 type structure with a lower average connectivity of 8, compared to the 12-connectivity of UiO-66. This lower connectivity may be attributed to the bulky side groups on L-1, which prevent a closer approach between the bridging units. The N2 adsorption isotherm of Zr-1 corresponds to a BET surface area of 893 m2 g−1, indicating that the lower average connectivity combined with the partial cleavage (ca. 10%) of the benzyl side groups has a beneficial effect on the material's porosity. The material retains its structural integrity after protonation with 4.0 M HCl to yield pZr-1, which has a BET surface area of 932 m2 g−1, a small increase compared to as synthesized Zr-1, partly attributed to the increased cleavage of the benzyl side groups (reaching ca. 30% after protonation). When pZr-1 is treated with aqueous solutions of TNP and DNP (10−3 M), we observe a darkening of the material's colour and the presence of analytes within the material is confirmed through 1H-NMR analysis. Fluorescence titrations of aqueous suspensions of pZr-1 with TNP and DNP lead to a strong quenching effect (92% and 86% by the end of the titration, respectively) with good signal linearity for analyte concentrations of up to ca. 250 ppb. We determined limits of detection and quantification of 0.011 and 0.037 μM for TNP, and 0.026 and 0.086 μM for DNP, respectively. Comparison with literature data places our material among the most efficient fluorescence sensors for the above analytes. It is important to emphasize that the excitation wavelength used in our studies (400 nm) is offset form the region of maximum absorption of DNP and TNP, thereby minimizing inner filter effects. We have confirmed this by observing insignificant alterations to the initial titration data after applying inner-filter corrections. Research on fluorescent metal–organic frameworks for the detection of nitroaromatic compounds is ongoing in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “1st Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment” (Project Number: 3371).Notes and references
- (a) V. Purohit and A. K. Basu, Chem. Res. Toxicol., 2000, 13, 673–692 Search PubMed; (b) J. D. Rodgers and N. J. Bunce, Water Res., 2001, 35, 2101–2111 CrossRef CAS; (c) P.-G. Rieger, H.-M. Meier, M. Gerle, U. Vogt, T. Groth and H.-J. Knackmuss, J. Biotech., 2002, 94, 101–123 CrossRef CAS; (d) Z. C. Symons and N. C. Bruce, Nat. Prod. Rep., 2006, 23, 845–850 RSC; (e) K.-S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250–272 CrossRef CAS PubMed; (f) P. Kovacic and R. Somanathan, J. Appl. Toxicol., 2014, 34, 810–824 CrossRef CAS PubMed.
- (a) D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS; (b) S. Singh, J. Hazard. Mater., 2007, 144, 15–28 CrossRef CAS PubMed; (c) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- (a) X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC; (b) W. P. Lustig, S. Mukherjee, N. D. Rudd, A. V. Desai, J. Li and S. K. Ghosh, Chem. Soc. Rev., 2017, 46, 3242–3285 RSC; (c) T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 CrossRef CAS PubMed; (d) L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115(15), 7840–7892 CrossRef CAS PubMed; (e) M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120(16), 8581–8640 CrossRef CAS PubMed.
- (a) J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 5321–5322 CrossRef CAS; (b) J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- (a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed; (b) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339–1386 CrossRef CAS PubMed; (c) O. P. Dimitriev, Chem. Rev., 2022, 122(9), 8487–8593 CrossRef CAS PubMed.
- S. Shanmugaraju and P. S. Mukherjee, Chem. Commun., 2015, 51, 16014–16032 RSC.
- (a) M. Gutiérrez, R. Navarro, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2017, 19, 16337–16347 RSC; (b) A. Sharma, D. Kim, J.-H. Park, S. Rakshit, J. Seong, G. H. Jeong, O.-H. Kwon and M. S. Lah, Commun. Chem., 2019, 2, 39 CrossRef; (c) G. Mercuri, G. Giambastiani and A. Rossin, Inorganics, 2019, 7, 144 CrossRef CAS; (d) D. J. Cerasale, D. C. Ward and T. L. Easun, Nat. Rev. Chem., 2022, 6, 9–30 CrossRef.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- (a) S. A. Diamantis, A. Margariti, A. D. Pournara, G. S. Papaefstathiou, M. J. Manos and T. Lazarides, Inorg. Chem. Front., 2018, 5, 1493–1511 RSC; (b) C. R. Martin, P. Kittikhunnatham, G. A. Leith, A. A. Berseneva, K. C. Park, A. B. Greytak and N. B. Shustova, Nano Res., 2021, 14, 338–354 CrossRef CAS; (c) C. R. Martin, K. C. Park, R. E. Corkill, P. Kittikhunnatham, G. A. Leith, A. Mathur, S. L. Abiodun, A. B. Greytak and N. B. Shustova, Faraday Discuss., 2021, 231, 266–280 RSC.
- (a) X. Fang, B. Zong and S. Mao, Nano-Micro Lett., 2018, 10, 64 CrossRef PubMed; (b) L. Liu, X. Chen, J. Qiu and C. Hao, Dalton Trans., 2015, 44, 2897–2906 RSC; (c) Y. Zhang, S. Yuan, G. Day, X. Wang, X. Yang and H.-C. Zhou, Coord. Chem. Rev., 2018, 354, 28–45 CrossRef CAS; (d) M.-L. Hu, S. A. A. Razavi, M. Piroozzadeh and A. Morsali, Inorg. Chem. Front., 2020, 7, 1598–1632 RSC; (e) T. Zhao, F. Zhang, J. Zhou and X. Zhao, Comments Inorg. Chem., 2021, 41, 100–132 CrossRef CAS.
- (a) T. K. Kim, J. H. Lee, D. Moon and H. R. Moon, Inorg. Chem., 2013, 52, 589–595 CrossRef CAS PubMed; (b) D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef CAS PubMed; (c) A. Azhdari Tehrani, H. Ghasempour, A. Morsali, G. Makhloufi and C. Janiak, Cryst. Growth Des., 2015, 15, 5543–5547 CrossRef; (d) M. Bagheri, M. Y. Masoomi, A. Morsali and A. Schoedel, ACS Appl. Mater. Interfaces, 2016, 8, 21472–21479 CrossRef CAS PubMed; (e) Q.-Y. Li, Z. Ma, W.-Q. Zhang, J.-L. Xu, W. Wei, H. Lu, X. Zhao and X.-J. Wang, Chem. Commun., 2016, 52, 11284–11287 RSC; (f) B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed; (g) A. Azhdari Tehrani, L. Esrafili, S. Abedi, A. Morsali, L. Carlucci, D. M. Proserpio, J. Wang, P. C. Junk and T. Liu, Inorg. Chem., 2017, 56, 1446–1454 CrossRef CAS PubMed; (h) L. Luconi, G. Mercuri, T. Islamoglu, A. Fermi, G. Bergamini, G. Giambastiani and A. Rossin, J. Mater. Chem. C, 2020, 8, 7492–7500 RSC; (i) G. Mercuri, M. Moroni, S. Galli, C. Piccirillo, A.-L. Capodilupo, G. Tuci, G. Giambastiani and A. Rossin, Inorg. Chem. Front., 2022, 9, 90–102 RSC.
- (a) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed; (b) L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- S. Rapti, D. Sarma, S. A. Diamantis, E. Skliri, G. S. Armatas, A. C. Tsipis, Y. S. Hassan, M. Alkordi, C. D. Malliakas, M. G. Kanatzidis, T. Lazarides, J. C. Plakatouras and M. J. Manos, J. Mater. Chem. A, 2017, 5, 14707–14719 RSC.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- (a) L. L. Keenan, H. A. Hamzah, M. F. Mahon, M. R. Warren and A. D. Burrows, CrystEngComm, 2016, 18, 5710–5717 RSC; (b) A. Helal, M. Nasiruzzaman Shaikh and M. Abdul Aziz, J. Photochem. Photobiol. A, 2020, 389, 112238 CrossRef CAS.
- A. D. Pournara, S. Rapti, A. Valmas, I. Margiolaki, E. Andreou, G. S. Armatas, A. C. Tsipis, J. C. Plakatouras, D. L. Giokas and M. J. Manos, J. Mater. Chem. A, 2021, 9, 3379–3387 RSC.
- Y. Feng, Q. Chen, M. Jiang and J. Yao, Ind. Eng. Chem. Res., 2019, 58, 17646–17659 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez–Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- G. E. Cmarik, M. Kim, S. M. Cohen and K. S. Walton, Langmuir, 2012, 28, 15606–15613 CrossRef CAS PubMed.
- (a) M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS; (b) S. Rapti, A. Pournara, D. Sarma, I. T. Papadas, G. S. Armatas, Y. S. Hassan, M. H. Alkordi, M. G. Kanatzidis and M. J. Manos, Inorg. Chem. Front., 2016, 3, 635–644 RSC.
- (a) K. Užarević, T. C. Wang, S.-Y. Moon, A. M. Fidelli, J. T. Hupp, O. K. Farha and T. Friščić, Chem. Commun., 2016, 52, 2133–2136 RSC; (b) S. Subudhi, G. Swain, S. P. Tripathy and K. Parida, Inorg. Chem., 2020, 59, 9824–9837 CrossRef CAS PubMed.
- (a) K. White, D. Chengelis, K. Gogick, J. Stehman, N. Rosi and S. Petoud, J. Am. Chem. Soc., 2009, 131(50), 18069–18071 CrossRef CAS PubMed; (b) A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef CAS PubMed; (c) Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC; (d) G. Collet, A. Hrvat, S. V. Eliseeva, C. Besnard, A. Kovalenko and S. Petoud, Chem. Commun., 2021, 57, 3351–3354 RSC.
- (a) M. A. Nasalevich, C. H. Hendon, J. G. Santaclara, K. Svane, B. van der Linden, S. L. Veber, M. V. Fedin, A. J. Houtepen, M. A. van der Veen, F. Kapteijn, A. Walsh and J. Gascon, Sci. Rep., 2016, 6, 23676 CrossRef CAS PubMed; (b) J. G. Santaclara, F. Kapteijn, J. Gascon and M. A. van der Veen, CrystEngComm, 2017, 19, 4118–4125 RSC.
- V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- J. Keizer, J. Am. Chem. Soc., 1983, 105, 1494–1498 CrossRef CAS.
- X. Zhang, G. Ren, M. Li, W. Yang and Q. Pan, Cryst. Growth Des., 2019, 19, 6308–6314 CrossRef CAS.
- B. Joarder, A. V. Desai, P. Samanta, S. Mukherjee and S. K. Ghosh, Chem. – Eur. J., 2015, 21, 965–969 CrossRef CAS PubMed.
- B. Wang, X.-L. Lv, D. Feng, L.-H. Xie, J. Zhang, M. Li, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 62046216 Search PubMed.
- (a) S. Azizian, J. Colloid Interface Sci., 2004, 276, 47–52 CrossRef CAS PubMed; (b) S. Douven, C. A. Paez and C. J. Gommes, J. Colloid Interface Sci., 2015, 448, 437–450 CrossRef CAS PubMed.
- L. Shi, N. Li, D. Wang, M. Fan, S. Zhang and Z. Gong, Trends Anal. Chem., 2021, 134, 116131 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc02494b |
| ‡ Caution: This work involves the study of nitroaromatic compounds which are toxic and potentially explosive. To avoid the risk of explosion, nitroaromatics should only be handled in small quantities and in aqueous solution. Gloves and appropriate eyes and face protection should be worn at all times. |
| This journal is © The Royal Society of Chemistry 2022 |