
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Additive transport in DNA molecular circuits†
Táňa
Sebechlebskᇧ
a,
Viliam
Kolivoška‡
 a,
Jakub
Šebera‡
ab,
Jiří
Fukal¶
b,
David
Řeha
c,
Miloš
Buděšínský
b,
Ivan
Rosenberg
b,
Lucie
Bednárová
b,
Jindřich
Gasior
a,
Gábor
Mészáros
d,
Magdaléna
Hromadová
*a and
Vladimír
Sychrovský
be
a,
Jakub
Šebera‡
ab,
Jiří
Fukal¶
b,
David
Řeha
c,
Miloš
Buděšínský
b,
Ivan
Rosenberg
b,
Lucie
Bednárová
b,
Jindřich
Gasior
a,
Gábor
Mészáros
d,
Magdaléna
Hromadová
*a and
Vladimír
Sychrovský
be
aJ. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Dolejškova 3, 18223 Prague, Czech Republic. E-mail: magdalena.hromadova@jh-inst.cas.cz
bInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, 166 10, Praha 6, Czech Republic
cCenter for Nanobiology and Structural Biology, Institute of Microbiology of the Czech Academy of Sciences, Zámek 136, 373 33 Nové Hrady, Czech Republic
dResearch Centre for Natural Sciences, Eötvös Lóránd Research Network, Magyar Tudósok krt. 2, H-1117, Budapest, Hungary
eDepartment of Electrotechnology, Faculty of Electrical Engineering, Czech Technical University, Technická 2, 166 27, Praha 6, Czech Republic
First published on 8th August 2022
Abstract
This work describes additive transport in DNA molecules due to a self-assembly of complementary single-stranded deoxyribonucleic acid chains, i.e. DNA hybridization. Charge transport properties in the DNA junctions at the single molecule level were studied experimentally by the break junction technique in an aqueous environment and theoretically including a non-equilibrium Green's function approach within the density functional based tight-binding method and molecular orbital calculations using density functional method and molecular dynamics simulations. Two types of anchoring groups, namely, amino and thiolate moieties were used to connect the single-stranded DNA (anchor-linker-3′-GGCACTCGG-5′-linker-anchor) to gold electrodes. Double-stranded DNA junctions were prepared by hybridization of single-stranded DNA with a complementary oligonucleotide chain (5′-CCGTGAGCC-3′) not containing linkers and anchoring groups. Three stable junction configurations were observed for both single-stranded and double-stranded DNA irrespective of the anchoring group, whereas junction conductance almost doubled upon DNA hybridization. Thiolate anchoring led to more robust and longer junction configurations compared to NH2 groups. Reasons for the observed conductance enhancement and the anchoring group effect on the overall conductance are being discussed.
Introduction
The deoxyribonucleic acid (DNA) was assumed to transport the electric charge since the resolution of its chemical structure. Early works using carefully dried double-stranded DNA (dsDNA) samples claimed its semiconducting properties due to the electronic orbital overlap of the bases along the DNA helix and postulated that the presence of water would increase its conducting properties.1,2 Later, an exponential increase of the bulk DNA conductivity with increasing sample humidity has been reported.3Nowadays, dsDNA as a naturally occurring biomolecule is regarded as a suitable platform for applications in single-molecule electronics due to its easily tunable structural and electronic properties.4 The dsDNA can perform not only as a molecular electronic element with insulating,5 semiconducting,6 metal-like conducting4a and superconducting7 properties, but also as a rectifier8 or a molecular switch.9 Recent discovery of a metallo-DNA, i.e. dsDNA involving site-specifically intercalated metal atoms or ions, further expanded the usability of DNA scaffolds for the construction of advanced molecular electronic elements.10
Scanning tunneling microscopy break junction (STM-BJ) technique has been a method of choice for determining single molecule conductance values of dsDNA in different environments.6,9,11–22 A selection of proper experimental conditions enabled STM-BJ studies of DNA duplexes in one of the three most common dsDNA forms: A, B or Z.9 The conductance was studied as a function of the varied molecular length and composition (i.e. number of base pairs, their type and position as well as the guanine content) or the presence of mismatches.
Single stranded oligonucleotides containing guanine bases were found to have the highest single molecule conductance compared to oligonucleotides consisting of equal number of either cytosine, thymine or adenosine bases.20 Double stranded DNA containing guanine–cytosine base pairs displayed higher single molecule conductance compared to that containing only thymine–adenine base pairs. It was also shown that the base pair mismatch lowers the dsDNA conductance due to a disrupted base pair stacking.10f,21,23 Other indirect methods employed the photochemical,24 biochemical25 and electro-chemical26 processes to study the charge transport (CT) in DNA.
Two principal CT mechanisms (tunnelling and hopping) have been invoked for explanation of dsDNA conductance properties.13a,27 Indeed, a multistep charge hopping over a long DNA path plays an important role in biochemical processes.6,28,29 The tunnelling, by contrast, dominates within much shorter distances. Both CT mechanisms have been used to explain theoretically the effect of DNA hybridization on its conductance properties.10,23,30–32 According to quantum chemical calculations, CT in dsDNA employs the HOMO/LUMO involving the π-electronic system of stacked base-pairs.9,10d,24d,33 On the contrary, in single-stranded DNA the conformational stability of the extended π-system of stacked nucleobases is unlikely. Therefore, the effect of stacking on CT upon DNA hybridization may be anticipated. The experimental verification of this hypothesis is rather compelling. The conductance of dsDNA increases as a result of more tightly packed base pairs upon the transition of B-form to the A-form DNA duplex.9,24d,34 To complete the story one also needs to consider another hypothesis which assumes CT through the outer-sphere of the sugar phosphodiester backbone of the DNA including tightly bound water molecules.4b,31a,f,35 In such a case the increased DNA conductance upon hybridization may be simply due to increased number of the conductance pathways. Indeed, dsDNA molecular conductance was substantially suppressed when a discontinuity (missing phosphate) was introduced on each DNA strand.31e One may then ask (i) which of the two hypotheses is correct upon DNA hybridization or (ii) if the base pair stacking as well as the backbone involvement are both needed to correctly describe the CT process. If (ii) is correct then one must consider the entire chemical structure of DNA molecule for a proper design of the DNA-based systems with desired CT properties. One indication that (ii) may be correct comes from a theoretical interpretation of the conductance differences between the A- and B-form of dsDNA.9 Indeed, it was necessary to include the backbone in the electronic structure calculations to reproduce the experimental results.
In spite of the fact that CT processes in dsDNA molecules have been studied by theoretical approaches quite extensively, the effect of hybridization on the CT process through individual DNA molecule remains still experimentally unconfirmed. This is related to the fact that the conductance of one single-stranded DNA molecule was too low to be measured by then available experimental techniques.28 Thus, the influence of individual bases on the CT upon hybridization has been addressed only after further development of the STM-BJ technique. In 2006 van Zalinge et al.20 studied the conductance properties of a series of homopolymeric oligonucleotides of different length and compared them to the results obtained for corresponding dsDNA structures both in the air and in the aqueous environment. Although the CT mechanism and the effect of DNA hybridization was not elucidated in this work it showed that single stranded DNA terminated at both ends by thiolate anchoring groups can provide measurable single molecule conductance values by the STM-BJ technique. This modification of single stranded DNA by anchoring groups at both ends inspired us and enabled us to investigate the effect of hybridization on the DNA conductance at the single molecule level.
Hereby we report systematic STM-BJ single molecule charge transport studies of single stranded and double stranded DNA molecules in their fully hydrated state (aqueous environment) with the goal of investigating the effect of DNA hybridization (dsDNA formation from two complementary ssDNA strands). Furthermore, our STM-BJ methodology36 enabled the determination of two important experimental parameters, namely, the single molecule conductance value and the most probable molecular junction (MJ) length. Presented statistical analysis of obtained CT parameters allowed more detailed characterization of the metal–DNA–metal MJs than previously reported.5,17,20,37 The non-equilibrium Green's function approach within the density functional based tight-binding method (NEGF38/DFTB39) in combination with molecular dynamics (MD)40 simulation have been used to illustrate the effects of the anchoring groups and geometry fluctuations on CT for single stranded DNA. DFTB method in connection with MD method was used for calculations of interaction energies between DNA and surface of gold electrodes. Furthermore, MD simulations were used to show possible MJ configurations of fully hydrated ssDNA and dsDNA molecule between two gold electrodes. Finally, molecular orbital calculations using density functional theory (DFT) were used for explanation of conductance difference between single stranded and double stranded DNA. For this task, the fluctuation of structure of single stranded and double stranded DNA in water environment was described by the MD simulation using DNA model without explicit inclusion of gold electrodes.
Results and discussion
Two single-stranded and two double stranded DNA molecules were used for STM-BJ single molecule conductance measurements. Chemical structures of these systems are given in Fig. S1 and S2 in ESI.† Single stranded DNA included 5′-GGCTCACGG-3′ oligonucleotide with either 5′-(CH2)6-NH2 and 3′-CH2CH(CH2OH)(CH2)4-NH2 (ssDNA-NH2) or 5′-(CH2)6-S- and 3′-(CH2)3-S- (ssDNA-S) anchoring groups, where 5′- and 3′-refer to the substituent position on deoxyribose. Molecule ssDNA-S was prepared in situ from a precursor containing 5′-(CH2)6-S-S-(CH2)6-OH and 3′-(CH2)3-S-S-(CH2)3-OH substituents after dissociation of a disulphide bond upon its contact with the gold surface.41,42 The complementary 5′-CCGTGAGCC-3′ oligonucleotide (ssDNA) was used for preparation of the double-stranded DNA molecules dsDNA-S and dsDNA-NH2. The dsDNA-NH2 duplex formation between ssDNA and ssDNA-NH2 and its stability at room temperature was confirmed by NMR and CD spectroscopy measurements (see Fig. S3–S6 in ESI†). The duplex formation between thiolate terminated single-stranded DNA and its complementary chain has been reported previously.43 Most of the previous experimental and theoretical works consider localization of HOMO on the bases (namely on the guanine) and regard such HOMO as the charge transporting pathway (hole transport) in DNA. Therefore, we selected amino and thiolate as the anchoring groups since it is well known that they also promote CT through HOMO of the molecular wire.44,45The STM-BJ method was used to determine the most probable single molecule conductance values of ssDNA-NH2, ssDNA-S, dsDNA-NH2 and dsDNA-S in the aqueous solution. The STM-BJ method is based on the repeated formation and breaking of the nanojunction between the STM tip and gold substrate in the absence and presence of studied molecules. During this process the current is monitored at a constant bias voltage applied between the tip and the substrate and the conductance is obtained using Ohm's law.46 The advantage of this technique is that both the gold–gold atomic contact conductance, so-called conductance quantum G0 = 77.5 μS, as well as the conductance corresponding to a single molecule trapped between the electrodes are being obtained in the same junction breaking process (see Fig. S7 in ESI†). Such junctions were formed and broken several hundreds of times. Recorded current–time traces were collected and treated statistically.36b Data are cumulatively presented as 1D logarithmic conductance and 2D logarithmic conductance–distance histograms. The characteristic conductance–distance behaviour can be obtained by constructing the master curves.44
Further experimental details concerning chemicals and materials, sample preparation, NMR, CD spectroscopic and STM-BJ measurements as well as detailed description of the methods used for theoretical calculations are given in ESI,† Sections S1–S7.
Fig. 1 shows 1D logarithmic conductance and 2D logarithmic conductance–distance histograms of ssDNA-NH2 (a), dsDNA-NH2 (b), ssDNA-S (c), and dsDNA-S (d), respectively. All molecules contain the same 5′-GGCTCACGG-3′ sequence wired to the gold electrodes by aliphatic linkers terminated by either the amino (ssDNA-NH2 and dsDNA-NH2) or thiolate (ssDNA-S and dsDNA-S) anchoring groups. The complementary ssDNA chain was used solely for the duplex formation (dsDNA-NH2 and dsDNA-S molecules) and did not contain any anchoring group. For each molecule, three peaks have been observed in 1D logarithmic conductance histogram. They are labelled high (H), medium (M) and low (L) based on the conductance values. The peak maxima based on the best Gaussian fits are summarized in Table 1. 2D logarithmic conductance–distance histograms in Fig. 1 represent the probability distribution of experimental pairs of [log(G/G0);Δz] values in the ensemble of the retraction curves. A conductance plateau (excluding the gold–gold atomic contacts appearing at log(G/G0) ≥ 0 as well as the noise level plateau at log(G/G0) ≈ −8) is observed only when the molecule bridges the electrodes. Up to three conductance plateaus were observed in this work for each studied molecule. For representative examples of individual logarithmic conductance–distance retraction curves see Fig. S7 (ESI†). The plateau length in 2D logarithmic conductance–distance histogram was determined statistically by the construction of the characteristic plateau length Δz histograms.47 The most probable characteristic plateau length Δz* value was obtained by the best Gaussian fit of the peak with the highest Δz value in the characteristic plateau length histogram (see Fig. S8–S11 in ESI†). Finally, the experimental MJ length zexp was obtained after the correction of Δz* for a snap-back distance. Further details on data analysis are provided in Section 5 of ESI.† The experimental MJ lengths are listed in Table 1.
 | ||
| Fig. 1 Experimental 1D logarithmic conductance (counts–log(G/G0)) and 2D logarithmic conductance–distance (log(G/G0)–Δz) histograms for (a) ssDNA-NH2 (b) dsDNA-NH2 (c) ssDNA-S and (d) dsDNA-S molecules. The master curves are shown as black dashed lines. | ||
| DNA | log(GH/G0) |
z
expH
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) [nm]
[nm] |
log(GM/G0) |
z
expM
[nm] |
log(GL/G0) |
z
expL
[nm] |
|---|---|---|---|---|---|---|
| a Experimental MJ length zexp = Δz* + 0.4 nm was obtained after correction of the most probable plateau length Δz* for a snap-back distance of 0.4 nm. The most probable plateau length Δz* values were obtained from the characteristic plateau length histograms shown in Fig. S8–S11 (ESI). | ||||||
| ssDNA-NH2 | −3.7 ± 0.4 | 0.8 ± 0.2 | −5.1 ± 0.5 | 1.6 ± 0.2 | −6.7 ± 0.3 | 2.0 ± 0.3 |
| dsDNA-NH2 | −3.5 ± 0.4 | 0.8 ± 0.2 | −4.8 ± 0.4 | 1.2 ± 0.2 | −6.4 ± 0.4 | 1.8 ± 0.3 |
| ssDNA-S | −4.2 ± 0.3 | 1.1 ± 0.2 | −5.5 ± 0.5 | 1.7 ± 0.4 | −6.9 ± 0.5 | 2.7 ± 0.3 |
| dsDNA-S | −4.1 ± 0.4 | 1.3 ± 0.4 | −5.3 ± 0.3 | 1.8 ± 0.5 | −6.7 ± 0.6 | 2.9 ± 0.6 |
Multiple conductance values have been reported for DNA molecular junction in the past.12,14,17,21 They represented integer multiples of a certain conductance value and were interpreted as conductances of junctions involving one, two or more molecules. In contrast to these reports, our work gives conductance values of distinct single molecule junction configurations differing by order(s) of magnitude from each other as well as in their length. A common feature for MJs involving a single DNA molecule is that their conductance value (GH, GM and GL) decreases exponentially with increasing MJ length, see Table 1 and Fig. 2. This fact strongly suggests that tunnelling is the dominant CT mechanism in reported systems containing either nine bases or nine base-pairs. Indeed, this CT mechanism was found to be operative within DNA molecules involving less than ca. twelve base pairs.48 For example, Hihath et al.9,34 observed tunnelling CT mechanism in a series of dsDNA molecules having an increasing number of guanine–cytosine base pairs. They obtained log(G/G0) value of −3.6 ± 0.2 for dsDNA containing nine base pairs comparable to CT characteristics of dsDNA molecules used in this work, see Table 1. Their dsDNA molecule consisted of the NH2–(CH2)3-3′-CCCGCGCCC-5′-(CH2)3–NH2 strand and complementary strand without the linkers and anchoring groups.
 | ||
| Fig. 2 Left: The correlation between log(G/G0) conductance and zexp junction length for (a) ssDNA-NH2 (black ●), dsDNA-NH2 (red ●); (b) ssDNA-S (black ●), dsDNA-S (blue ●). The error bars indicate the standard deviations of log(G/G0) and zexp values. Right: The conductance G ratio of dsDNA and ssDNA for H, M and L junction state for molecules terminated with (a) NH2– and (b) S– anchoring groups. | ||
Fig. 2 further demonstrates the effect of anchoring groups on log(G/G0) and zexp values. DNA molecules (both single stranded as well as double stranded) form longer MJ configurations when thiolates are employed as anchoring groups, which can be explained by higher mechanical stability of the junction due to a strong covalent bonding between sulphur and gold atoms.49 The NH2 anchoring groups are more directional but their interaction with gold is weaker.44c Thus, the MJs terminated by NH2 anchoring groups break on average at much shorter length than thiol-based molecules, compare Fig. 2(a) and (b). The available experimental evidence confirms that breaking of MJs based on thiolate-terminated molecules requires three times higher force compared to amino-anchored ones.49 Qualitatively different MJ evolution is seen also from the master curve analysis (black lines) in Fig. 1. All three MJ configurations (H, M, L) of ssDNA-NH2 and dsDNA-NH2 molecules display master curves of slightly decreasing logarithmic conductance with increasing junction length (black dashed lines in Fig. 1(a) and (b)), whereas length independent trends are observed for thiol-anchored DNA molecules (Fig. 1(c) and (d)). This observation is consistent with previously reported differences in the MJ breaking process for organic molecular wires terminated by these two types of anchoring groups.44 It is generally accepted that the anchoring groups can control not only the strength of the electronic coupling between molecules and metallic leads, but also the energetic alignment of molecular orbitals involved in CT through the molecule and Fermi levels of surrounding electrodes.
In order to get deeper insight into the MJ evolution and breaking process we scrutinized and categorized individual logarithmic conductance–distance retraction curves according to the occurrence of molecular plateaus. Exemplary logarithmic conductance–distance curves are shown in Fig. S7 (ESI†). All possible combinations of plateau types in individual logarithmic conductance–distance retraction curves corresponding to three MJ configurations (H, M and L) have been identified for each studied molecule. Each ensemble contained retraction curves with no plateau, with one plateau only (H, M, L), two plateaus (H + M, H + L, M + L) and three plateaus (H + M + L), respectively. A percentual distribution of curves belonging to such defined categories is given for ssDNA-NH2, dsDNA-NH2, ssDNA-S and dsDNA-S molecules in Fig. S12 of ESI.† Taking into consideration only junctions with plateaus (i.e. excluding the category of curves showing no plateau) one arrives at the conclusion that H, M and L states are almost equally probable (see Table S1, ESI,† highlighted part in orange colour) independent of DNA being single or double stranded or having different type of anchoring groups. Retraction curves that end up (MJ finally breaks off) in the L state are observed with the highest probability followed by curves that end up in the M state and the smallest number represent curves showing only the shortest H state of the junction (see Table S1 of ESI,† highlighted part in light blue colour). Similar trend was observed for detection probability of curves showing one, two and three plateaus (MJ configurations), see Table S1 (ESI†), highlighted part in light green colour. The highest number of retraction curves contains one plateau, followed by two and three plateaus. Comparison between single and double stranded DNA molecules indicates that after the DNA hybridization curves with only one plateau increase in number at the expense of curves with three plateaus. This increase is more pronounced for dsDNA-S compared to dsDNA-NH2. The number of curves where the last state before junction breaking is L increases for dsDNA-NH2 and decreases for dsDNA-S upon hybridization, which may be a consequence of the decreased flexibility of molecular structure upon hybridization as well as different mechanical stability of the MJ. One may relate this different stability to the anchoring groups. One should remind the reader that ssDNA-S molecules form systematically shorter H, M and L junction configurations than dsDNA-S molecules (Fig. 2(b)). This is not the case for ssDNA-NH2 and dsDNA-NH2, where the trend is just opposite (Fig. 2(a)). Molecules dsDNA-NH2 form MJs with shorter length than their single stranded counterparts. Importantly, the conductance values increase upon hybridization for DNA molecules terminated by both types of the anchoring groups (Fig. 2(a) and (b), right panel).
A theoretical model of single stranded DNA containing two guanine bases connected to the gold electrodes via linkers terminated by thiolate (ssDNA(GG)-S) or amino (ssDNA(GG)-NH2) anchoring groups identical to those used in STM-BJ measurements performed in this work (Fig. S13 and S14, ESI†) was developed to study the effect of anchoring groups on the DNA conductance. Molecular conductance was calculated with the DFTB39/NEGF38 approach. The effect of molecular dynamics was included. For more computational details see Section 6 of ESI.† The average interaction energy Eint between ssDNA(GG)-S and Au108 electrodes was by 44.9 kcal mol−1 more negative compared to Eint of NH2- anchored molecules (Fig. S15 of ESI†). The junction length differences found experimentally during the MJ evolution process are consistent with computed differences in the interaction energies of these two anchoring groups with the gold surface. This model served also as a benchmark for understanding of the effect of geometrical fluctuations within one MJ configuration on the distribution of the conductance values. The MJ was studied by MD throughout 10 ns time period providing eleven consecutive MJ configuration snapshots. Within this time window the original stacking of 2 guanine bases was partially disrupted (Fig. S16, ESI†), whereas NEGF-calculated conductance values based on DFTB/MD snapshots varied substantially (almost two orders of magnitude) among individual snapshots (see Fig. S17, ESI†). Results of DFTB/NEGF calculations clearly illustrate the essential contribution of geometric fluctuations towards the experimentally-obtained conductance peak widths (see Fig. S17, ESI†). Finally, DFTB/NEGF calculations support the notion that the difference in molecular conductance values between ssDNA-S and ssDNA-NH2 molecules is governed rather by the length (distance between two gold electrodes) of individual experimentally observed MJs (dependent on the type of the anchoring groups) than by the electronic properties of the anchoring groups themselves.
The experimentally obtained single molecule conductance was enhanced upon hybridization for DNA molecules terminated by both types of anchoring groups, see Fig. 2. Higher conductance increase observed for NH2- anchored DNA is only apparent due to the systematically smaller MJ length values compared to the S anchoring, compare data in Table 1 and right graphs in Fig. 2. Indeed, dsDNA-NH2 forms shorter MJ compared to ssDNA-NH2 molecular junction, whereas the opposite trend is observed for S anchored DNA molecules. Based on purely geometrical considerations, the GL conductance of dsDNA-S represents most likely the MJ configuration with double-helix chain oriented perpendicularly to the surface plane of the gold substrate. This statement is based on the fact that the estimated length of the B-form of dsDNA50 amounts to ca. 2.72 nm (eight steps multiplied by 0.34 nm), considering the molecule without any linkers and anchoring groups. The length of DNA molecules including linkers and thiolate anchors roughly corresponds to zexpL value of 2.9 ± 0.6 nm obtained for dsDNA-S and is also very close to zexpL value observed for ssDNA-S molecule (2.7 ± 0.3 nm). In analogy with the β conductance decay parameter obtained from G ∼ e−βL dependence (tunnelling CT mechanism) reported for dsDNA molecules of different base-pair composition,17,20,34,48 we have determined β values considering H, M and L states of ssDNA-S and dsDNA-S junctions from the slopes in Fig. 2(b). An identical value β = 3.7 ± 0.2 nm−1 was obtained for both thiol-based molecules. This observation may serve as additional evidence that the oligonucleotide chains in ssDNA-S and dsDNA-S MJs share similar spatial arrangement. This value compares well with β parameters17,34,48 obtained from the variation of single molecule length by changing the number of nucleotides. As discussed above, ssDNA-NH2 and dsDNA-NH2 molecules form much shorter MJs compared to ssDNA-S and dsDNA-S molecules (related to reduced bond strength between the anchor and gold). As a result, we did not find meaningful to associate the slope in Fig. 2(a) for MJs of NH2-terminated DNA molecules with β parameter. Nonetheless, this experimental observation (shorter MJs) may also indicate a qualitatively different characteristic configuration of ssDNA-NH2 and dsDNA-NH2 molecules in the junction. To explore the effect of hybridization on the single molecule conductance and MJ evolution, we have further evaluated GdsDNA/GssDNA ratios for all three experimentally observed MJ states (H, M and L). Such ratios determined for molecules terminated by both types of anchoring groups are summarized in Fig. 2 right. For thiol-terminated molecules this ratio is always lower than 2. However, for these molecules one can further account for the effect of different zexp length on GdsDNA/GssDNA ratio by employing G ∼ e−βL expression and recalculating GdsDNA/GssDNA ratio for one zexp = L since the same β parameter was found experimentally for both ssDNA-S and dsDNA-S MJs. Such corrected GdsDNA/GssDNA ratio amounts to 2.7 ± 0.5. In any case one can conclude that the conductance of studied DNA molecules approximately doubles upon the DNA hybridization in the CT tunnelling regime.
Three distinct conductance states indicate the existence of different DNA configurations and possible transitions between them during the MJ evolution. Thus, as a first step in our theoretical characterization of these systems we have performed MD simulations for ssDNA-NH2 and dsDNA-NH2 molecules in the aqueous solution (see Section 6 of ESI† for details). Fig. 3 shows the representative MD snapshots for these molecules. For the sake of simplicity, water molecules are not shown. The hypothetical CT pathway lengths indicated by numbers in between two configuration types in Fig. 3 represent the range of experimentally-observed MJ length values (zexp) given in Table 1. A blue sphere centred on one of the two NH2 anchoring groups (see top left snapshot in Fig. 3) relates to each number through its radius. Blue-coloured parts of the DNA molecule are those found within this sphere for each given pathway length. For ssDNA-NH2, the starting configuration derived from a B-form DNA duplex (Fig. 3(a) left) swiftly transfers to a “cyclic” one (Fig. 3(a) right) that is sustained during the rest of MD simulation and is therefore considered in further discussion. The fluctuations within the anchoring groups and those within terminal 3′ and 5′ phosphates follow the same pattern (see Fig. S22, ESI†). A CT pathway for MJs with zexp larger than 1.1 nm (i.e. corresponding to experimentally observed M and L states, see Table 1) involves almost the entire ssDNA-NH2 (blue coloured part of the molecule). Nevertheless, partially buried anchoring groups indicate that the MJ formation through both NH2 terminal groups may not be straightforward. Fig. S23 (ESI†) clearly indicates the proximity of 5′ and 3′ phosphates in such single-stranded DNA system(s). The existence of the “cyclic” DNA in solution does not exclude the possibility of molecular anchoring only to one electrode either.
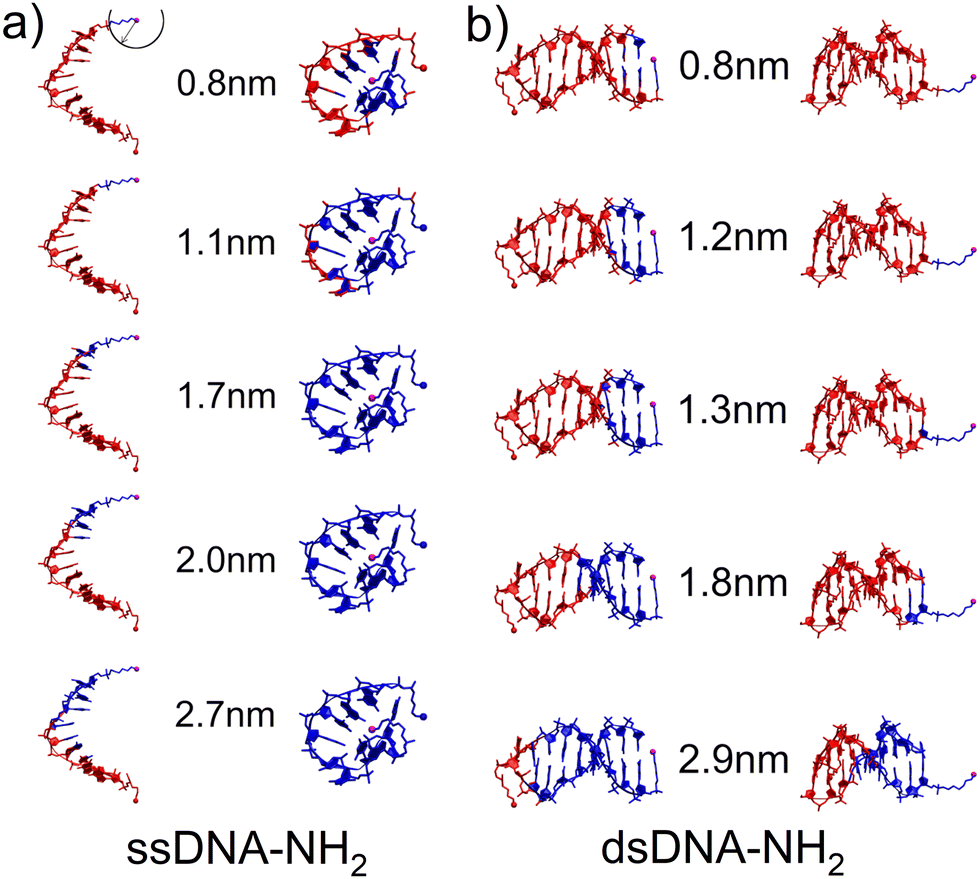 | ||
| Fig. 3 Hypothetical CT pathway domains indicated by blue-colored atoms for characteristic configurations of (a) ssDNA-NH2 and (b) dsDNA-NH2 calculated with MD in the aqueous solution. The domains include all atoms within a sphere of the radius corresponding to the experimental MJ length values given by numbers. Sphere is centered on the nitrogen atom of one of the NH2 anchoring groups (magenta ball) as indicated in the upper-left corner. (a) “Open” duplex-like (left) and “cyclic” (right) configuration for ssDNA-NH2. (b) “Nucleobase-like” (left) and “backbone-like” (right) orientation of linkers bearing NH2 anchoring group for dsDNA-NH2. | ||
The dsDNA-NH2, by contrast, behaves regularly like a canonical duplex (see Fig. 3(b) and Fig. S22, ESI†), where the hypothetical CT pathway is affected largely by a specific orientation of the linkers bearing anchoring groups (Fig. 3(b)). A closer look on structurally dynamical behaviour of the linkers and anchoring groups in dsDNA-NH2 unveiled two principal orientations, where the predominant one labelled as “nucleobase-like” refers to the parallel orientation of the linker-NH2 group (see Fig. 3(b) left and Fig. S24–S26, ESI†). The other orientation denoted as “backbone-like” involves the linker and anchoring group bent out of the duplex, see Fig. 3(b) right. Relatively free variation of the linker orientation thus may be anticipated in the experiment owing to its substantial flexibility. For dsDNA-NH2 junctions zexp values vary from 0.8 to 1.8 nm (H to L state, see Table 1). These numbers are represented by four out of five configurations depicted in Fig. 3(b). For the “backbone-like” configuration, the hypothetical CT pathway involves only the NH2- anchoring group and the alkyl linker, without the involvement of nucleotides. We consider such MJ configuration as highly unlikely. On the contrary, the “nucleobase-like” configuration involves gradually increasing number of nucleotides in the hypothetical CT pathway. We find such MJ configuration as much more realistic. We further presume that for both ssDNA-NH2 and dsDNA-NH2 molecules, the existence of three distinct MJ states (H, M and L) and sharp transitions among them (see Fig. S7, ESI†) reflect increasing number of nucleotides involved in the CT pathway in the course of the MJ elongation. Furthermore, to obtain the ratio between conductance of double and single stranded DNA we calculated molecular orbital energies using DFT method and geometries based on MD snapshots taken from previously mentioned MD simulations of ssDNA-NH2 and dsDNA-NH2 molecules in aqueous solution (see Fig. S27 and S28, ESI†). Calculated transmission functions are depicted in Fig. S29 (ESI†). These calculations provided the ratio 2.7 between conductances of double and single stranded DNA.
Further, MD simulations of MJ configurations of ssDNA-S and dsDNA-S molecules were conducted in the aqueous solution in the presence of the gold electrodes (see Section 6 of ESI†). The distance between the surface planes of gold clusters representing electrodes was set to the respective experimental MJ length values reported in Table 1 for H, M and L configurations. The MD simulations were performed by stepwise closing of the MJ to achieve the desired zexp values. Fig. 4 (water molecules are not visualized) shows that simulated MJ configuration corresponding to the experimental L state preserves the original conformation of both ssDNA-S and dsDNA-S systems. Only after a decrease of the zexp distance to that corresponding to H state the tendency to squeeze out the complementary chain in dsDNA-S is predicted. It cannot be verified that squeezing of DNA strands indeed occurs in the H state of experimentally formed MJs, but such configuration may partially explain the lower GdsDNA/GssDNA ratio observed for the H state (see Fig. 2 right). Based on these computations one may say that thus obtained predictions support the claim regarding the importance of the base pairing interactions in the overall CT process. This theoretical observation together with the fact that the experimentally obtained single molecule conductance approximately doubles upon hybridization (involving two backbone chains) could be in favour of the statement that both base-pair formation as well as an increased number of identical sugar phosphodiester backbone chains contribute to the observed conductance changes. To further corroborate the hypothesis that the sugar phosphodiester backbone plays an important role in the CT through the DNA molecule, we have performed control STM-BJ experiments in which the single stranded DNA of the same oligonucleotide sequence as used in this work had only one anchor at 5′ position. In particular, we studied CT properties of three molecules: single stranded H2N–(CH2)6-5′-GGCTCACGG-3′ and H2N–(CH2)6-5′-CCGAGTGCC-3′ and the double stranded DNA formed by hybridization of these two chains. Such prepared dsDNA contains one NH2 anchoring group on each of its complementary strands. The anchoring groups are located on the opposite side of the duplex providing spatially favorable arrangement for being contacted by the substrate and by the tip. STM-BJ histograms obtained from these measurements are summarized in Section 7 of ESI.† No single molecule conductance features were observed for these three additionally inspected systems indicating that the presence of the anchoring group on each end of one DNA strand is critical for obtaining functional MJs. The realization of MJs via hybridization of two strands each bearing only one anchoring group failed too. Such finding supports our hypothesis that the sugar phosphodiester backbone must be involved in the CT through the DNA molecule. To confirm this hypothesis, we calculated the difference in charge transport properties between ssDNA(GG)-S and ssDNA(abasic)-S junction using DFTB/NEGF method. The results show that ssDNA(GG)-S junction is indeed more effective in charge transport than ssDNA(abasic)-S junction, however, calculated ssDNA(abasic)-S conductance value is relatively close to that calculated for ssDNA(GG)-S junction. This result implies that sugar phosphodiester backbone chain can contribute to the charge transport pathway in DNA, for more details see Section 6 of the ESI.†
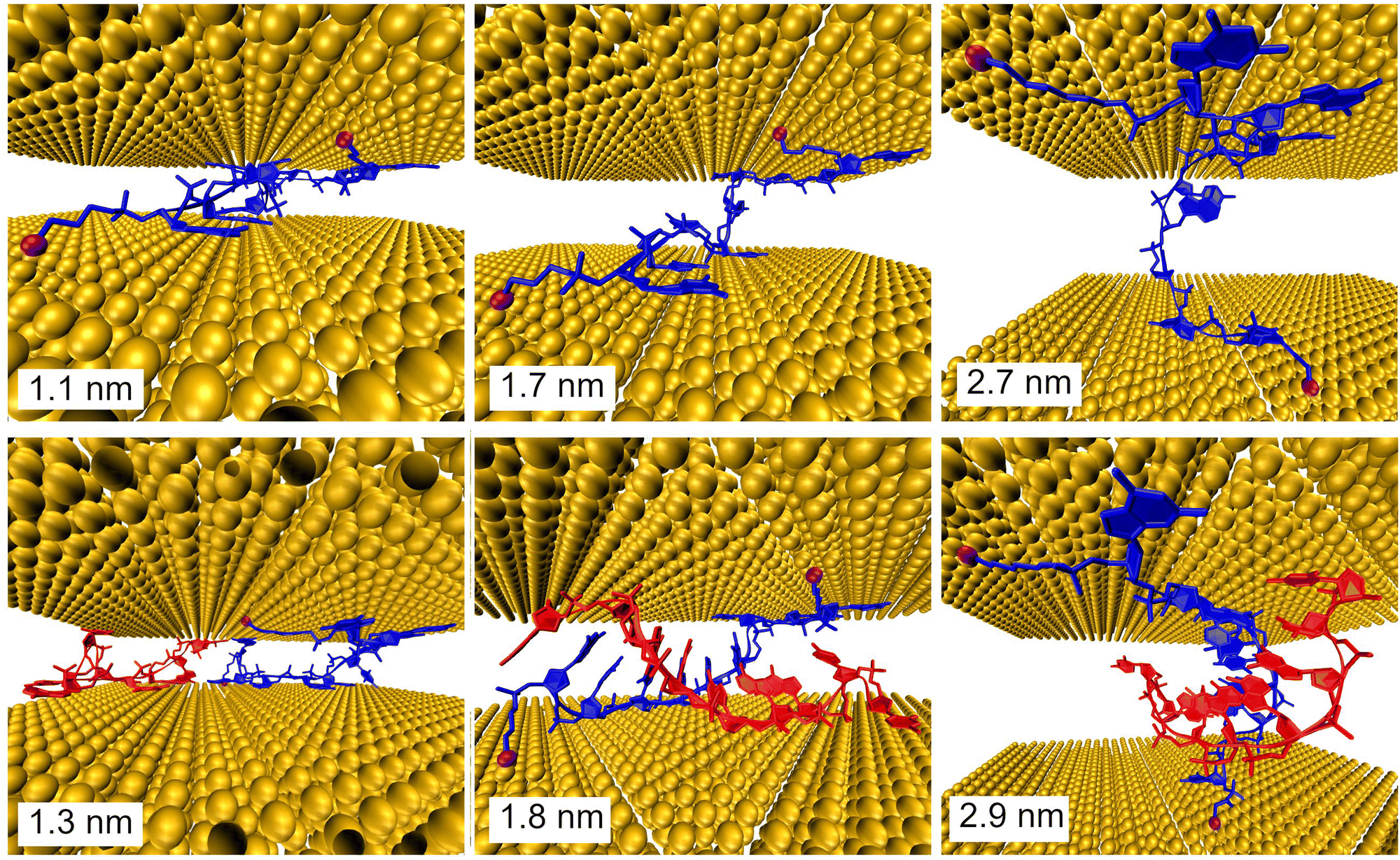 | ||
| Fig. 4 Configurations of the ssDNA-S (upper part) and dsDNA-S (lower part) molecules within gold electrodes calculated with MD. The indicated distances between electrodes in MD simulations corresponded to the experimentally obtained MJ length zexp values for the H (left), M (middle) and L (right) junction configuration. The S- anchoring groups are indicated as magenta balls. | ||
It is important to remind the reader that three distinct conductance states with roughly similar occurrence probability were observed during the MJ evolution irrespective of the number of DNA chains (one versus two) and of the type of the anchoring groups (S- versus NH2-). This observation suggests that such states are related to the MJ configurations involving the interactions between the electrodes and the sugar phosphodiester backbone (rather than directly bases or base-pairs). Presumably, distinct states are due to geometrically and energetically favourable MJ configurations that differ in the number of “electrode free” nucleotides in their entirety (i.e. not having a direct contact with the electrodes) involved in the CT pathway of increasing length in the course of the MJ evolution.
Conclusions
The conductance properties of single stranded DNA molecules containing at both backbone ends either thiolate ssDNA-S or amino ssDNA-NH2 anchoring groups have been obtained at the single molecule level by STM-BJ measurements in the aqueous environment. The evidence of the conductance enhancement upon hybridization with its complementary oligonucleotide chain ssDNA was clearly demonstrated. The conductance almost doubled after the hybridization process reflecting additive transport in DNA molecular circuits. These findings compare well with recently observed current doubling in the (gold)–semiconductor (DNA) Schottky junctions containing 0.5 mm thick layers of single stranded or double stranded DNA.51 Three distinct MJ configurations (H, M, L) were observed for both single stranded and double stranded DNA irrespective of the anchoring group used. Thiolate anchoring led to longer MJ configurations upon hybridization in all three states compared to NH2 groups, where the tendency was just the opposite. Quantum mechanical calculations supported our findings pointing to the differences in the interaction energies as well as in the DNA configuration. The upmost double enhancement of the DNA conductance due to hybridization is thus consistent with doubled number of CT pathways through DNA molecules. A control experiment with only one amino anchoring group on each strand of dsDNA-NH2 (opposite sides) failed to detect any single molecule junction conductance within the available conductance range of our STM-BJ setup. Based on the combined experimental and theoretical evidence we are able to postulate that sugar phosphodiester backbone chains can contribute to the charge transport pathway and the π-system of stacked nucleobases involving the base-pair formation is important factor in the explanation of CT through double and single stranded DNA molecules.Author contributions
Táňa Sebechlebská: STM-BJ investigation, data curation and analysis, Viliam Kolivoška: STM-BJ investigation, data curation and analysis, writing – review & editing, funding acquisition, Jakub Šebera: investigation, project administration, funding acquisition, DFTB and DFTB/NEGF calculations, Jiří Fukal: molecular dynamics simulations, NMR calculations, David Řeha: parametrization of molecular dynamics simulations of gold surface and DNA linkers, Miloš Buděšínský: data curation and analysis, NMR measurement, Ivan Rosenberg: data curation and analysis, Lucie Bednárová: data curation and analysis, CD spectroscopy, Jindřich Gasior: formal analysis, validation, Gábor Mészáros: formal analysis and validation, Magdaléna Hromadová: investigation, funding acquisition, writing of original draft, writing – review & editing, Vladimír Sychrovský: investigation, project administration, funding acquisition, writing of original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (project 18-14990S) and the Czech Academy of Sciences (RVO: 61388963 and 61388955). Computational resources were supplied by MetaCentrum computational centre.Notes and references
- D. D. Eley and D. I. Spivey, Trans. Faraday Soc., 1962, 58, 411 RSC.
- D. Han, Ha, H. Nham, K.-H. Yoo, H.-M. So, H.-Y. Lee and T. Kawai, Chem. Phys. Lett., 2002, 355, 405 CrossRef.
- T. Kleine-Ostmann, C. Jordens, K. Baaske, T. Weimann, M. H. de Angelis and M. Koch, Appl. Phys. Lett., 2006, 88, 102102 CrossRef.
- (a) H. W. Fink and C. Schonenberger, Nature, 1999, 398, 407 CrossRef CAS PubMed; (b) J. C. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642 CrossRef CAS PubMed; (c) I. Bâldea, Molecular Electronics: An Experimental and Theoretical Approach, Pan Stanford, 2016 CrossRef; (d) K. Wang, J. Funct. Biomater., 2018, 9, 8 CrossRef PubMed.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775 CrossRef CAS PubMed.
- D. Porath, A. Bezryadin, S. de Vries and C. Dekker, Nature, 2000, 403, 635 CrossRef CAS PubMed.
- A. Y. Kasumov, M. Kociak, S. Guéron, B. Reulet, V. T. Volkov, D. V. Klinov and H. Bouchiat, Science, 2001, 291, 280 CrossRef CAS PubMed.
- L. Kekedy-Nagy and E. E. Ferapontova, Angew. Chem., Int. Ed., 2019, 58, 3048 CrossRef CAS PubMed.
- J. M. Artés, Y. H. Li, J. Q. Qi, M. P. Anantram and J. Hihath, Nat. Commun., 2015, 6, 8870 CrossRef PubMed.
- (a) Y. Tanaka, J. Kondo, V. Sychrovsky, J. Sebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343 RSC; (b) J. Muller, Eur. J. Inorg. Chem., 2008, 3749 CrossRef; (c) P. Auffinger and E. Ennifar, Nat. Chem., 2017, 9, 932 CrossRef CAS PubMed; (d) I. Kratochvilova, M. Golan, M. Vala, M. Sperova, M. Weiter, O. Pav, J. Sebera, I. Rosenberg, V. Sychrovsky, Y. Tanaka and F. M. Bickelhaupt, J. Phys. Chem. B, 2014, 118, 5374 CrossRef CAS PubMed; (e) I. Kratochvilova, K. Kral, M. Buncek, A. Viskova, S. Nespurek, A. Kochalska, T. Todorciuc, M. Weiter and B. Schneider, Biophys. Chem., 2008, 138, 3 CrossRef CAS PubMed; (f) I. Kratochvilova, T. Todorciuc, K. Kral, H. Nemec, M. Buncek, J. Sebera, S. Zalis, Z. Vokacova, V. Sychrovsky, L. Bednarova, P. Mojzes and B. Schneider, J. Phys. Chem. B, 2010, 114, 5196 CrossRef CAS PubMed; (g) E. Toomey, J. Xu, S. Vecchioni, L. Rothschild, S. Wind and G. E. Fernandes, J. Phys. Chem. C, 2016, 120, 7804 CrossRef CAS; (h) S. Roy, H. Vedala, A. D. Roy, D. H. Kim, M. Doud, K. Mathee, H. K. Shin, N. Shimamoto, V. Prasad and W. B. Choi, Nano Lett., 2008, 8, 26 CrossRef CAS PubMed; (i) C. L. Guo, K. Wang, E. Zerah-Harush, J. Hamill, B. Wang, Y. Dubi and B. Q. Xu, Nat. Chem., 2016, 8, 484 CrossRef CAS PubMed; (j) D. Reha, A. A. Voityuk and S. A. Harris, ACS Nano, 2010, 4, 5737 CrossRef CAS PubMed; (k) K. Siriwong and A. A. Voityuk, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 780 CAS.
- L. Zhang, J. Zhang and J. Ulstrup, Curr. Opin. Electrochem., 2017, 4, 166 CrossRef CAS.
- R. Venkatramani, K. L. Davis, E. Wierzbinski, S. Bezer, A. Balaeff, S. Keinan, A. Paul, L. Kocsis, D. N. Beratan, C. Achim and D. H. Waldeck, J. Am. Chem. Soc., 2011, 133, 62 CrossRef CAS PubMed.
- (a) L. Xiang, J. L. Palma, Ch Bruot, V. Mujica, M. A. Ratner and N. J. Tao, Nat. Chem., 2015, 7, 221 CrossRef CAS PubMed; (b) L. Xiang, J. L. Palma, Y. Li, V. Mujica, M. A. Ratner and N. J. Tao, Nat. Commun., 2017, 8, 14471 CrossRef CAS PubMed.
- (a) J. Hihath, F. Chen, P. Zhang and N. J. Tao, J. Phys.: Condens. Matter, 2007, 19, 215202 CrossRef; (b) J. Hihath, B. Xu, P. M. Zhang and N. J. Tao, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16979 CrossRef CAS PubMed; (c) J. Hihath, S. Guo, P. Zhang and N. J. Tao, J. Phys.: Condens. Matter, 2012, 24, 164204 CrossRef PubMed.
- M. W. Shinwari, M. J. Deen, E. B. Starikov and G. Cuniberti, Adv. Funct. Mater., 2010, 20, 1865 CrossRef.
- P. T. Bui, T. Nishino, H. Shiigi and T. Nagaoka, Chem. Commun., 2015, 51, 1666 RSC.
- B. Xu, P. Zhang, X. Li and N. Tao, Nano Lett., 2004, 4, 1105 CrossRef CAS.
- (a) C. Nogues, S. R. Cohen, S. S. Daube and R. Naaman, Phys. Chem. Chem. Phys., 2004, 6, 4459 RSC; (b) C. Nogues, S. R. Cohen, S. Daube, N. Apter and R. Naaman, J. Phys. Chem. B, 2006, 110, 8910 CrossRef CAS PubMed.
- S. M. Iqbal, G. Balasundaram, S. Ghosh, D. E. Bergstrom and R. Bashir, Appl. Phys. Lett., 2005, 86, 153901 CrossRef.
- H. van Zalinge, D. J. Schiffrin, A. D. Bates, W. Haiss, J. Ulstrup and R. J. Nichols, ChemPhysChem, 2006, 7, 94 CrossRef CAS PubMed.
- (a) E. Wierzbinski, R. Venkatramani, K. L. Davis, S. Bezer, J. Kong, Y. Xing, E. Borguet, C. Achim, D. N. Beratan and D. H. Waldeck, ACS Nano, 2013, 7, 5391 CrossRef CAS PubMed; (b) R. Venkatramani, E. Wierzbinski, D. H. Waldeck and D. N. Beratan, Faraday Discuss., 2014, 174, 57 RSC.
- X. Guo, A. A. Gorodetsky, J. Hone, J. K. Barton and C. Nuckolls, Nat. Nanotechnol., 2008, 3, 163 CrossRef CAS PubMed.
- M. H. Lee, G. Brancolini, R. Gutiérrez, R. Di Felice and G. Cuniberti, J. Phys. Chem. B, 2012, 116, 10977 CrossRef CAS PubMed.
- (a) C. J. Murphy, M. R. Arkin, Y. Jenkins, N. D. Ghatlia, S. H. Bossmann, N. J. Turro and J. K. Barton, Science, 1993, 262, 1025 CrossRef CAS PubMed; (b) C. J. Murphy, M. R. Arkin, N. D. Ghatlia, S. Bossmann, N. J. Turro and J. K. Barton, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 5315 CrossRef CAS PubMed; (c) F. D. Lewis, T. F. Wu, Y. F. Zhang, R. L. Letsinger, S. R. Greenfield and M. R. Wasielewski, Science, 1997, 277, 673 CrossRef CAS PubMed; (d) I. Kratochvilova, M. Vala, M. Weiter, M. Sperova, B. Schneider, O. Pav, J. Sebera, I. Rosenberg and V. Sychrovsky, Biophys. Chem., 2013, 180, 127 CrossRef PubMed; (e) B. Giese, J. Amaudrut, A. K. Kohler, M. Spormann and S. Wessely, Nature, 2001, 412, 318 CrossRef CAS PubMed.
- (a) D. B. Hall, R. E. Holmlin and J. K. Barton, Nature, 1996, 382, 731 CrossRef CAS PubMed; (b) E. Meggers, D. Kusch, M. Spichty, U. Wille and B. Giese, Angew. Chem., Int. Ed., 1998, 37, 460 CrossRef CAS; (c) I. Saito, T. Nakamura, K. Nakatani, Y. Yoshioka, K. Yamaguchi and H. Sugiyama, J. Am. Chem. Soc., 1998, 120, 12686 CrossRef CAS.
- (a) S. O. Kelley, N. M. Jackson, M. G. Hill and J. K. Barton, Angew. Chem., Int. Ed., 1999, 38, 941 CrossRef CAS; (b) T. Liu and J. K. Barton, J. Am. Chem. Soc., 2005, 127, 10160 CrossRef CAS PubMed.
- Y. A. Berlin, A. L. Burin and M. A. Ratner, Chem. Phys., 2002, 275, 61 CrossRef CAS.
- H. Cohen, C. Nogues, R. Naaman and D. Porath, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11589 CrossRef CAS PubMed.
- (a) M. E. Nunez, D. B. Hall and J. K. Barton, Chem. Biol., 1999, 6, 85 CrossRef CAS PubMed; (b) B. Giese and M. Spichty, ChemPhysChem, 2000, 1, 195 CrossRef CAS PubMed.
- (a) R. G. Endres, D. L. Cox and R. R. P. Singh, Rev. Mod. Phys., 2004, 76, 195 CrossRef CAS; (b) K. Senthilkumar, F. C. Grozema, C. F. Guerra, F. M. Bickelhaupt, F. D. Lewis, Y. A. Berlin, M. A. Ratner and L. D. A. Siebbeles, J. Am. Chem. Soc., 2005, 127, 14894 CrossRef CAS PubMed; (c) T. Kubar, R. Gutierrez, U. Kleinekathofer, G. Cuniberti and M. Elstner, Phys. Status Solidi B, 2013, 250, 2277 CrossRef CAS; (d) K.-I. Dedachi, T. Natsume, T. Nakatsu, S. Tanaka, Y. Ishikawa and N. Kurita, Chem. Phys. Lett., 2007, 436, 244 CrossRef CAS PubMed; (e) D. Řeha, W. Barford and S. Harris, Phys. Chem. Chem. Phys., 2008, 10, 5436 RSC.
- (a) U. Diederichsen, Angew. Chem., Int. Ed. Engl., 1997, 36, 2317 CrossRef CAS; (b) C. Bruot, L. M. Xiang, J. L. Palma, Y. Q. Li and N. J. Tao, J. Am. Chem. Soc., 2015, 137, 13933 CrossRef CAS PubMed; (c) S. O. Kelley, R. E. Holmlin, E. D. A. Stemp and J. K. Barton, J. Am. Chem. Soc., 1997, 119, 9861 CrossRef CAS; (d) S. O. Kelley, E. M. Boon, J. K. Barton, N. M. Jackson and M. G. Hill, Nucleic Acids Res., 1999, 27, 4830 CrossRef CAS PubMed; (e) R. Zhuravel, H. Huang, G. Polycarpou, S. Polydorides, P. Motamarri, L. Katrivas, D. Rotem, J. Sperling, L. A. Zotti, A. B. Kotlyar, J. C. Cuevas, V. Gavini, S. S. Skourtis and D. Porath, Nat. Nanotechnol., 2020, 15, 836 CrossRef CAS PubMed; (f) E. Beall, S. Ulku, C. Liu, E. Wierzbinski, Y. Zhang, Y. Bae, P. Zhang, C. Achim, D. N. Beratan and D. H. Waldeck, J. Am. Chem. Soc., 2017, 139, 6726 CrossRef CAS PubMed.
- M. Wolter, M. Elstner and T. Kubař, J. Chem. Phys., 2013, 139, 125102 CrossRef PubMed.
- T. Uchiyama, T. Miura, H. Takeuchi, T. Dairaku, T. Komuro, T. Kawamura, Y. Kondo, L. Benda, V. Sychrovsky, P. Bour, I. Okamoto, A. Ono and Y. Tanaka, Nucleic Acids Res., 2012, 40, 5766 CrossRef CAS PubMed.
- Y. H. Li, J. M. Artes, J. Qi, I. A. Morelan, P. Feldstein, M. P. Anantram and J. Hihath, J. Phys. Chem. Lett., 2016, 7, 1888 CrossRef CAS PubMed.
- J. M. Warman, M. P. de Haas and A. Rupprecht, Chem. Phys. Lett., 1996, 249, 319 CrossRef CAS.
- (a) V. Kolivoška, M. Mohos, I. V. Pobelov, S. Rohrbach, K. Yoshida, W. Hong, Y. C. Fu, P. Moreno-García, G. Mészáros, P. Broekmann, M. Hromadová, R. Sokolová, M. Valášek and Th Wandlowski, Chem. Commun., 2014, 50, 11757 RSC; (b) J. Šebera, V. Kolivoška, M. Valášek, J. Gasior, R. Sokolová, G. Mészáros, W. Hong, M. Mayor and M. Hromadová, J. Phys. Chem. C, 2017, 121, 12885 CrossRef; (c) Š. Nováková Lachmanová, J. Šebera, V. Kolivoška, J. Gasior, G. Mészáros, G. Dupeyre, P. P. Lainé and M. Hromadová, Electrochim. Acta, 2018, 264, 301 CrossRef; (d) J. Šebera, T. Sebechlebská, Š. Nováková Lachmanová, J. Gasior, P. Moreno Garcia, G. Mészáros, M. Valášek, V. Kolivoška and M. Hromadová, Electrochim. Acta, 2019, 301, 267 CrossRef.
- (a) M. Ratner, Nat. Nanotechnol., 2013, 8, 378 CrossRef CAS PubMed; (b) Y. Okahata, T. Kobayashi, K. Tanaka and M. Shimomura, J. Am. Chem. Soc., 1998, 120, 6165 CrossRef CAS; (c) Y. Zhang, R. H. Austin, J. Kraeft, E. C. Cox and N. P. Ong, Phys. Rev. Lett., 2002, 89, 198102 CrossRef CAS PubMed; (d) Y. H. Li, J. M. Artes and J. Hihath, Small, 2016, 12, 432 CrossRef CAS PubMed.
- S. Datta, Superlattices Microstruct., 2000, 28, 253 CrossRef CAS.
- G. Seifert, J. Phys. Chem. A, 2007, 111, 5609 CrossRef CAS PubMed.
- (a) D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701 CrossRef CAS PubMed; (b) H. J. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43 CrossRef CAS; (c) S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson and D. van der Spoel, Bioinformatics, 2013, 29, 845 CrossRef CAS PubMed.
- C. D. Bain, H. A. Biebuyck and G. M. Whitesides, Langmuir, 1989, 5, 723 CrossRef CAS.
- X. D. Cui, A. Primak, X. Zarate, J. Tomfohr, O. F. Sankey, A. L. Moore, T. A. Moore, D. Gust, G. Harris and S. M. Lindsay, Science, 2001, 294, 571 CrossRef CAS PubMed.
- (a) T. Ohshiro and M. Maeda, Chem. Commun., 2010, 46, 2581 RSC; (b) T. Harashima, Y. Hasegawa, S. Kaneko, Y. Jono, S. Fujii, M. Kiguchi and T. Nishino, Chem. Sci., 2021, 12, 2217 RSC.
- (a) W. Hong, D. Z. Manrique, P. Moreno-García, M. Gulcur, A. Mishchenko, C. J. Lambert, M. R. Bryce and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 2292 CrossRef CAS PubMed; (b) P. Moreno-García, M. Gulcur, D. Z. Manrique, T. Pope, W. Hong, V. Kaliginedi, C. Huang, A. S. Batsanov, M. R. Bryce, C. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2013, 135, 12228 CrossRef PubMed; (c) F. Chen, X. Li, J. Hihath, Z. Huang and N. J. Tao, J. Am. Chem. Soc., 2006, 128, 15874 CrossRef CAS PubMed.
- K. W. Hipps, Science, 2001, 294, 536 CrossRef CAS PubMed.
- M. Hromadová and V. Kolivoška, in Encyclopedia of Interfacial Chemistry: Surface Science and Electrochemistry, ed. K. Wandelt, Elsevier, Amsterdam, 2018, vol. 5, pp. 271–280 Search PubMed.
- I. V. Pobelov, M. Mohos, K. Yoshida, V. Kolivoska, A. Avdic, A. Lugstein, E. Bertagnolli, K. Leonhardt, G. Denuault and B. Gollas, Nanotechnology, 2013, 24, 115501 CrossRef PubMed.
- Y. Q. Li, L. M. Xiang, J. L. Palma, Y. Asai and N. J. Tao, Nat. Commun., 2016, 7, 11294 CrossRef CAS PubMed.
- (a) B. Xu, X. Xiao and N. J. Tao, J. Am. Chem. Soc., 2003, 125, 16164 CrossRef CAS PubMed; (b) M. Frei, S. V. Aradhya, M. Koentopp, M. S. Hybertsen and L. Venkataraman, Nano Lett., 2011, 11, 1518 CrossRef CAS PubMed; (c) I. V. Pobelov, K. Primdal Lauritzen, K. Yoshida, A. Jensen, G. Mészáros, K. W. Jacobsen, M. Strange, T. Wandlowski and G. C. Solomon, Nat. Commun., 2017, 8, 15931 CrossRef CAS PubMed.
- H. F. Lodish, A. Berk, Ch Kaiser, M. Krieger, M. P. Scott, A. Bretscher, H. L. Ploegh and P. T. Matsudaira, Molecular cell biology, Macmillan, New York, 2008 Search PubMed.
- S. M. A. Daraghma, S. Talebi and V. Periasamy, Eur. Phys. J. E: Soft Matter Biol. Phys., 2020, 43, 40 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Chemicals and materials; sample preparation; NMR, CD and STM break junction measurements; theoretical calculations; STM break junction results for DNA chains with one anchoring group. See DOI: https://doi.org/10.1039/d2tc01219g |
| ‡ These authors contributed equally. |
| § Present address: Department of Physical and Theoretical Chemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Mlynská dolina, Ilkovičova 6, 84215 Bratislava, Slovakia. |
| ¶ Present address: Department of Physical Chemistry, Faculty of Science, Palacký University Olomouc, tr. 17. listopadu 1192/12, 771 46 Olomouc, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2022 |