
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Critical analysis of self-doping and water-soluble n-type organic semiconductors: structures and mechanisms†
Lewis M.
Cowen
*a,
Peter A.
Gilhooly-Finn
ab,
Alexander
Giovannitti
 c,
Garrett
LeCroy
c,
Harry
Demetriou
d,
William
Neal
b,
Yifan
Dong
a,
Megan
Westwood
a,
Sally
Luong
e,
Oliver
Fenwick
e,
Alberto
Salleo
c,
Sandrine
Heutz
d,
Christian B.
Nielsen
b and
Bob C.
Schroeder
*a
c,
Garrett
LeCroy
c,
Harry
Demetriou
d,
William
Neal
b,
Yifan
Dong
a,
Megan
Westwood
a,
Sally
Luong
e,
Oliver
Fenwick
e,
Alberto
Salleo
c,
Sandrine
Heutz
d,
Christian B.
Nielsen
b and
Bob C.
Schroeder
*a
aDepartment of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: lewis.cowen.17@ucl.ac.uk; b.c.schroeder@ucl.ac.uk
bDepartment of Chemistry, Queen Mary University of London, Mile End Road, London, E1 4NS, UK
cDepartment of Materials Science and Engineering, Stanford University, Stanford, CA 94305, USA
dDepartment of Materials and London Centre for Nanotechnology, Imperial College London, London SW7 2AZ, UK
eSchool of Engineering and Materials Science, Queen Mary University of London, London, E1 4NS, UK
First published on 24th May 2022
Abstract
Self-doping organic semiconductors provide a promising route to avoid instabilities and morphological issues associated with molecular n-type dopants. Structural characterization of a naphthalenetetracarboxylic diimide (NDI) semiconductor covalently bound to an ammonium hydroxide group is presented. The dopant precursor was found to be the product of an unexpected base catalyzed hydrolysis, which was reversible. The reversible hydrolysis had profound consequences on the chemical composition, morphology, and electronic performance of the doped films. In addition, we investigated the degradation mechanism of the quaternary ammonium group and the subsequent doping of NDI. These findings reveal that the products of more than one chemical reaction during processing of films must be considered when utilizing this promising class of water-soluble semiconductors.
Introduction
The introduction of charges on to organic semiconductors through doping with an external molecular species is vital for their use in various applications. The requirement for low ionization potentials (IP) in n-type dopants means that their application has been slow when compared to their p-type counter-parts.1 Organic photovoltaics (OPV),2 thermoelectrics (OTE)3,4 and field effect transistors (OFET)5 are all reliant upon, or their performance can be improved by, efficient doping of the n-type component. A more thorough understanding of the chemical structures, transformations and charge transfer processes undergone between n-type semiconductors and dopants is therefore important for future commercial success of these various fields.One major obstacle to efficient n-type doping systems has been the instability of dopants and doped semiconductors. Dopants with small solid-state ionization potentials (IP), typically less than 4.0 eV, are capable of reducing O2 to O2− and H2O to HO− under atmospheric conditions. Similarly, doped n-type semiconductors require very large electron affinities (EA) to prevent oxidation of the excited state.6,7 Problems are therefore encountered in their handling and experimental studies are mostly limited to glove-box work. To avoid this, successful n-type dopants are normally generated from a stable precursor. For example, stable 18 electron dimers of metallocenes are often cleaved to give the 19 electron metallocene as dopant.8,9 Similarly, 2,3-dihydro-1H-benzoimidazoles (DMBI-H)10–13 and triaminomethanes (TAM)14 are not active dopants but donate hydrides which reduce organic semiconductors.
Quaternary ammonium halide dopant precursors, covalently bound to a perylenetetracarboxylic diimide (PDI) aromatic acceptor, have been designed in an effort to produce n-type organic semiconductors which experience minimal morphological disruption upon doping. Reducing the stable ammonium halide precursor with sodium metal leads to the zwitterionic doped semiconductor. Blends of the zwitterionic doped species and a neutral PDI derivative exhibited conductivities of 10 mS cm−1.15,16 The introduction of covalently bound quaternary ammonium groups is an attractive prospect due to their charged nature, allowing for processing from more environmentally friendly solvents such as water without the use of additives.17 Furthermore the use of orthogonal solvent systems facilitates device fabrication, as miscibility between dopant and semiconductor in the solid state is avoided.18–20 Quaternary ammonium hydroxide moieties have been bound to rylene diimides and, upon dehydration of aqueous films, absorption spectra indicating the presence of charge carriers were recorded.21 An ion-exchange column was used to exchange a halide precursor for the hydroxyl counterion, shown as PDI-OH in Fig. 1. The resultant aqueous solution was found to be stable under ambient conditions and formed conductive homogeneous films upon dehydration. It was suggested that the driving force behind doping was the loss of solvation energy around the hydroxyl anion upon film formation, subsequently the negative charge was transferred to the more polarizable PDI core. By altering the alkyl spacer length, n, between the PDI core and the quaternary ammonium moiety, PDI–OH has exhibited maximum electrical conductivities, σ, of 0.5 S cm−1.22 A mechanism of doping has been investigated involving the demethylation of the ammonium, leaving a dimethylamine group bound to the PDI sidechain.23 The resultant dimethylamine was suggested to be the doping moiety. This was evidenced by a growth in X-ray photoelectron spectroscopy (XPS) signal for NMe2 nitrogen upon film annealing while the NMe3+ signal diminished. Photo-24–26 and thermal-induced27 electron transfers between bound tertiary amines and rylene diimide semiconductors have previously been observed.
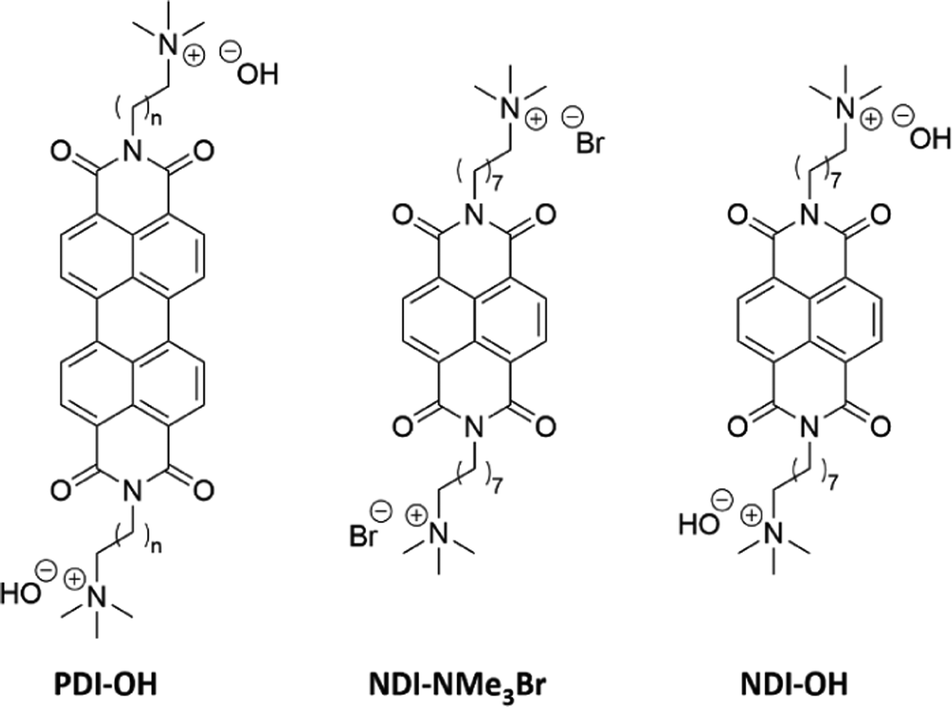 | ||
| Fig. 1 Structure of previously studied PDI–OH21–23 as well as the halide precursor NDI-NMe3Br and NDI-OH reported here. | ||
It is possible however that the quaternary ammonium end group degrades via alternative routes. Previously degradation has been shown to favor an E2 Hofmann elimination when β-hydrogens are present and sterically available for abstraction.28–31 This would result in a tertiary amine, trimethylamine (TMA), as well as a terminal alkene on the semiconductor side chain.
In order to further elucidate the chemical pathways through which quaternary amines act as n-type dopants to bound rylene diimides, we synthesized the compound N,N′-bis(8-(trimethyl-ammoniumbromide)octylene)-1,4,5,8-naphthalenetetracarboxylic diimide (NDI-NMe3Br, Fig. 1). As the aim of this study is to gain deeper insights into the underlying doping chemistry of quaternary amine functionalized rylene diimides, rather than to maximize electrical conductivity, NDI was chosen as the semiconducting building block rather than PDI. In case of unexpected chemical reactions during the doping mechanism, the NDI core will favor the formation of a lesser number of isomers, thereby facilitating the structural characterization compared to PDI derivatives. The halide counterion was exchanged for a hydroxide via ion-exchange resin to give an aqueous solution of the dopant precursor, N,N′-bis(8-(trimethylammoniumhydroxide)octylene)-1,4,5,8-naphthalene-tetracarboxylic diimide (NDI-OH, Fig. 1). Using an NDI aromatic core, rather than PDI, allows for increased solubility and simplified analysis of solution spectroscopic measurements at the expense of a decreased conjugation length in the semiconducting core. This trade-off between potentially decreased electronic performance, in NDI compared to PDI, and ease of analysis was considered worthwhile, since the aim of this study was primarily focused on structural characterization of NDI-OH and determination of the role of the quaternary ammonium group in doping, rather than optimizing electrical conductivity.
A combination of nuclear magnetic resonance (NMR) and infra-red (IR) spectroscopies, as well as mass-spectrometry, have been used to confirm the structure of the dopant precursor for the first time as well as analyse the product of doping upon drying. These structural characterizations revealed that dynamic reactions involving the NDI imide group play a part in the composition of doped films. An insight into how the complex reactions involving NDI and quaternary ammonium groups affects film morphology was investigated through atomic force microscopy (AFM) and grazing incidence wide angle X-ray scattering (GIWAXS) measurements. In combination with this ultraviolet-visible (UV-vis) absorption spectroscopy, electron paramagnetic resonance (EPR) spectroscopy and four-point probe conductivity measurements, have been used to help probe both the mechanism and efficiency of intramolecular charge transfer.
Results and discussion
Synthesis and characterisation of NDI-OH
Two synthetic routes to the quaternary ammonium halides 5 and NDI-NMe3Br are outlined in Scheme 1. Napthalenetetracarboxylic dianhydride (1) was reacted with ammonium acetate to give naphthalenetetracarboxylic diimide (2). A Mitsunobu type reaction was then used to give the N-bromooctyl functionalized product 3. The initial target was the quaternary ammonium iodide product 5, however the dimethylamine intermediate 4 was found to be unstable, likely through an intramolecular photoinduced electron transfer.245 was therefore synthesized via a one-pot reaction in which the SN2 substitution to give 4 was followed immediately by quaternization with methyl iodide. This made the separation of pure 5 from the reaction mixture difficult and the quaternary ammonium halide target was changed to NDI-NMe3Br. Quaternization via this route was achieved by reaction of 3 with the salt trimethylamine hydrochloride under basic conditions to yield NDI-NMe3Br. The presence of predominantly bromide counterion, rather than chloride, was confirmed by X-ray fluorescence (XRF) spectroscopy and results are summarised in Table S1 of the ESI.† Bromine was detected at a concentration of 94.364% and chlorine at 3.422%. Elements lighter than sodium are difficult to detect by XRF so are not included in the results.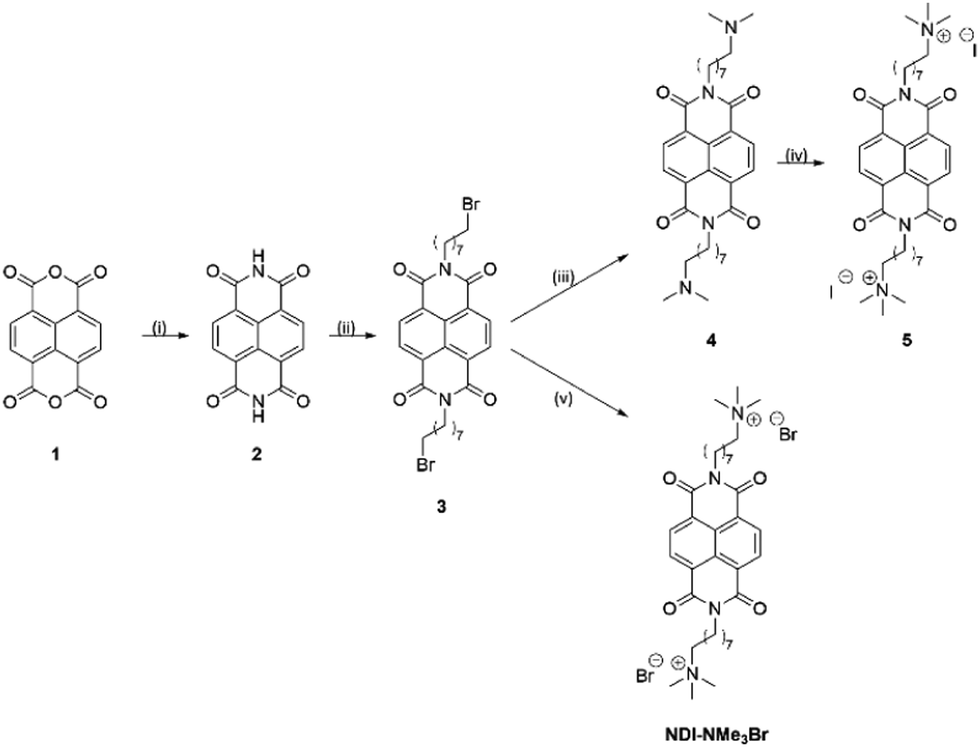 | ||
| Scheme 1 Synthesis of quaternary ammonium halides 5 and NDI-NMe3Br. Reagents and conditions: (i) NH4OAc, AcOH, reflux 1 h, 72% (ii) 8-bromooctan-1-ol, DEAD, PPh3, THF, 14 h, 21% (iii) NHMe2, THF, reflux 16 h, (iv) MeI, CHCl3, reflux 2 h, 44% combined yield with (iii). (v) NMe3·HCl, NaHCO3, MeCN, 80 °C, 12 h, 51%. | ||
Previously quaternary ammonium hydroxides, such as NDI-OH, have been generated by eluting an aqueous solution of a corresponding ammonium halide through a column of hydroxy-form ion exchange resin.21–23 The resultant aqueous solution has not previously been subjected to any structural analysis, however. To elucidate the chemical structure of NDI-OH we first prepared a sample by passing NDI-NMe3Br through a column of hydroxy-form ion exchange resin using deuterium oxide as eluent. It was then possible to perform NMR experiments on the resultant solution. Two key identifying regions of the solution 1H NMR spectrum of NDI-OH are shown in Fig. 2 and are compared to the same regions for NDI-NMe3Br. A singlet corresponding to the four equivalent aromatic protons, normally observed in symmetrical NDI compounds, was seen in NDI-NMe3Br at 8.24 ppm. This singlet however was not seen in NDI-OH but instead four doublets were observed at 8.51, 8.48, 7.89 and 7.86 ppm. The appearance of these doublets suggests a loss of equivalence in the NDI aromatic hydrogens and the loss of a plane of symmetry in the molecule. The loss of symmetry is also supported by the observation of additional signals at 3.35 ppm and 3.25 ppm in the spectrum for NDI-OH. Two further singlets are also observed in the region between 3.04 and 3.02 ppm indicating the presence of additional N-methyl hydrogen environments.
 | ||
| Fig. 2 (a) Hydrolysis of NDI-NMe3Br to give a mixture of products. (b) Solution 1H NMR spectra of NDI-NMe3Br (bottom) and hydrolysed NDI-OH mixture (top) in deuterium oxide. Inset shows signals attributed to the doubly hydrolysed syn (7) and anti (8) products. | ||
When exposed to solutions of aqueous base, the imide group of NDI derivatives are susceptible to ring-opening hydrolysis.32 This observation provides a rationale for the differences in NMR spectra between NDI-NMe3Br and NDI-OH. The measured pH of an aqueous solution of NDI-OH after eluting through the ion-exchange column was 9.5. When exposed to the basic ion-exchange resin NDI-NMe3Br underwent hydrolysis at one or both of its imide groups, the process is summarized in Fig. 2. The majority product, which gives rise to the doublets between 8.52 and 7.85 ppm, is the mono-amide (6). Two isolated singlets are observed for the syn-product (7) at 7.71 and 7.67 ppm, whereas the anti-product (8) displays two pairs of equivalent hydrogens which give rise to an AB-quartet centered on 7.69 ppm (J = 7.4 Hz).
Full NMR characterization of NDI-OH, including 1H, 13C and homonuclear 2D NMR (Fig. S2 to S8, ESI†), further support the claim that NDI-OH in solution is a mixture of products of base catalyzed hydrolysis. In addition to this electron-spray ionization mass spectrometry (ESI-MS), shown in Fig. S1 (ESI†), gives rise to peaks at 636 m/z and 312 m/z, corresponding to the singly and doubly charged product 6.
Finally, hydrolysis was also observed in absorption spectra. Fig. 3a shows a comparison between the solution absorption spectra of NDI-NMe3Br and NDI-OH and the results are summarized in Table 1. The spectrum of NDI-NMe3Br closely matches previous NDI derivatives with two absorption maxima at 363 and 383 nm.32,36 For NDI-OH a single absorption band with a small shoulder was observed with maxima at 352 nm. Similar absorption spectra have been observed for 1,8-naphthalamides37 and have previously been assigned as mono-hydrolysed products when naphthalenediimides were treated with aqueous base.32 The loss of an imide ring in NDI-OH, when compared to NDI-NMe3Br, may also lead to a decrease in the electron affinity (EA).36 This is supported by the results of solution cyclic voltammetry (CV) measurements, shown in Fig. S17 (ESI†) and results are summarized in Table 1. NDI-NMe3Br shows a fully reversible reduction with a half potential (E1/2) of −0.56 V. From E1/2, and using Ag/Ag+ as reference half-cell, an EA of 4.16 eV was estimated for NDI-NMe3Br. The reduction of NDI-OH observes quasi-reversibility and therefore the EA was estimated from the onset of reduction (Eonset) at −0.91 V. An estimated EA of 3.80 eV was derived for NDI-OH, a decrease of 0.36 eV when compared to NDI-NMe3Br. This destabilization of the LUMO, as a result of ring-opening hydrolysis, may have a detrimental effect on the NDI-OH aromatic core's ability to act as an electron acceptor.
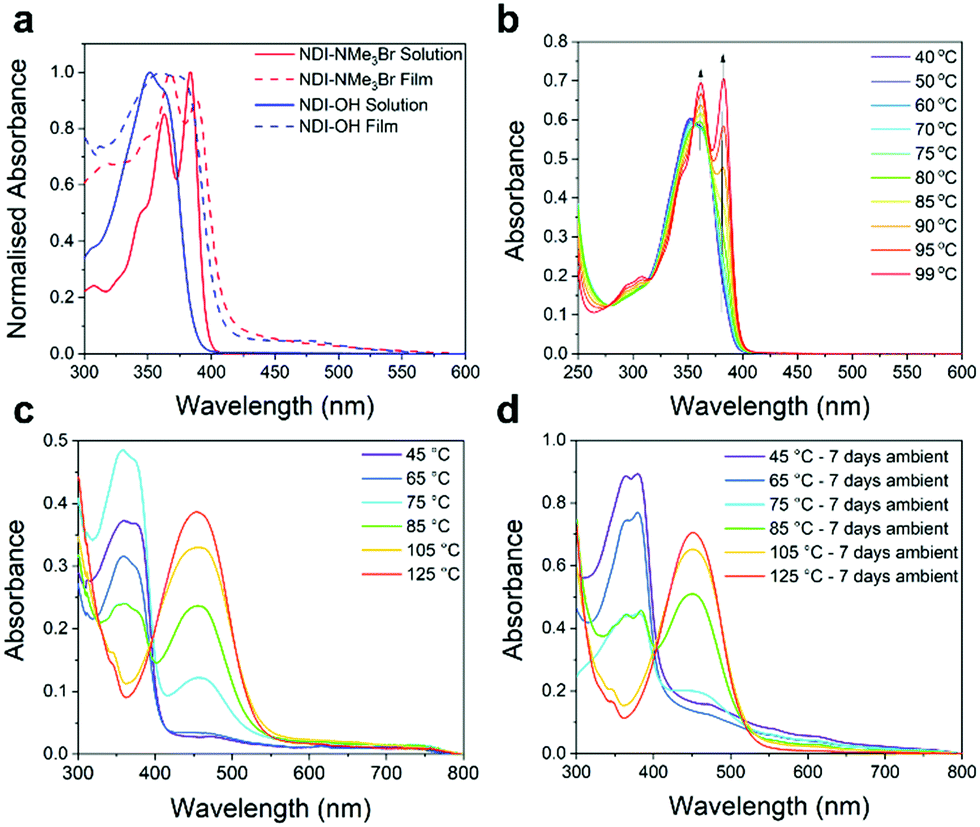 | ||
| Fig. 3 (a) UV-vis. absorption spectra of aqueous solutions of NDI-NMe3Br (solid red) and NDI-OH (solid blue) and thin films of NDI-NMe3Br (dashed red) and NDI-OH (dashed blue). Thin films were made by drop-casting from aqueous solutions at 45 °C. (b) UV-vis. absorption spectra of a solution of NDI-OH heated between temperatures of 40 °C and 99 °C. (c) UV-vis. absorption spectra of NDI-OH thin-films made by drop-casting a 5 mg mL−1 solution at temperatures between 45 °C and 125 °C. (d) UV-vis. absorption spectra of NDI-OH thin-films made by drop-casting a 5 mg mL−1 solution at temperatures between 45 °C and 125 °C then left under ambient conditions for 7 days. | ||
| Compound | E 1/2 (V) | E onset (V) | λ max (nm) | E g (eV) | EA (eV) | IPf (eV) |
|---|---|---|---|---|---|---|
| a From solution cyclic voltammetry in water using glassy carbon working electrode, platinum wire counter electrode and Ag/Ag+ reference electrode. Values are reported from the 4th cycle and measured at 100 mV s−1. b From solution UV-vis absorption spectroscopy in water. c Optical bandgap estimated from absorption band onset of solution UV-vis spectra in Fig. 3a. d Estimated using the equation EA = 4.71 + E1/2. e Estimated using the equation EA = 4.71 + Eonset.33–35 f Estimated using equation IP = EA + Optical band gap. | ||||||
| NDI-NMe3Br | −0.56 | n/a | 383 386 | 3.12 | 4.16d | 7.28 |
| NDI-OH | n/a | −0.91 | 352 | 3.22 | 3.80e | 7.02 |
Doped structures of NDI-OH
Previous studies of tethered quaternary ammonium salts have triggered doping either by spin-coating21 or drop-casting22,23 the aqueous solution followed by annealing at temperatures of 120 °C or above. The resultant thin films then exhibited maximum electrical conductivities of up to 0.5 S cm−1 and red-shifted absorption features indicative of polaron type energy levels within the band gap. To confirm the presence of similar radical type charged species in NDI-OH, the aqueous solution was drop-cast and annealed for 40 minutes on pre-heated glass substrates at a range of temperatures between 45 °C and 125 °C. The absorption spectra of thin films are shown in Fig. 3c. At lower drop-casting temperatures (<65 °C), the absorption spectra closely resemble those of NDI-OH in solution with one major absorption band at 350 nm. This indicates that films formed at lower drop-casting temperatures are primarily composed of the ring-opened compounds 6, 7 and 8.After drop-casting at higher temperatures (>75 °C) a second absorption feature, with maxima at 458 nm, was observed. As the drop-casting temperature was increased this longer wavelength band intensified while the band at 350 nm continuously decreased in intensity. At 125 °C the shorter wavelength band was completely quenched. The absorption at 458 nm closely matches the previously reported absorbance of NDI radical anions generated both chemically and electrochemically which suggests charge transfer on to the NDI core has occurred.38,39 However, we cannot in good confidence assign the absorption band at 458 nm solely to the generation of radical anions, as this should have a much more profound impact on the materials charge transport properties discussed below. In our opinion it is more likely that a significant part of this absorption band originates from further, and currently unidentified, chemical changes in the thin films, whose absorption is overshadowing the underlying polaron absorption band, known to arise at similar wavelengths.
Absorption spectra of NDI-OH drop-cast films which were left under ambient conditions for seven days (Fig. 3d) show that at higher drop-casting temperatures the longer wavelength absorbance persisted, providing further evidence that NDI radical anions are unlikely the only cause of this absorption band since doped n-types are often air sensitive. It is widely believed that for n-doped semiconductors to avoid oxidation by oxygen in air they require a LUMO level of less than −4.0 eV.7,38,39 In addition NDI radical anions have been shown to be notoriously difficult to stabilize with only a few successful examples reported in which the NDI aromatic core was functionalized with highly electron withdrawing groups.27,40–42 Although the electrochemically determined EA of NDI-OH was 3.80 eV, this was measured in an aqueous solution and it should not be assumed that doped thin films will have the same EA in the solid state.
In order to investigate the origin of the additional absorption band further, UV-vis absorption spectra were also obtained for an aqueous solution of NDI-OH which was heated to 99 °C (b). At no point was an absorption at 458 nm observed indicating that our hypothesis of chemical defects related to the ring-closure reaction in solid-state is a possibility. Instead as the temperature was increased two absorption maxima at 362 nm and 383 nm develop, which are consistent with the presence of fully ring-closed NDI ring-systems (see NDI-NMe3Br, Fig. 3a). This suggests that a reversal of the base catalysed hydrolysis, shown in Fig. 2a, may occur at elevated temperatures in solution.
To determine if a ring-closing reverse hydrolysis reaction has an effect on the chemical composition of doped films, three films were drop-cast at 45 °C, 85 °C and 125 °C and then redissolved in deuterium oxide for solution NMR analysis. The 1H NMR regions of the three redissolved NDI-OH films are compared with the solution spectra of NDI-NMe3Br and NDI-OH in Fig. S9–S13 (ESI†). After drop-casting at 45 °C, a singlet at 8.29 ppm was observed, which is indicative of the reformation of four equivalent aromatic protons consistent with a ring-closed NDI. A similar singlet was observed in spectra for the 85 °C and 125 °C films (8.13 and 8.01 ppm respectively), both shifted further up-field. COSY NMR spectra of the 85 °C drop-cast film redissolved in D2O (Fig. S14 and S15, ESI†) also shows no clear correlation between the reformed singlet and any other signal. This suggests the proton only couples to equivalent protons as is the case in symmetrical NDIs.
The integrated area of these singlets increases with temperature when compared to the peaks for the mono-hydrolysed product 6. At drop-cast temperatures of 85 °C and higher the characteristic AB quartet and singlets, at approximately 7.70 ppm, for the two doubly hydrolysed products, 7 and 8, were no longer observed.
The appearance, and subsequent increase in integrated peak area with drop-casting temperature, of an aromatic singlet is consistent with the reformation of the NDI ring-system through a temperature induced reversal of hydrolysis. It is therefore clear that the dynamic nature of the hydrolysis reaction led to a significant difference in the chemical composition of the films drop-cast at different temperatures. This will have to be accounted for when considering the morphology and electronic performance of the doped films. The up-field shift of the reformed aromatic peak with increasing temperature is slightly more difficult to rationalize, however. NDIs n-doped by amines have exhibited a broadening then disappearance of the NDI aromatic proton signal.25,27 This was attributed to the formation of paramagnetic radical anions on the NDI. The shift up-field, observed in NDI-OH thin-films, instead indicates an increase in electron shielding through increased electron density around these aromatic protons.
Electrical conductivity of doped films
Measurement of electrical conductivity, σ, can be a useful way of judging the practical performance of doped semiconducting films. An increase in σ should also help to confirm that the changes in absorption spectra seen in Fig. 3c are due to polaron type charge carriers. In addition, conductivity measurements will provide insights into the varying chemical compositions of the films described above and how such compositional changes impact charge transport. With σ being a product of mobility, μ, and the charge carrier density, n, it can be a useful first indication of how the dynamic structure of NDI-OH is affecting the doping efficiency and the film morphology. Fig. 4 shows a plot of electrical conductivities as a function of drop-casting temperature used for film formation. Conductivities were measured by four-point probe in the Van der Pauw geometry at room temperature under ambient conditions.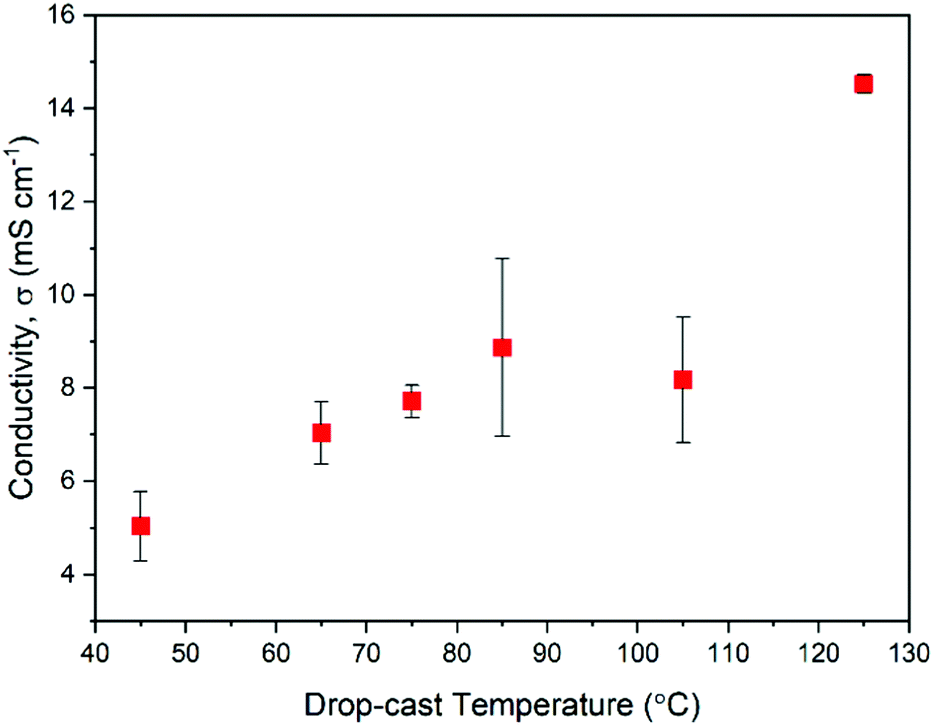 | ||
| Fig. 4 Plot of thin-film conductivities of NDI-OH against the temperature of drop-casting used to form the film. Measurements were made at room temperature in air. Error bars were determined from film thickness measurements. | ||
Films formed at drop-casting temperatures between 45 °C to 85 °C showed a linear increase in σ from 5.0 mS cm−1 to 8.9 mS cm−1. The film at 105 °C however showed a slight decrease in conductivity to 8.2 mS cm−1 before increasing to 14.5 mS cm−1 when deposited at 125 °C. I–V curves obtained from the films (Fig. S22, ESI†) also show better ohmic behavior agreeing with the improved conductivity at higher drop-cast temperatures.
The drop in conductivity at 105 °C is initially difficult to explain. A film morphology less favorable for charge transport can lead to a decrease in mobility which in turn may be caused by the mixed chemical composition. It has also been shown that intermolecular distances can impact the generation of radical anions in NDIs bound to tertiary amines.24 It is possible that both the hydrolysis reaction and the ammonium breakdown played a part in leading to an unfavorable morphology for charge transport and doping in films drop-cast at 105 °C. It was therefore decided that a study of the film morphology was necessary to gain a further insight into the morphological effects on electronic performance.
Morphology of doped films
Charge carrier mobility, and therefore conductivity through the doped film, is dependent on the film morphology. Furthermore, the doping mechanism and efficiency can be largely reliant on the alignment and interactions between the dopant and the semiconductor, both of which could vary greatly with film morphology.43,44 To first image the surface morphology of the films drop-cast at different temperatures atomic force microscopy (AFM) was employed. AFM images of the six films are shown in Fig. 5. Between the drop-casting temperatures of 45 °C and 75 °C, large crystallites (approx. 2 μm in length) were observed. At 85 °C a mixed surface morphology was seen which consisted of some of the larger crystallites separated by more homogeneous and fibrous domains. At 105 °C and 125 °C the surface morphology was almost completely fibrous and seemingly continuous.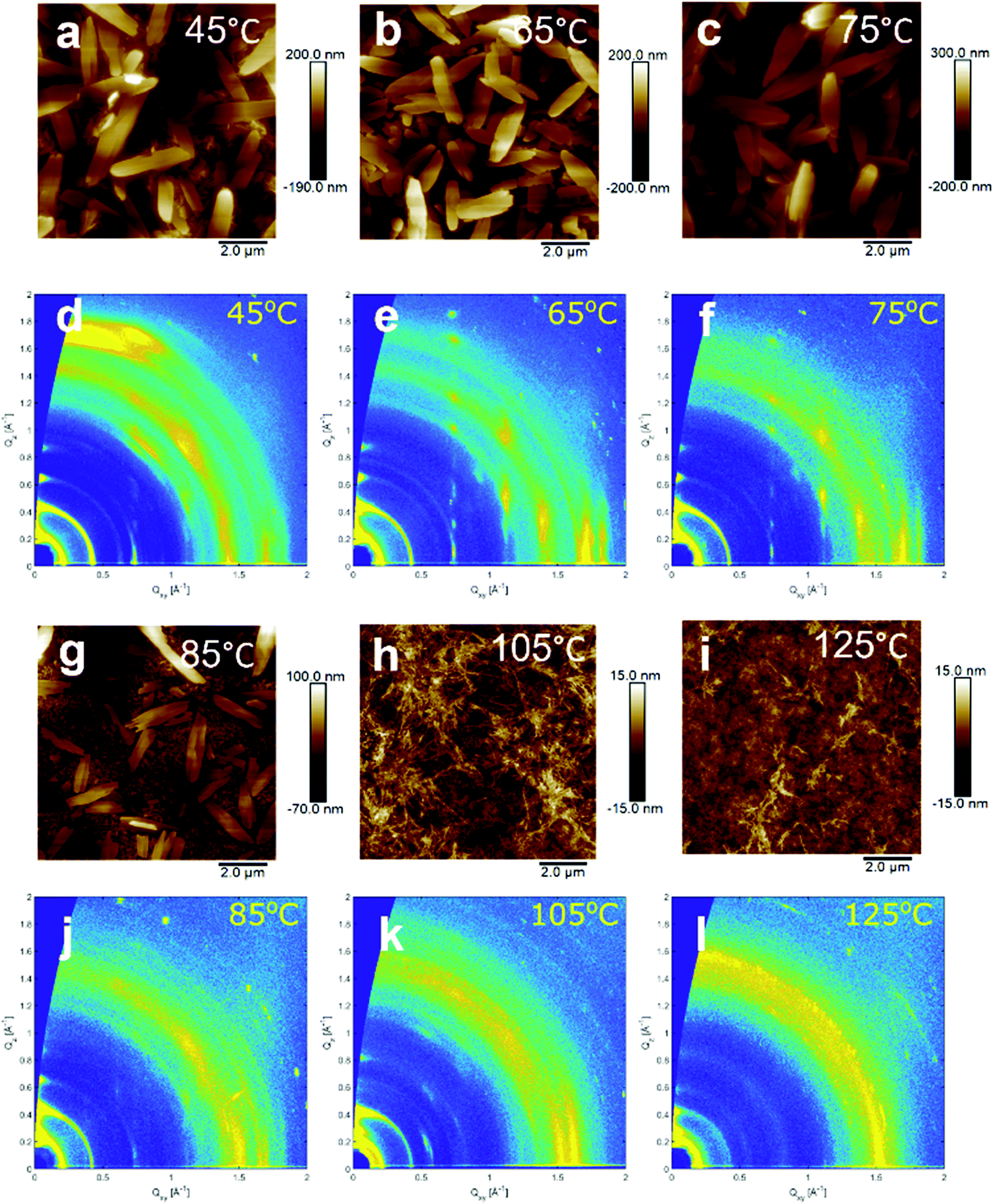 | ||
| Fig. 5 AFM images (a, b, c, g, h and i) and 2D GIWAXS measurements (d, e, f, j, k and l) of NDI-OH films drop-cast at temperatures between 45 °C and 125 °C. | ||
In order to determine if the bulk morphology was changing in a similar fashion, grazing incidence wide-angle X-ray scattering (GIWAXS) was performed. 2D-scattering profiles of NDI-OH are shown in Fig. 5. The films drop-cast at 65 °C and 75 °C showed intense and well-defined Bragg peaks. This suggests good alignment in the out-of-plane direction and indicates that the large crystallites observed in AFM images are present in the bulk. Films drop-cast at temperatures higher than 85 °C generally do not display the same crystalline order, with a loss of the Bragg peaks centred at Qxy = 0.74 Å−1. A new set of broad and intense Bragg peaks at Qxy = 0.55 Å−1 were seen at drop-cast temperatures of 105 °C. This new crystalline structure is less well-textured than those seen at lower temperatures and again reflects the rise in fibrous surface morphologies seen in AFM images at temperatures above 85 °C. It is clear that both the crystalline order and the surface morphology of NDI-OH are drastically changed by the drop-casting temperature. This is likely due to the changing chemical composition of the film, as evidenced by NMR. Changes in morphology after doping, even in pure molecular semiconductors, are also well reported in literature.45–47 Charge transport regimes are known to differ depending on the packing of molecules within the crystal structure, with the favourable overlap of transfer integrals (J) and higher degrees of order leading to increased mobilities.48 The linear increase in conductivity seen between 45 °C and 75 °C can, at least partially, be explained by an increase in the crystallinity. The decrease in conductivity between 85 °C and 105 °C may be due to the disappearance of highly ordered crystalline regions between these two temperatures, although the large amount of grain boundaries between crystalline domains can be a high energetic barrier for charge carriers to cross.49
Electron paramagnetic resonance spectroscopy
An indication of the mobility component of the conductivity can be given by the morphological experiments above. To investigate the extent of doping, the number of polaronic unpaired electrons must be determined. Unpaired spins can be investigated through electron paramagnetic resonance (EPR) spectroscopy, where the presence of any diamagnetic species will not directly contribute to the observed signal. Generation of an anionic radical species in annealed NDI-OH thin-films is indicated by the single, sharp peak observed, shown in Fig. 6a. The g-factor for this film peak, and others in the series, was found to be anisotropic and closely match that of a free electron (giso = 2.002 ± 0.001). No hyperfine splitting could be resolved, likely an effect of self-exchange between neutral and anionic radical NDI-OH species.50,51 A significantly weaker signal was observed from an NDI-NMe3Br thin film annealed at 45 °C compared to NDI-OH annealed at an analogous temperature (Fig. S18, ESI†).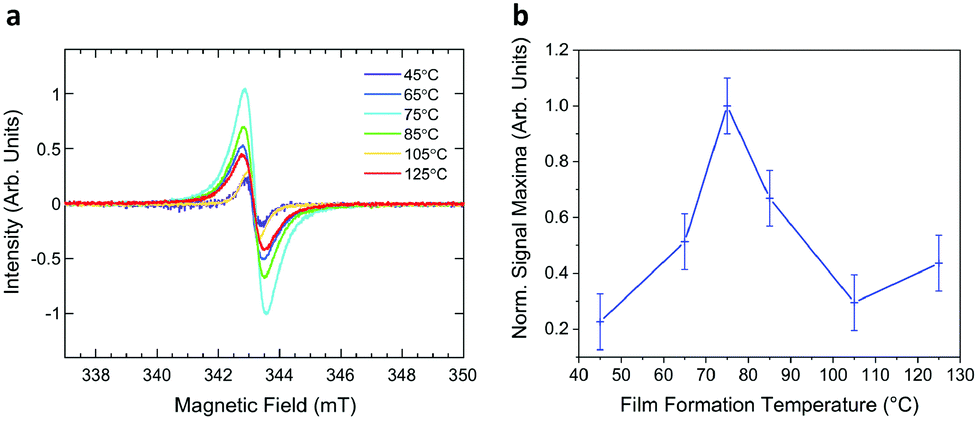 | ||
| Fig. 6 (a) EPR spectrum of NDI-OH thin-films drop cast at temperatures between 45 °C and 125 °C. (b) General trend of radical signal intensities in NDI-OH thin films drop cast at temperatures between 45 °C and 125 °C, normalised to the maximum intensity observed. Measurements were taken after storage in ambient conditions for 24 h. | ||
The greatest signal intensity after 24 hours storage under ambient conditions occurred by drop casting NDI-OH at 75 °C, with general trends in signal intensity corroborating those observed by conductivity measurements. Fig. 6b shows signal intensity increase until 75 °C, where a decrease occurs and a minimum in intensity is present at 105 °C, followed by an increase at 125 °C. Such trends in radical presence also mirror morphological changes caused by annealing, shown in Fig. 5. Charge carrier mobility is less of a significant factor for EPR measurements, impacting linewidths of signals,52 whereas charge carrier concentration will affect signal intensity. This explanation does not fully explain the minimum at 105 °C however, with competing chemical and morphological processes occurring simultaneously.
Identification of the doping species
The structure of the doping species and the specific chemical transformations which have occurred to generate the dopant are yet to be confirmed. With the successful doping of NDI derivatives by tertiary amines already reported,24–27 it seems likely that the quaternary ammonium moiety degrades via one of two pathways. Previously it has been suggested that PDI–OH degraded by the SN2 demethylation to generate a pendant dimethylamine moiety on the PDI core.23 The degradation via an E2 Hofmann elimination to generate trimethylamine (TMA) as a dopant to rylene diimides has not been previously studied. Both computational and experimental studies have shown that the Hofmann pathway is generally favoured if β-hydrogens are available to be abstracted however.28–31 An overview of a study in which the product of Hofmann elimination, TMA, was used to dope a neutral NDI derivative is hereby described and is briefly discussed in the ESI.†To determine the likelihood that TMA can act as a dopant for NDI molecular semiconductors; thin films of NDI-TEG doped by one equivalent of TMA were made (structures in Fig. 7). Films were made by drop-casting from an aqueous solution of NDI-TEG and TMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio). The ethylene glycol sidechains on NDI-TEG were used to allow solubility in water and simulate the polar environment created by charged NDI-OH. Initially UV-vis absorbance spectra of films formed by drop-casting at temperatures between 45 °C and 125 °C (Fig. S23 and S24, ESI†) showed only a broad absorption with maxima at approximately 350 nm.
:1 molar ratio). The ethylene glycol sidechains on NDI-TEG were used to allow solubility in water and simulate the polar environment created by charged NDI-OH. Initially UV-vis absorbance spectra of films formed by drop-casting at temperatures between 45 °C and 125 °C (Fig. S23 and S24, ESI†) showed only a broad absorption with maxima at approximately 350 nm.
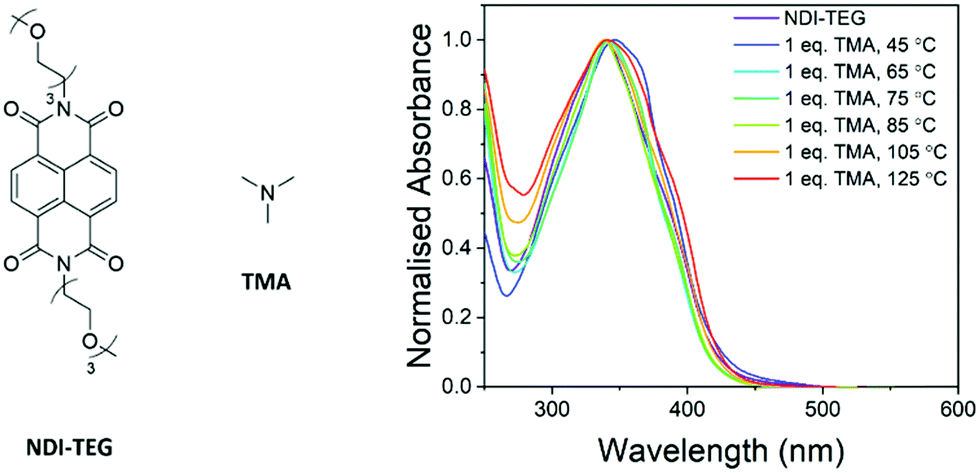 | ||
| Fig. 7 Structures of NDI-TEG and dopant TMA (left). UV-vis absorption spectra of thin-films from blends of NDI-TEG and 1 equivalent trimethylamine drop-cast at temperatures between 45 °C and 125 °C (right). Solutions of NDI-TEG used for film formation were made basic by running through an ion-exchange column prior to addition of TMA and drop-casting. | ||
To rule out the possibility that processing films of NDI-OH from a basic solution was having an effect on the identity of the dopant, solutions of NDI-TEG were also run through an ion-exchange column before addition of one equivalent of TMA. The absorbance spectra for these solutions are shown in Fig. 7 and show one absorption with maxima at 350 nm. Similar attempts to dope NDI-TEG with triethylamine were also made in order to rule out evaporation of trimethylamine from the film during drop-casting due to its low boiling point (2.9 °C). These studies are expanded upon further in the ESI.† Doping with both trimethylamine and triethylamine showed no absorption at 450 nm which was present in films of NDI-OH drop-cast at higher temperatures.
The lack of any evidence for doping in absorption spectra for tertiary amine doped NDI-TEG suggests that the active dopant in films of NDI-OH is not trimethylamine. In turn this adds weight to the argument that the quaternary ammonium hydroxide group decomposes via a demethylation23 rather than a Hofmann elimination. With additional factors at play, such as the hydrolysis reaction, it is hard to definitively say that the active doping species is the bound dimethylamine product of demethylation. The NMR spectra of NDI-OH films shown in Fig. S9–S16 (ESI†) show that at higher drop-casting temperatures unidentified chemical species, beyond the products of ring-opening hydrolysis and the ring-closing reverse reaction, are present. In addition to this the ESI† shows an expansion of these NMR studies into NDI-OH and at least two different transformations can be seen to the NMe3 protons of NDI-OH after drop-casting.
Conclusion
This study provides for the first time, detailed insights into the film formation chemistry when processing quaternary amine tethered rylene diimides. It has been shown that a reversible base catalyzed hydrolysis reaction contributes significantly to a mixed chemical composition of quaternary ammonium hydroxide self-dopants when processed as films. Through AFM and GIWAXS it could be seen that this has a pronounced effect on the film morphology. Producing films of NDI-OH at lower temperatures lead to a more crystalline and ordered film morphology which in turn gave a linear increase in electrical conductivity between drop-casting temperatures of 45 °C and 85 °C. Between 85 °C and 105 °C a drop in conductivity was observed which was accompanied by a change to a more fibrous and less ordered morphology. Preliminary EPR results also suggested that the spin concentration approximately followed the rise in conductivity at lower temperatures and fell between 75 °C and 105 °C.The nature of the relationship between the film morphology, doping efficiency and the chemical composition of the films was further complicated by at least two ongoing chemical reactions at the point of film processing, the imide hydrolysis and quaternary ammonium breakdown. Gaining a more thorough understanding of these two processes, and the possibility of further chemical changes during processing, is key to identifying the active doping species in bound quaternary ammonium hydroxide n-doping systems. The water solubility and long-term ambient stability this class of materials have exhibited is rare amongst n-type organics. Although this study revealed that their mechanism of operation likely relies on a more complicated interplay of factors than assumed; beginning to control and utilize these factors will pave the way for a broader application of these promising self-dopant materials in organic electronic devices.
Author contributions
L. M. C. and B. C. S. designed the study. L. M. C., M. W., Y. D. and B. C. S. synthesized and characterised the materials. P. A. G., W. N., S. L., O. F. and C. B. N. performed the conductivity and AFM measurements. A. G., G. L. and A. S. collected and analysed the GIWAXS data. H. D. and S. H. performed the EPR experiments. All authors contributed to discussion of results and wrote the manuscript. O. F., A. S., S. H., C. B. N. and B. C. S. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. C. acknowledges financial support from the EPSRC (EP/N509577/1). B. C. S. acknowledges the UK Research and Innovation for Future Leaders Fellowship no. MR/S031952/1. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-76SF00515. O. F. is funded by a Royal Society University Research Fellowship (UF140372 & URF/R/201013). S. H. acknowledges funding for the SPIN-lab (EP/P030548/1), and both H. D. and S. L. are funded by the EPSRC through grant EP/L016702/1.Notes and references
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1703063 CrossRef PubMed.
- Y. Lin, Y. Firdaus, M. I. Nugraha, F. Liu, S. Karuthedath, A.-H. Emwas, W. Zhang, A. Seitkhan, M. Neophytou, H. Faber, E. Yengel, I. McCulloch, L. Tsetseris, F. Laquai and T. D. Anthopoulos, Adv. Sci., 2020, 7, 1903419 CrossRef CAS PubMed.
- L. M. Cowen, J. Atoyo, M. J. Carnie, D. Baran and B. C. Schroeder, ECS J. Solid State Sci. Technol., 2017, 6, N3080–N3088 CrossRef CAS.
- W. Zhao, J. Ding, Y. Zou, C.-a Di and D. Zhu, Chem. Soc. Rev., 2020, 49, 7210–7228 RSC.
- B. Lüssem, C.-M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed.
- D. M. de Leeuw, M. M.-J. Simenon, A. R. Brown and R. E.-F. Einerhand, Synth. Met., 1997, 87, 53–59 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed.
- S. Guo, S. B. Kim, S. K. Mohapatra, Y. Qi, T. Sajoto, A. Kahn, S. R. Marder and S. Barlow, Adv. Mater., 2012, 24, 699–703 CrossRef CAS PubMed.
- J. Avila, M.-G. La-Placa, E. Longhi, M. Sessolo, S. Barlow, S. R. Marder and H. J. Bolink, J. Mater. Chem. A, 2019, 7, 25796–25801 RSC.
- P. Wei, J. H. Oh, G. Dong and Z. Bao, J. Am. Chem. Soc., 2010, 132, 8852–8853 CrossRef CAS PubMed.
- M. Lu, H. T. Nicolai, G.-J. A.-H. Wetzelaer and P. W.-M. Blom, Appl. Phys. Lett., 2011, 99, 173302 CrossRef.
- B. D. Naab, S. Guo, S. Olthof, E. G.-B. Evans, P. Wei, G. L. Millhauser, A. Kahn, S. Barlow, S. R. Marder and Z. Bao, J. Am. Chem. Soc., 2013, 135, 15018–15025 CrossRef CAS PubMed.
- Y. Zeng, W. Zheng, Y. Guo, G. Han and Y. Yi, J. Mater. Chem. A, 2020, 8, 8323–8328 RSC.
- C.-Y. Yang, Y.-F. Ding, D. Huang, J. Wang, Z.-F. Yao, C.-X. Huang, Y. Lu, H.-I. Un, F.-D. Zhuang, J.-H. Dou, C.-a Di, D. Zhu, J.-Y. Wang, T. Lei and J. Pei, Nat. Commun., 2020, 11, 3292 CrossRef CAS PubMed.
- B. A. Gregg and R. A. Cormier, J. Am. Chem. Soc., 2001, 123, 7959–7960 CrossRef CAS PubMed.
- B. A. Gregg, S.-G. Chen and H. M. Branz, Appl. Phys. Lett., 2004, 84, 1707–1709 CrossRef CAS.
- A. Rahmanudin, R. Marcial-Hernandez, A. Zamhuri, A. S. Walton, D. J. Tate, R. U. Khan, S. Aphichatpanichakul, A. B. Foster, S. Broll and M. L. Turner, Adv. Sci., 2020, 7, 2002010 CrossRef CAS PubMed.
- P. Luo, K. An, L. Ying, G. Li, C. Zhu, B. Fan, F. Huang and Y. Cao, J. Mater. Chem. C, 2020, 8, 5273–5279 RSC.
- F. Peng, J. Xu, Y. Zhang, R. He, W. Yang and Y. Cao, Polym. Chem., 2019, 10, 1367–1376 RSC.
- A. M. Gaikwad, Y. Khan, A. E. Ostfeld, S. Pandya, S. Abraham and A. C. Arias, Org. Electron., 2016, 30, 18–29 CrossRef CAS.
- T. H. Reilly, A. W. Hains, H.-Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef CAS.
- B. Russ, M. J. Robb, F. G. Brunetti, P. L. Miller, E. E. Perry, S. N. Patel, V. Ho, W. B. Chang, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Adv. Mater., 2014, 26, 3473–3477 CrossRef CAS PubMed.
- B. Russ, M. J. Robb, B. C. Popere, E. E. Perry, C.-K. Mai, S. L. Fronk, S. N. Patel, T. E. Mates, G. C. Bazan, J. J. Urban, M. L. Chabinyc, C. J. Hawker and R. A. Segalman, Chem. Sci., 2016, 7, 1914–1919 RSC.
- Y. Matsunaga, K. Goto, K. Kubono, K. Sako and T. Shinmyozu, Chem. – Eur. J., 2014, 20, 7309–7316 CrossRef CAS PubMed.
- S. B. Schmidt, T. Biskup, X. Jiao, C. R. McNeill and M. Sommer, J. Mater. Chem. C, 2019, 7, 4466–4474 RSC.
- D. Powell, E. V. Campbell, L. Flannery, J. Ogle, S. E. Soss and L. Whittaker-Brooks, Mater. Adv., 2021, 2, 356–365 RSC.
- S. B. Schmidt, M. Hönig, Y. Shin, M. Cassinelli, A. Perinot, M. Caironi, X. Jiao, C. R. McNeill, D. Fazzi, T. Biskup and M. Sommer, ACS Appl. Polym. Mater., 2020, 2, 1954–1963 CrossRef CAS.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399-400, 49–59 CrossRef CAS.
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CrossRef CAS.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- M. B. Kim and D. W. Dixon, J. Phys. Org. Chem., 2008, 21, 731–737 CrossRef CAS.
- Q. Sun, H. Wang, C. Yang and Y. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- J. Hou, Z.-a Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS PubMed.
- Y. Li in Organic Optoelectronic Materials. Lecture Notes in Chemistry, ed. B. Capenter, P. Ceroni, B. Kirchner, K. Landfester, J. Leszczynski, T.-Y. Luh, C. Mahlke, N. C. Polfer, R. Salzer, Springer, Cambridge, 2015, ch. 2, p. 41 Search PubMed.
- T. C. Barros, S. Brochsztain, V. G. Toscano, P. B. Filho and M. J. Politi, J. Photochem. Photobiol., A, 1997, 111, 97–104 CrossRef CAS.
- V. Wintgens, P. Valat, J. Kossanyi, L. Biczok, A. Demeter and T. Bérces, J. Chem. Soc., Faraday Trans., 1994, 90, 411–421 RSC.
- S. Guha and S. Saha, J. Am. Chem. Soc., 2010, 132, 17674–17677 CrossRef CAS PubMed.
- J. Li, X. Pang, Y. Wang, Y. Che and J. Zhao, Catal. Today, 2014, 224, 258–262 CrossRef CAS.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004–12010 CrossRef CAS PubMed.
- S. Kumar and P. Mukhopadhyay, Green Chem., 2018, 20, 4620–4628 RSC.
- K. Kang, S. Watanabe, K. Broch, A. Sepe, A. Brown, I. Nasrallah, M. Nikolka, Z. Fei, M. Heeney, D. Matsumoto, K. Marumoto, H. Tanaka, S.-i Kuroda and H. Sirringhaus, Nat. Mater., 2016, 15, 896–902 CrossRef CAS PubMed.
- J. E. Cochran, M. J.-N. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS.
- J. Gao, J. D. Roehling, Y. Li, H. Guo, A. J. Moulé and J. K. Grey, J. Mater. Chem. C, 2013, 1, 5638–5646 RSC.
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef PubMed.
- R. A. Schlitz, F. G. Brunetti, A. M. Glaudell, P. L. Miller, M. A. Brady, C. J. Takacs, C. J. Hawker and M. L. Chabinyc, Adv. Mater., 2014, 26, 2825–2830 CrossRef CAS PubMed.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- L. G. Kaake, P. F. Barbara and X. Y. Zhu, J. Phys. Chem. Lett., 2010, 1, 628–635 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331–342 RSC.
- R. L. Ward and S. I. Weissman, J. Am. Chem. Soc., 1957, 79, 2086–2090 CrossRef CAS.
- H. Matsui, Y. Takeda and S. Tokito, Org. Electron., 2019, 75, 105432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemicals and instrument details, detailed synthetic procedures, structural characterisation, NMR spectra, cyclic voltammetry, EPR spectra, GIWAXS data, electrical characterization and an overview of the NDI-TEG and tertiary amine doping study. See DOI: https://doi.org/10.1039/d2tc01108e |
| This journal is © The Royal Society of Chemistry 2022 |