
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMore complex than originally thought: revisiting the origins of the relaxation processes in dimethylammonium zinc formate†
Paulina
Peksa
 *a,
Andrzej
Nowok
a,
Filip
Formalik
bc,
Jan K.
Zaręba
*d,
Justyna
Trzmiel
c,
Anna
Gągor
e,
Mirosław
Mączka
e and
Adam
Sieradzki
*a
*a,
Andrzej
Nowok
a,
Filip
Formalik
bc,
Jan K.
Zaręba
*d,
Justyna
Trzmiel
c,
Anna
Gągor
e,
Mirosław
Mączka
e and
Adam
Sieradzki
*a
aDepartment of Experimental Physics, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: adam.sieradzki@pwr.edu.pl; paulina.peksa@pwr.edu.pl
bDepartment of Micro, Nano, and Bioprocess Engineering, Faculty of Chemistry, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
cDepartment of Theoretical Physics, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
dAdvanced Materials Engineering and Modelling Group, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: jan.zareba@pwr.edu.pl
eInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, Box 1410, 50-950 Wrocław, Poland
First published on 30th March 2022
Abstract
Metal formates are a subclass of coordination polymers that is renowned for the rich phase transition behavior arising from the complex interplay of molecular dynamics of organic guests and the surrounding coordination net. This contribution challenges current consensus on the origins of the relaxation processes present in the low temperature phase of dimethylammonium (DMA+) zinc formate [(CH3)2NH2][Zn(HCOO)3] (DMAZn). Thus far, it was believed that below 156 K the order–disorder structural phase transition leads to the reduction of the crystal structure symmetry from hexagonal to monoclinic, as well as causes nearly complete freezing of the DMA+ cation. Herein, we assign the crystal symmetry of low-temperature phase DMAZn to triclinic (P1), based on the observed splitting of Bragg peaks into six different components originating from six ferroelastic domains. Noncentrosymmetry of the triclinic phase of DMAZn is confirmed with second harmonic generation measurements. Full reversibility of phase transition between triclinic (noncentrosymmetric) and trigonal (centrosymmetric) crystal phases allowed for the demonstration of nonlinear optical switching of the SHG-on–SHG-off type. In turn, a new set of experimental and theoretical data on cation dynamics shows that in the low temperature phase the flipping of organic cations does not completely freeze as previously thought. Dielectric measurements on the DMAZn sample synthesized in an electric field display enhanced intensity of two relaxation phenomena in dielectric spectra. The origin of these processes, previously misinterpreted as methyl group rotations, has been explored with the use of DFT calculations. It was found that the high- and low-frequency processes can be attributed to the hopping between stable and metastable positions of the DMA+ cation, respectively.
1. Introduction
Coordination polymers and metal–organic frameworks (MOFs) featuring the perovskite ABX3 architecture have been extensively investigated in the past few decades. While the diversity of dense MOFs is generally overwhelming, one of the simplest perovskite-like MOF subfamilies are metal formates. Metal formates are compounds characterized by the generic formula [A][M(HCOO)3] (in which A and M stand for a monoprotonated molecular alkylammonium cation and a divalent metal center, respectively) whose hallmark feature is the diversity of physical properties they display, related to changes in the structural, dielectric, optical and magnetic responses triggered by a temperature change.1–4 It is thus not surprising that these materials have been around for some time due to their multiferroic characteristics and non-trivial structural phase transition behaviors especially those of the order–disorder nature.2,5,6 Currently, the library of characterized metal formates is dominated by analogs comprising dimethylammonium cations ([(CH3)2NH2]+ referred to as DMA+) as organic guests. Indeed, in combination with a wide selection of metal centers, e.g., Zn, Mn, Cu, and Ni, dozens of scientific studies reporting different aspects of DMA+ cation dynamics have emerged, and their number is still growing.2,3,5–23 As a part of our ongoing interest in the mechanistic aspects of phase transitions in metal formates in this work we revisit 3D zinc formate hosting DMA+ cations, i.e. [(CH3)2NH2][Zn(HCOO)3] (DMAZn). Without any exaggeration, this compound was and still is a hotspot for the metal-formate community, as it boasts a rich record of disparate interpretations of its electrical properties. Much of the controversy revolves around the electrical properties of a phase transition that takes place at Tc ∼156 K.2,3,10,19,21,22 For instance, it was initially assumed to be an antiferroelectric order–disorder structural transformation initiated by the ordering of the hydrogen atoms in NH2.3 However, later studies have hypothesized that it would rather be a phase transition from a high temperature paraelectric to a low temperature ferroelectric phase.19 In this scenario, the phase transition mechanism has been attributed to the alignment of the hydrogen bonds between DMA+ and the formate group with concomitant buckling of the metal-formate coordination net.20,21 On the other hand, as of now, the notion of DMAZn proper ferroelectricity is challenged. This is due to the fact that there is no direct proof confirming proper ferroelectric behavior in terms of the results of electric polarization hysteresis loop measurements.24The molecular mechanism of this phase transition has been extensively studied using a number of experimental methods such as differential scanning calorimetry (DSC), Raman spectroscopy, X-ray diffraction (XRD), broadband dielectric spectroscopy (BDS), nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) spectroscopies.2,3,14,19,21,22,24–28 Current consensus is that the low temperature phase of this compound crystallizes in the non-centrosymmetric, monoclinic Cc phase, whereas the high-temperature phase is of trigonal symmetry (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c) and shows dynamic disorder of organic cations. Below the temperature of phase transition, the flipping of the DMA+ cation between three equivalent positions was supposed to disappear, leading to a complete ordering of these cations.9,11,29 This effect was believed to be associated with the distortion of the framework, collectively rendering a long-range electrical order.29 At first, it was thought that dielectric spectroscopy studies support the hypothesis of complete freezing of molecular motions in the monoclinic phase of DMAZn,2,13,16,19,21,26i.e., the dipolar relaxation was detected only for the high-temperature phase (described as the thermally activated Arrhenius process, with an activation energy of 0.28 eV).21 Nevertheless, traces of dipolar relaxation response have recently been observed for the low-temperature phase, albeit with a significantly smaller amplitude, compared to the high temperature process.25 The activation energy was estimated to be 0.29 eV for the relaxation process observed at high frequencies25 and this energy value is close to the one obtained for DMA+ cation flips in the high-temperature phase. Another relaxation process observed in the lower frequency range, with an activation energy of 0.2 eV, was attributed to the rotations of the methyl groups of the DMA+ cation.25 However, as discussed in ref. 24, this assignment cannot be backed up by NMR and EPR measurements, as the results of both showed a much smaller activation energy of methyl group rotation (∼0.1 eV).13,22 Taken together, despite quite a broad set of experimental data, diverging interpretations of the phase transition mechanism and dynamics of the associated phases are apparent. This calls for detailed reinvestigation of dipolar properties of the title compound using alternative approaches.
c) and shows dynamic disorder of organic cations. Below the temperature of phase transition, the flipping of the DMA+ cation between three equivalent positions was supposed to disappear, leading to a complete ordering of these cations.9,11,29 This effect was believed to be associated with the distortion of the framework, collectively rendering a long-range electrical order.29 At first, it was thought that dielectric spectroscopy studies support the hypothesis of complete freezing of molecular motions in the monoclinic phase of DMAZn,2,13,16,19,21,26i.e., the dipolar relaxation was detected only for the high-temperature phase (described as the thermally activated Arrhenius process, with an activation energy of 0.28 eV).21 Nevertheless, traces of dipolar relaxation response have recently been observed for the low-temperature phase, albeit with a significantly smaller amplitude, compared to the high temperature process.25 The activation energy was estimated to be 0.29 eV for the relaxation process observed at high frequencies25 and this energy value is close to the one obtained for DMA+ cation flips in the high-temperature phase. Another relaxation process observed in the lower frequency range, with an activation energy of 0.2 eV, was attributed to the rotations of the methyl groups of the DMA+ cation.25 However, as discussed in ref. 24, this assignment cannot be backed up by NMR and EPR measurements, as the results of both showed a much smaller activation energy of methyl group rotation (∼0.1 eV).13,22 Taken together, despite quite a broad set of experimental data, diverging interpretations of the phase transition mechanism and dynamics of the associated phases are apparent. This calls for detailed reinvestigation of dipolar properties of the title compound using alternative approaches.
The main aim of the present paper is to systematize the knowledge of the title compound and to cast more light on its dielectric response, in particular dipolar relaxation processes. To this end, we have performed electric field-assisted crystallization of [(CH3)2NH2][Zn(HCOO)3] (DMAZn_E) and compared its properties with those of the same compound but synthesized in a traditional way (DMAZn). Utilizing the broadband dielectric technique, we show that, contrary to a common belief, the organic DMA+ cation does not completely freeze at the low-temperature phase. Furthermore, we reveal the complex nature of the phase transition. In particular, we dielectrically prove the formation of the pretransitional clusters of the low-temperature phase even much above Tc. Based on the single-crystal X-ray crystallographic studies of the monodeuterated analogue [(CH3)2NH2][Zn(DCOO)3] (DMAZnD) we show that in the low-temperature phase it is noncentrosymmetric, and crystallizes in triclinic space group P1. Assignment of the acentric setting for this phase has been corroborated with the use of SHG measurements. Application-wise, this contribution also reports on temperature-driven SHG-on–SHG-off nonlinear optical switching in DMAZn, which could be achieved thanks to the reversibility of the phase transition between triclinic (acentric) and trigonal (centrosymmetric) crystal phases.
2. Experimental
2.1 Synthesis
Three kinds of materials were prepared: (1) reference [(CH3)2NH2][Zn(HCOO)3] (DMAZn) synthesized in a traditional way, (2) [(CH3)2NH2][Zn(HCOO)3] (DMAZn_E) obtained as a product of electric field-assisted crystallization, and (3) dimethylammonium zinc formate with deuterated formate [(CH3)2NH2][Zn(DCOO)3] (DMAZnD).The following reagents were used during synthesis of the reference DMAZn: ZnCl2, methanol, and a 2.0 M solution of dimethylamine in methanol and formic acid. A solution containing 10 mL of methanol, 5 mL of 2.0 M solution of dimethylamine in methanol and 4 mL of HCOOH was placed at the bottom of a glass tube. To this solution, 15 mL of a methanol solution containing 0.99 mmol of ZnCl2 was gently added.21 The second material (DMAZn_E) was synthesised in the same way, but an external electric field of 10 kV was additionally applied during the whole synthesis process. In both cases, crystals were harvested after 4 days.
In order to obtain DMAZnD 2 mL of a 2.0 M solution of dimethylamine in methanol, 1 mL of formic-d acid and 10 mL of N,N-dimethylformamide were added to 10 mL of methanol and mixed. In the next step, 10 mL of methanol solution containing 1 mmol of ZnCl2 was added, mixed, and left at room-temperature in a sealed polypropylene container. The crystals were harvested after 48 hours, washed three times with methanol, and dried at room-temperature.17 Only DMAZn was used for the second-harmonic generation measurement. DMAZn_E was used in thermal and dielectric measurements. DMAZnD was used only for thermal measurements and X-ray diffraction studies in order to obtain results of better quality.
2.2 Differential scanning calorimetry (DSC)
Differential scanning calorimetry (DSC) measurements were performed using a high-resolution Mettler Toledo DSC-1 calorimeter at a scanning rate of 5 K min−1. The measurements were performed in the temperature range of 130–270 K upon heating and cooling. Nitrogen was used as a purging gas.2.3 Single-crystal X-ray diffraction
The diffraction data were collected for DMAZnD using a Xcalibur diffractometer equipped with CCD Atlas camera and Mo Kα radiation source. CrysAlisPro 1.171.38.46 (Rigaku Oxford Diffraction, 2015) was used for data processing. An empirical absorption correction was applied using spherical harmonics implemented in Scale3 Abspack scaling algorithm. The low-temperature structure was solved at 100 K by intrinsic phasing using SHELXT and refined in SHELXL-9730 on two dominating domains using hklf 5 refinement. The crystal structure has been deposited in the CCDC data base with deposition number: 2114953. Table S1 (ESI†) gathers the most important crystal, collection and refinement data.2.4 Temperature-resolved second-harmonic generation (TR-SHG) and SHG-on–SHG-off switching
Second-harmonic generation (SHG) studies were performed using a laser system based on a Quantronix Integra-C regenerative amplifier providing 800 nm laser pulses of ∼130 fs length and operating at a repetition rate of 1 kHz. Prior to measurements, the single crystals of DMAZn were crushed with a spatula, fixed between microscope glass slides (forming tightly packed layers) and sealed. The employed measurement setup operates in the reflection mode. Specifically, the laser beam was directed onto the sample at 45 degrees to the sample surface and was unfocused. Emission collecting optics consisted of a Ø25.0 mm plano-convex lens of focal length 25.4 mm (Thorlabs) mounted to a 400 μm 0.22 NA glass optical fiber (Ocean Optics) and was placed along the normal to the sample surface. The distance between collection lens and the sample was equal to 30 mm. No polarizer was put in front of the collection lens. Scattered pumping radiation was suppressed with the use of a 700 nm shortpass dielectric filter (FESH0700, Thorlabs).All emission spectra were recorded by an Ocean Optics Flame T spectrograph (200 μm slit). Temperature control of the sample was performed using the Linkam LTS420 Heating/Freezing Stage. The TR-SHG study (from 173 K to 133 K for cooling run, and from 133 K to 173 K for heating run with a 2–3 K step) was performed using an 800 nm laser beam of 0.42 W cm−2 power density. Temperature switching of SHG was performed by setting the temperature at 145 K and 157 K in an alternate manner, and at these temperature points the DMAZn sample was irradiated using the same beam parameters as those used for the TR-SHG study.
2.5 Dielectric properties
The powdered sample of DMAZn_E was pressed between 5 mm diameter discs under a hydraulic press (1 GPa). A pellet was made since the obtained single crystals were not big enough to perform direction-dependent dielectric measurements on them. The dielectric properties at ambient pressure were measured as a function of frequency and temperature using a broadband impedance Novocontrol Alpha analyzer. The small signal of an amplitude of 1 V was applied across the sample. The dielectric measurements were carried out over a wide frequency range (1 Hz–1 MHz) every 1 K over the temperature range of 300 to 130 K. Measurements in an electric field were performed by applying a constant external voltage of 40 V to the investigated sample and measurements of broadband dielectric spectroscopy were performed simultaneously. Dielectric measurement was performed for the DMAZn_E sample. Additionally, for DMAZn and DMAZn_E samples, the measurements were taken without and with the application of an external electric field of 40 V (DMAZn40V and DMAZn_E40V).2.6 Density functional theory (DFT) calculations
Structures used for computations were obtained from the optimization of the positions of ions taken from the experimental XRD structures. The structure optimization was performed using the PAW30–32 method as implemented in the VASP 5.4.4 package. PBE33 density functional with the addition of Grimme D3(BJ) dispersion correction34 was used. As previously shown35 it is critical to properly describe the structure of metal–organic frameworks (MOFs) and related materials. A convergence test was performed to find the value of cutoff energy (900 eV) and the k-point set. The calculations were assumed to be converged when an increase of either energy cutoff or number of k-points does not change the absolute value of each element of the stress tensor by more than 0.1 GPa. For the self-consistent cycle (electronic minimization) the convergence criterion was set to 10−6 eV and for geometry optimization (ionic minimization, conjugate gradient algorithm) to 10−2 eV Å−1. Optimization was performed for both volume and ionic position, without symmetry constraint. The dipole moment of the DMA+ cation was calculated using the following formula:where N denotes the number of atoms in the cation, μatomici represents the atomic dipole moment for i-th atom, qi is the partial charge of i-th atom, and ri = (xi, yi, zi) is the position of i-th with respect to the position of center of mass of the cation. Atomic dipole moments and partial charges were calculated using the DDEC6 method,36 as implemented in the Chargemol program. All visualizations of molecular and crystal structures were generated with VESTA software.37
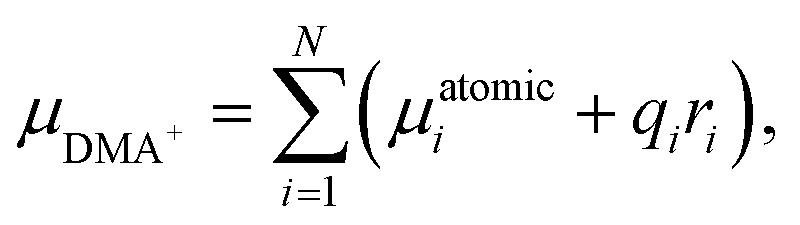
2.7 SS-NEB
To calculate the minimum energy path (MEP) of the DMA+ cation rotation we used the solid-state nudge elastic band method (SS-NEB)38 as implemented in Transition State Atomistic Simulation Environment (TSASE), an extension of Atomistic Simulation Environment (ASE).39 For SS-NEB optimization we used FIRE algorithm,40 and force convergence was set to 10−1 eV Å−1.3. Results and discussion
3.1 Thermal properties
Characterization of the synthesized materials was started with DSC analysis. It aimed at checking the purity of the synthesized products and determining whether the application of an external electric field during crystallization affects the thermal properties. Therefore, DMAZn, DMAZn_E and DMAZnD materials were examined calorimetrically between 130 and 270 K.As presented in Fig. 1, similar thermograms were registered for both DMAZn and DMAZn_E. They are characterized by a single symmetrical heat anomaly with the onset at 164 K and 159 K upon heating and cooling, respectively. This outcome largely mirrors that reported for non-deuterated DMAZn3,6,21 and confirms the first-order nature of the phase transition. Thus, one can state that an external electric field does not affect crystallization, i.e., does not lead to the formation of another polymorph. In other words, DMAZn and DMAZn_E are the same materials in terms of thermal properties. In fact, this conclusion is well supported by Fig. S1 (ESI†), which showcases changes in the heat capacity (ΔCp) and entropy (ΔS) associated with structural phase transition.
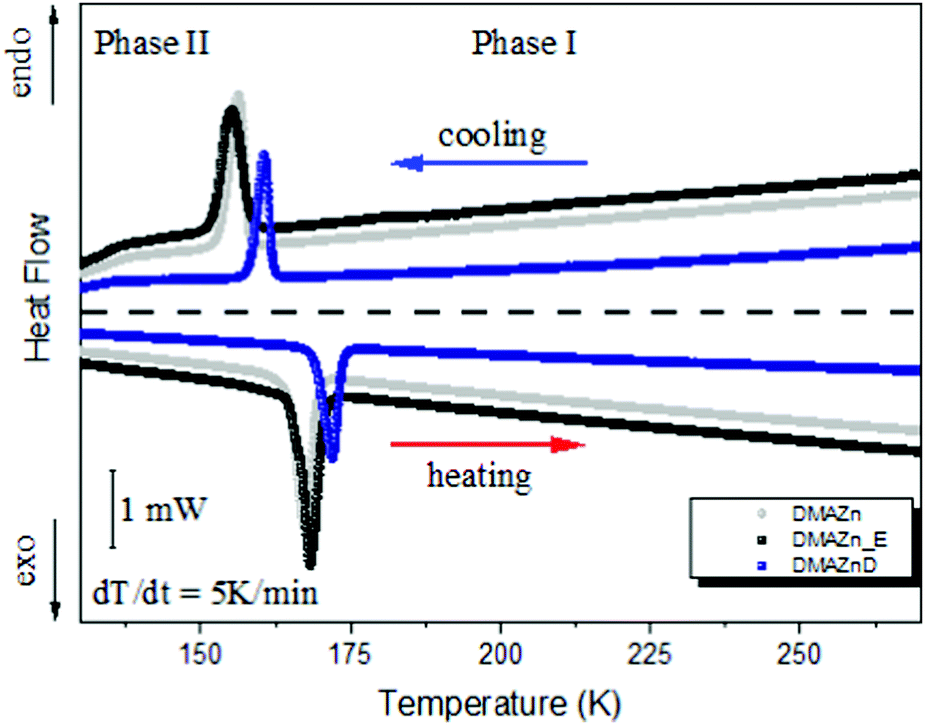 | ||
| Fig. 1 DSC traces for DMAZn, DMAZn_E and DMAZnD between 130 K and 270 K for the cooling and heating run. | ||
As can be seen, ΔS is of similar value for both materials, i.e., ∼4 J mol−1 K−1 (see Table 1). This parameter can be associated with a number of distinguishable positions of the DMA+ cation in the ordered and disordered phases. Therefore, the entropy difference can also be estimated from structural data using the formula ΔS = R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(N2/N1), where N1 and N2 are the numbers of distinguishable positions of the DMA+ cation. The N2 parameter is equal to 3, as each DMA+ cation occupies three symmetry-equivalent positions in the high-temperature phase. On the other hand N1 = 1 because DMA+ is believed to be structurally ordered at low temperatures. Consequently, the entropy difference for the studied samples is expected to be 9.1 J mol−1 K−1. In turn, the experimentally estimated value (ΔS ∼4 J mol−1 K−1)17,21 is significantly smaller compared to the value predicted from the structural order changes. This result suggests that the phase transition is more complex than can be expected from the 3-fold order–disorder model.17,21
ln(N2/N1), where N1 and N2 are the numbers of distinguishable positions of the DMA+ cation. The N2 parameter is equal to 3, as each DMA+ cation occupies three symmetry-equivalent positions in the high-temperature phase. On the other hand N1 = 1 because DMA+ is believed to be structurally ordered at low temperatures. Consequently, the entropy difference for the studied samples is expected to be 9.1 J mol−1 K−1. In turn, the experimentally estimated value (ΔS ∼4 J mol−1 K−1)17,21 is significantly smaller compared to the value predicted from the structural order changes. This result suggests that the phase transition is more complex than can be expected from the 3-fold order–disorder model.17,21
| DMAZn | DMAZn_E | DMAZnD | |
|---|---|---|---|
| Superscripts c and h denote cooling and heating cycles, respectively and av denotes average. | |||
| Onset of Tc (K) | 159c, 164h | 159c, 164h | 162c, 169h |
| ΔCp (J mol−1 K−1) | 222av | 186av | 277av |
| ΔS (J mol−1 K−1) | 4.3 | 4.0 | 4.4 |
Deuteration of the formate linkers slightly changes the thermal properties (see Fig. 1). Namely, the phase transition temperature onset is shifted towards higher temperatures for DMAZnD (in line with previous reports17), i.e., 162 K upon cooling and 169 K upon heating. However, first-order character of the phase transition and its mechanism remain invariant. The latter conclusion is evident when comparing the entropy change which is also close to 4 J mol−1 K−1 (see Table 1).
3.2 Single-crystal X-ray diffraction
In order to understand the particularly complex behavior of DMAZn, first, we provide some so-far reported structural information on this hybrid compound and its chemically-related analogues. Indeed, extant X-ray diffraction experiments provide extensive body of data on the crystal architectures of [(CH3)2NH2][M(HCOO)3] (M: Zn, Mn, Co, and Ni) room-temperature phases. According to the literature, all these materials crystallize in a trigonal Rc centrosymmetric space group at room temperature and possess a perovskite-like structure.2,3,41–43 Octahedral coordination spheres of metal atoms placed at the corners of distorted square blocks are exclusively completed by formate oxygen atoms; furthermore, neighboring atoms are connected via formate linkers (HCOO−) in an anti–anti configuration mode. The dynamics of DMA+ cations themselves is a critical factor shaping the phase behavior of [(CH3)2NH2][M(HCOO)3] hybrids. Indeed, the DMA+ cations reside in the cavities and are anchored through N–H⋯O hydrogen bonds to the metal-formate scaffold. However, it is the relative weakness of these hydrogen bonds that enables the amine molecules to easily rotate around a 3-fold axis (the axis led through carbon atoms of amine molecule), which allows for flipping between three distinct, yet symmetry equivalent positions. These motions are thermally activated, the main consequence of which is that upon cooling the hopping between the three sites is essentially blocked, and the DMA+ is trapped in one of three positions allowed at high temperature. It has been established that [(CH3)2NH2][M(HCOO)3] (M = Zn, Mn, Co, and Ni) compounds undergo a phase transition, yet the structural determination of low-temperature phases poses a challenge. The significant symmetry reduction from Rc to the low symmetry phase is associated with the emergence of ferroelastic domains (see Fig. S2, ESI†), convoluting the analysis of X-ray diffraction data. In this context it comes as no surprise that the unambiguous crystal structure model of low-temperature phase has been so far reported for [(CH3)2NH2][Mn(HCOO)3] only.11,44 In this case the symmetry of the low-temperature phase of this compound was reduced to the monoclinic polar Cc space group, characterized by a minor deformation of the metal-formate substructure and fully ordered DMA+ cations. Furthermore, during the course of extended studies on phase transitions in metal-formates, including Mn and Ni analogues with deuterated amines,6 and doped21 and pure Zn-formates,45 it emerged that the deformation of the metal-formate scaffold below Tc depends not only on sample composition, but also on the sample's thermal history and treatment. The extent of the structure deformation has a particular impact on the separation of Bragg diffraction peaks from different domains. Usually, the deformations of the metal-formate framework after phase transition were rather insignificant so that the diffracted intensities showed pseudo-trigonal symmetry and the structure could be refined in the paraelastic trigonal Rc space group with R factors similar to those recorded for high-temperature phases. The X-ray crystallographic data presented here for DMAZnD, a deuterated analog of DMAZn, display a different behavior i.e. there is evident separation of Bragg peaks pointing to its underlying low crystal symmetry.
Fig. 2 shows diffracted X-ray intensities recorded at 100 K for the deuterated DMAZnD. It can be clearly seen that Bragg peaks split into six different components originating from six ferroelastic domains, which can be explained only by a symmetry reduction to the triclinic system.46 Several DMAZnD samples have been examined for that property, and indeed, the splitting occurred in either case below Tc. Backed up by TR-SHG results, which show a non-zero signal below Tc (vide infra), we were confident to refine the structure in the non-centrosymmetric P1 space group indexing the unit cell in the primitive cell of the Rc, with lattice parameters a = 8.1573(9) Å, b = 8.1491(7) Å, c = 8.8244(10) Å, α = 61.343 (10)°, β = 62.557 (11)°, and γ = 60.057(10)°. Taking into account that the deuterated DMAZnD and non-deuterated DMAZn are considered as isomorphs, one can ascribe the P1 symmetry also to the low-temperature phase in DMAZn (in contrast to previous reports on this compound, in which the symmetry of the low-temperature phase was considered as Cc). One can thus conclude that the low-temperature phase behavior of dimethylammonium zinc formates (deuterated and non-deuterated) differs from the picture known for the Mn analogue. It is worth adding that similar symmetry reduction from trigonal to triclinic has also been reported for methylhydrazinium metal formates47 and mixed-valence iron(ii)–iron(iii) metal formates.48
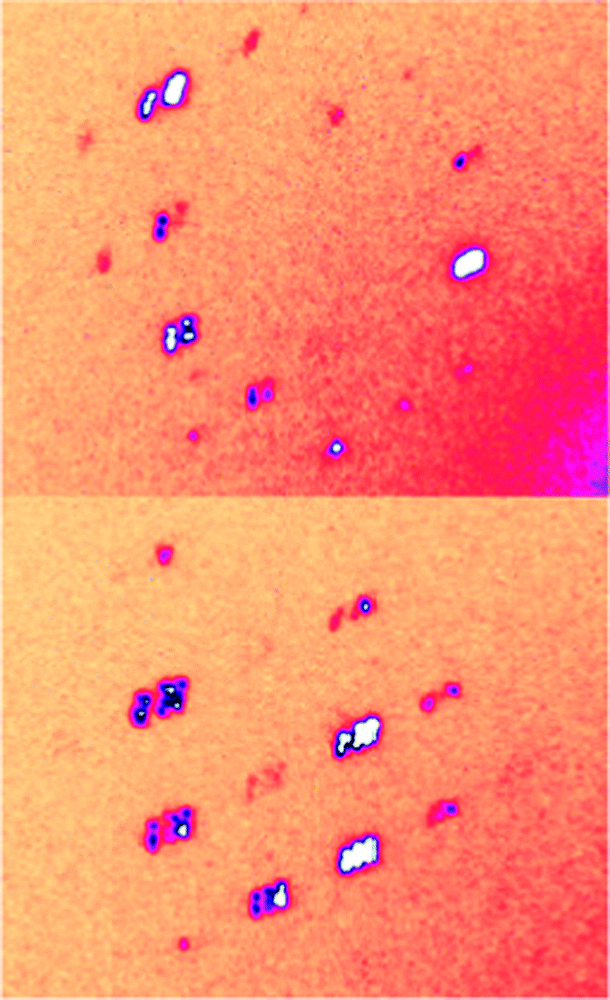 | ||
| Fig. 2 The diffracted X-ray intensities in DMAZnD collected at the low-temperature phase, at 100 K. The splitting of Bragg peaks can be clearly observed. | ||
The main difference between the monoclinic Cc and P1 models concerns the number of inequivalent crystal cavities and amines. Fig. 3 compares the placement and the bonding of DMA+ in both models, whereas Tables S2 and S3 (ESI†) present the geometry of the hydrogen bonds. The atom numbering scheme is shown in Fig. S3 (ESI†). In the Cc symmetry, each cavity is the same, and each cavity adopts one, ordered amine which is symmetry equivalent with all amines in the structure. In the triclinic P1 symmetry, there are two crystallographically distinct cavities and two inequivalent amines which interact with the formate framework through hydrogen bonds of similar strength. In the high-temperature phase of DMAZnD (trigonal, Rc) all Zn–O distances are the same (2.104 Å at 180 K), the octahedral distortion parameter is equal to 0, and bond angle variance σ = 1.9 deg.2,49 Symmetry lowering to the triclinic system brings two inequivalent Zn positions with distorted octahedral disposition. As all atoms sit on C1 symmetry sites, all Zn–O distances are inequivalent and range between 2.068(16) Å and 2.139(15) Å. Bond angle variance rises to 6.3 and 5.0 deg for both Zn1 and Zn2, respectively.
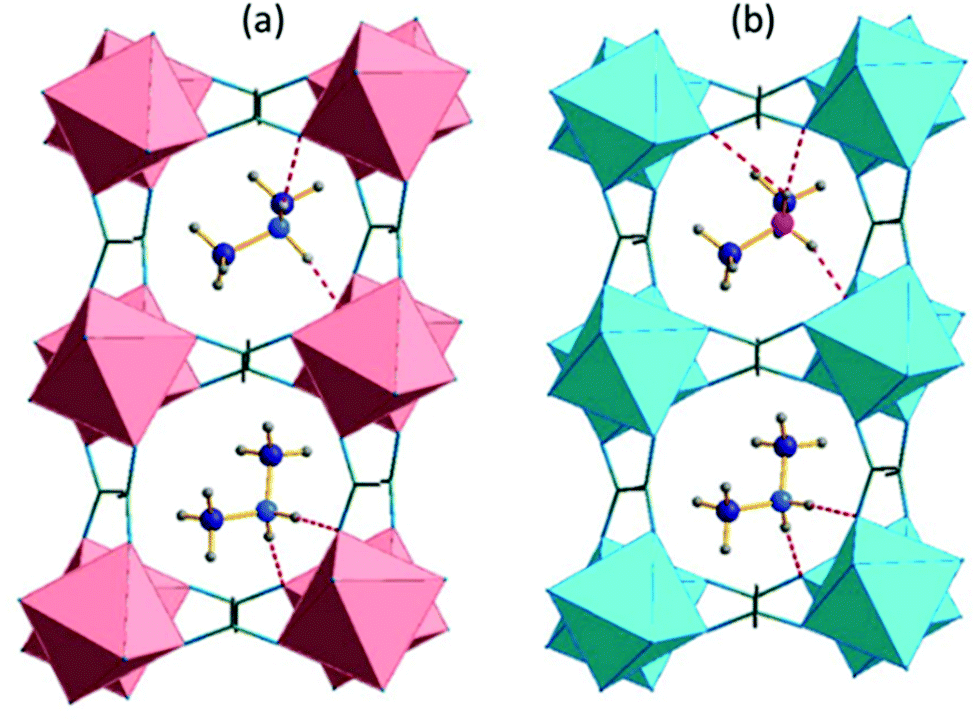 | ||
| Fig. 3 The crystal structure of the low-temperature phase (a) [(CH3)2NH2]Mn(HCOO)3, 100 K, the monoclinic Cc model taken from ref. 43; and (b) [(CH3)2NH2]Zn(DCOO)3 triclinic, P1. The inequivalent nitrogen positions are marked by blue and fuchsia. | ||
3.3 Temperature-resolved second-harmonic generation (TR-SHG) and SHG-on–SHG-off switching
Metal–organic hybrid compounds which feature at least two distinct temperature-dependent crystal phases, among which at least one is noncentrosymmetric, have generated substantial interest as prospective quadratic nonlinear optical switches.41–43,50–53 Bearing in mind that X-ray crystallography indicated structural noncentrosymmetry of the low-temperature phase II of DMAZn, we have sought to spectroscopically verify this property by means of SHG measurements; based on this we have also aimed to assess prospects for this material in quadratic NLO switching.To this end, we have probed the presence of SHG activity around the phase transition temperature Tc through irradiation of the DMAZn microcrystalline powder with 800 nm femtosecond laser pulses in the 133–173 K range. Fig. S4, ESI,† contains all obtained emission spectra, while Fig. 4a displays integral intensities of SHG signals (λSHG = 400 nm) plotted as a function of temperature for cooling and heating cycles. It is evident that cooling below 149 K leads to the formation of a noncentrosymmetric phase, as can be judged from the increase of the SHG activity below that temperature point. Conversely, subsequent heating cycle leads to the abrupt decay of the SHG response, which happens at about 153 K.
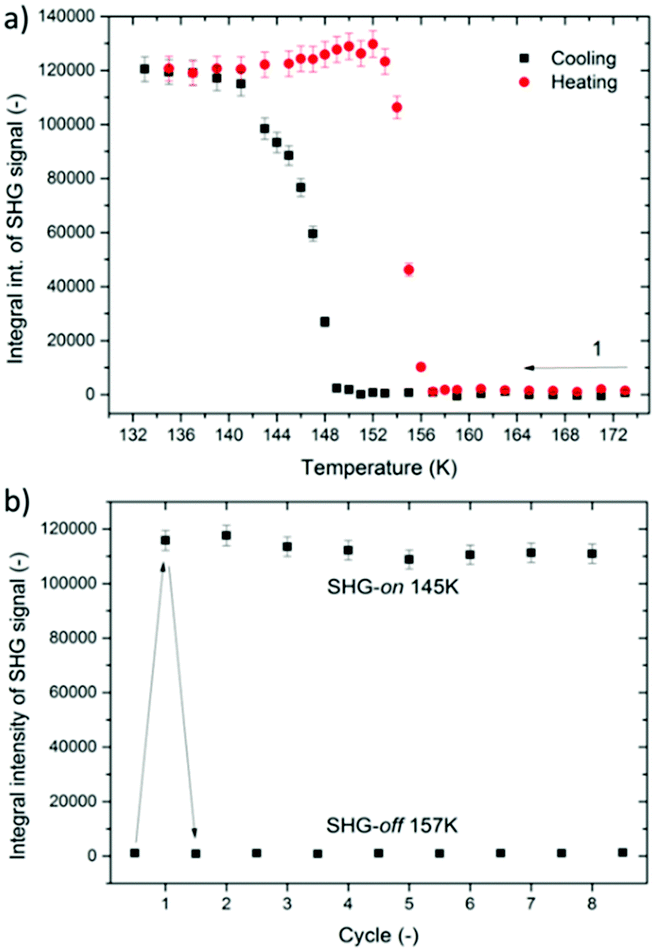 | ||
| Fig. 4 (a) Plots of integral intensity of the SHG signal of DMAZn for heating and cooling cycles. (b) Plot of integral intensities of SHG signals obtained during switching experiment between 157 K and 145 K. | ||
It should also be noted that the SHG temperature response of the same pattern has been found for the Mn analogue, [(CH3)2NH2][Mn(HCOO)3].44 Overall, the obtained TR-SHG plot demonstrates a well-shaped 8 K-wide hysteresis loop confirming the postulated first-order character of the studied phase transition. Full reversibility of the phase transition and relatively narrow temperature hysteresis are quintessential characteristics for SHG switching functionality; nevertheless, to explore the switching behavior more we have checked whether the phase transition between the noncentrosymmetric and centrosymmetric phase is reversible over a period of several cooling and heating cycles. This was done in a separate SHG switching experiment in which the temperature of the sample was toggled between boundary temperatures of 145 K (SHG-on) and 157 K (SHG-off). A plot of integral intensities of SHG signals is presented in Fig. 4b. It is apparent that SHG maintains its activity for at least across 8 cooling–heating runs. Accordingly, one can conclude that DMAZn features reversible SHG-on–SHG-off switching in the low temperature range. It should be noted that the present literature contains a large set of quadratic NLO switches of the metal–organic structure with varied switching temperatures.41,54–57
3.4 Dielectric properties
BDS experiments were performed on both DMAZn21 and DMAZn_E samples. After the initial standard procedure of dielectric spectra collection, a DC voltage of about 52 V mm−1 was applied and the measurements were repeated for both samples under the same frequency and temperature conditions. In other words, DMAZn and DMAZn_E materials were measured without and with an external high electric field on. Such a procedure was selected since numerous experiments revealed that a sufficiently high electric field may affect the dielectric phenomena in a material, leading to a better understanding of processes standing behind them. Collected ε′ and ε′′ spectra of the complex dielectric permittivity ε* = ε′− iε′′, as well as exemplary fitting procedures are presented in Fig. 5 and Fig. S5 (ESI†). The spectra are highly similar for DMAZn and DMAZn_E materials independent of the field conditions. Therefore, further discussion is made on DMAZn_E measured under a DC voltage of about 52 V mm−1. At first glance, a single dipolar relaxation process is visible above Tc (further named as process I) in line with previous reports for DMAZn measured with the DC electric field off.21 The real part of dielectric permittivity (ε′) exhibits a smeared step-like feature which shifts towards higher temperatures with increasing frequency. The corresponding dielectric loss (ε′′) curves show a bell-shape anomaly (Fig. 5a). However, more in-depth analysis in the frequency domain revealed a much more complex image than previously expected and reported. As presented in Fig. 5b, the dielectric anomalies at temperatures higher or equal to 188 K can be well described with a single Havriliak–Negami (HN) fitting function: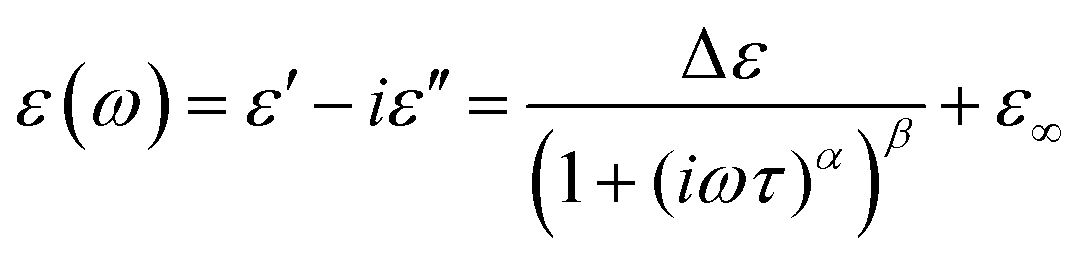 . In this formula Δε denotes dielectric increment, τ is the so-called Havriliak–Negami relaxation time, ε∞ is the high-frequency limit of the dielectric permittivity and α and β are shape parameters. Surprisingly, this situation changes below 188 K in the high-temperature phase (Fig. 5c). Here, the dielectric anomalies cannot be described satisfactorily with even the most general single HN function. This feature gets steadily more apparent when approaching Tc. For example, a significant kink separated by roughly 1 decade from the loss maximum characterizes the loss spectrum at 174 K (see Fig. 5c). Consequently, two fitting functions are needed for its description. This indicates that the loss peaks below 188 K are a combination of two relaxation processes (I and II) of different origins. The dielectric response of DMAZn and DMAZn_E becomes more complex in the transient region around Tc (i.e. 158–164 K), where the coexistence of two phases is apparent (see Fig. S6, ESI†). The amplitude of the most prominent relaxation process from the high-temperature phase starts decreasing below 162 K. Eventually, it vanishes around 150 K and becomes substituted by two dipolar relaxation processes. These relaxations dominate all low-temperature spectra (i.e., below 150 K). Their traces were previously demonstrated for single crystals.25 Therefore, it is reasonable to assume that they are not related to any defect sites in the crystal lattice. In other words, the DMA+ cage cations are expected not to freeze completely below the phase transition temperature. To get a more in-depth insight into the dynamic nature of DMA+ in both low- and high-temperature phases, a temperature plot of relaxation times was prepared and analyzed. Relaxation times, τmax, were determined based on the fitting function adjustment to the experimental spectra and were calculated from the formula:
. In this formula Δε denotes dielectric increment, τ is the so-called Havriliak–Negami relaxation time, ε∞ is the high-frequency limit of the dielectric permittivity and α and β are shape parameters. Surprisingly, this situation changes below 188 K in the high-temperature phase (Fig. 5c). Here, the dielectric anomalies cannot be described satisfactorily with even the most general single HN function. This feature gets steadily more apparent when approaching Tc. For example, a significant kink separated by roughly 1 decade from the loss maximum characterizes the loss spectrum at 174 K (see Fig. 5c). Consequently, two fitting functions are needed for its description. This indicates that the loss peaks below 188 K are a combination of two relaxation processes (I and II) of different origins. The dielectric response of DMAZn and DMAZn_E becomes more complex in the transient region around Tc (i.e. 158–164 K), where the coexistence of two phases is apparent (see Fig. S6, ESI†). The amplitude of the most prominent relaxation process from the high-temperature phase starts decreasing below 162 K. Eventually, it vanishes around 150 K and becomes substituted by two dipolar relaxation processes. These relaxations dominate all low-temperature spectra (i.e., below 150 K). Their traces were previously demonstrated for single crystals.25 Therefore, it is reasonable to assume that they are not related to any defect sites in the crystal lattice. In other words, the DMA+ cage cations are expected not to freeze completely below the phase transition temperature. To get a more in-depth insight into the dynamic nature of DMA+ in both low- and high-temperature phases, a temperature plot of relaxation times was prepared and analyzed. Relaxation times, τmax, were determined based on the fitting function adjustment to the experimental spectra and were calculated from the formula: 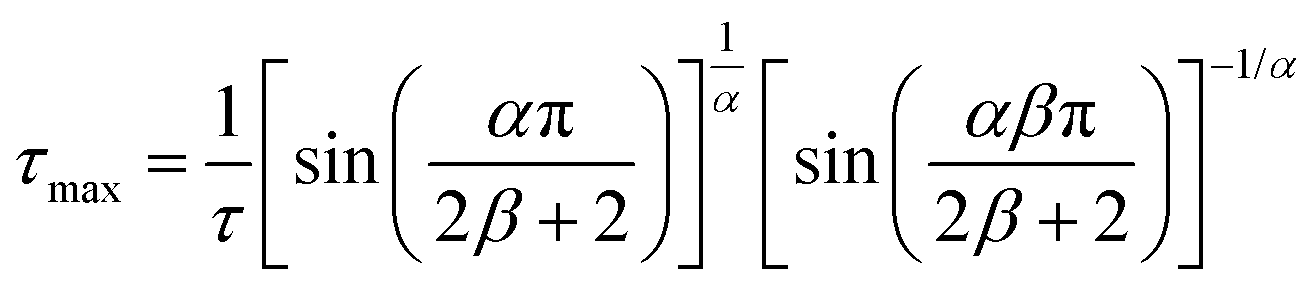 . Due to the highly similar dielectric response of DMAZn and DMAZn_E measured both with and without the dc field, the same fitting procedure was used for each case. Namely, single, two, and three term functions were used depending on the temperature region. Loss peaks above 188 K were described using the most-general single Havriliak–Negami formalism since their both low- and high-frequency slopes are asymmetrically broadened (Fig. 5b). In turn, two Cole–Cole (CC) functions (with the shape parameter β = 1) were selected for dielectric spectra parametrization between 188 and 166 K (Fig. 5c). Two well-resolved low-temperature processes characteristic for the low-temperature phase were parametrized with two HN functions (Fig. 5e). Finally, the model encompassing three fitting functions (two HN and one CC) was used in the transient region (158–164 K) due to overlapping relaxations from the low- and high-temperature phases (Fig. 5d). Shape parameters of these functions were kept fixed and were taken from the dielectric spectra analysis from other temperature ranges. As expected, similar temperature dependences of τmax, were obtained for DMAZn and DMAZn_E independent of the field conditions. As presented in Fig. 6 for the exemplary DMAZn_E measured with an applied dc field, they are linear for all processes, allowing us to use the classical Arrhenius law in a logarithm form for their characterization:
. Due to the highly similar dielectric response of DMAZn and DMAZn_E measured both with and without the dc field, the same fitting procedure was used for each case. Namely, single, two, and three term functions were used depending on the temperature region. Loss peaks above 188 K were described using the most-general single Havriliak–Negami formalism since their both low- and high-frequency slopes are asymmetrically broadened (Fig. 5b). In turn, two Cole–Cole (CC) functions (with the shape parameter β = 1) were selected for dielectric spectra parametrization between 188 and 166 K (Fig. 5c). Two well-resolved low-temperature processes characteristic for the low-temperature phase were parametrized with two HN functions (Fig. 5e). Finally, the model encompassing three fitting functions (two HN and one CC) was used in the transient region (158–164 K) due to overlapping relaxations from the low- and high-temperature phases (Fig. 5d). Shape parameters of these functions were kept fixed and were taken from the dielectric spectra analysis from other temperature ranges. As expected, similar temperature dependences of τmax, were obtained for DMAZn and DMAZn_E independent of the field conditions. As presented in Fig. 6 for the exemplary DMAZn_E measured with an applied dc field, they are linear for all processes, allowing us to use the classical Arrhenius law in a logarithm form for their characterization: 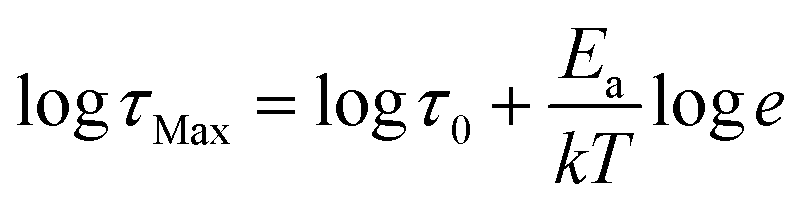 . In this formula Ea and τ0 denote the activation energy and attempt time, respectively, and k is the Boltzmann constant. The determined Ea values for this material are summarized in Table 2. As can be seen, they are similar to the reference DMAZn measured under various electric field conditions and DMAZn_E investigated without an external DC field. For the most prominent high-temperature process I, Ea takes the value around 0.28 eV, in agreement with previous reports on DMAZn. Therefore, its origin can be ascribed to hopping of the DMA+ cation between three symmetry-equivalent positions as indicated in the literature.2,21,24,26 Processes II and III (which are visible as a kink in the high-temperature spectra and well-resolved loss peak in low-temperature spectra, respectively) are characterized by similar Ea equal to 0.25 and 0.23 eV. Moreover, Fig. 6 suggests that process II is a continuation of relaxation III. Such a situation may occur, e.g., due to formation of pretransitional clusters above the phase transition temperature. In fact, this phenomenon was evidenced by solid-state NMR studies19 on DMAZn but was never reported in the dielectric response. According to the NMR experiments, pretransitional clusters of the low-temperature phase start forming below 210 K, i.e. the Burns temperature for DMAZn19,58,59 in agreement with our finding. Therefore, it is reasonable to assume that processes II and III have the same origin. The last low-temperature process IV was parametrized by Arrhenius law with Ea around 0.2 eV. It was postulated that at temperatures below 156 K the rotation of the DMA+ cation becomes frozen. NMR measurements22 suggested that this behaviour is related to the ordering of the NH2 group. The character of the second dipolar relaxation process, yet in the case of analogue compound [(CH3)2NH2][Co(HCOO)3], was assigned to the CH3 group rotation. Recently, CH3 group rotation was also suggested as the source of dipolar relaxation in the low-temperature phase of DMAZn.13 The molecular dynamics of the DMA+ cations in the cyano-bridged frameworks was investigated at low temperatures.15,50 Based on NMR results, two possible origins of the movement have been proposed. The first model assumes that the DMA+ cation flips 180° around the
. In this formula Ea and τ0 denote the activation energy and attempt time, respectively, and k is the Boltzmann constant. The determined Ea values for this material are summarized in Table 2. As can be seen, they are similar to the reference DMAZn measured under various electric field conditions and DMAZn_E investigated without an external DC field. For the most prominent high-temperature process I, Ea takes the value around 0.28 eV, in agreement with previous reports on DMAZn. Therefore, its origin can be ascribed to hopping of the DMA+ cation between three symmetry-equivalent positions as indicated in the literature.2,21,24,26 Processes II and III (which are visible as a kink in the high-temperature spectra and well-resolved loss peak in low-temperature spectra, respectively) are characterized by similar Ea equal to 0.25 and 0.23 eV. Moreover, Fig. 6 suggests that process II is a continuation of relaxation III. Such a situation may occur, e.g., due to formation of pretransitional clusters above the phase transition temperature. In fact, this phenomenon was evidenced by solid-state NMR studies19 on DMAZn but was never reported in the dielectric response. According to the NMR experiments, pretransitional clusters of the low-temperature phase start forming below 210 K, i.e. the Burns temperature for DMAZn19,58,59 in agreement with our finding. Therefore, it is reasonable to assume that processes II and III have the same origin. The last low-temperature process IV was parametrized by Arrhenius law with Ea around 0.2 eV. It was postulated that at temperatures below 156 K the rotation of the DMA+ cation becomes frozen. NMR measurements22 suggested that this behaviour is related to the ordering of the NH2 group. The character of the second dipolar relaxation process, yet in the case of analogue compound [(CH3)2NH2][Co(HCOO)3], was assigned to the CH3 group rotation. Recently, CH3 group rotation was also suggested as the source of dipolar relaxation in the low-temperature phase of DMAZn.13 The molecular dynamics of the DMA+ cations in the cyano-bridged frameworks was investigated at low temperatures.15,50 Based on NMR results, two possible origins of the movement have been proposed. The first model assumes that the DMA+ cation flips 180° around the  axis. The second approach
describes the movement in the transitional phase, where the observed spectral changes are attributed to the oscillatory fluctuations.15 The evidence for the explanation of molecular dynamics was investigated by neutron scattering experiments.50 These results indicate that in the low-temperature phase there exist the rotation of the methyl group around the C–N bond and π-flips. Additionally, the proton nuclear magnetic resonance results indicate molecular motion, the oscillation of the N–CH3 bonds around the axis of the electric dipole, or the fluctuation of the NH2 group around the C2 axis.50 However, it is worth formulating the question whether in the case of DMAZn the methyl group rotation does really induce the change of dipole moment high enough that could be visible in the dielectric spectra? At present this hypothesis lacks both experimental and theoretical verification. Thus, in order to resolve this ambiguity we have performed electronic structure calculations of various structural scenarios that could (or could not) contribute to the observed dielectric relaxation in the low temperature phase.
axis. The second approach
describes the movement in the transitional phase, where the observed spectral changes are attributed to the oscillatory fluctuations.15 The evidence for the explanation of molecular dynamics was investigated by neutron scattering experiments.50 These results indicate that in the low-temperature phase there exist the rotation of the methyl group around the C–N bond and π-flips. Additionally, the proton nuclear magnetic resonance results indicate molecular motion, the oscillation of the N–CH3 bonds around the axis of the electric dipole, or the fluctuation of the NH2 group around the C2 axis.50 However, it is worth formulating the question whether in the case of DMAZn the methyl group rotation does really induce the change of dipole moment high enough that could be visible in the dielectric spectra? At present this hypothesis lacks both experimental and theoretical verification. Thus, in order to resolve this ambiguity we have performed electronic structure calculations of various structural scenarios that could (or could not) contribute to the observed dielectric relaxation in the low temperature phase.
 | ||
| Fig. 5 (a) Dielectric permittivity as a function of temperature for the selected frequencies (the color background represents a range of temperature for the relaxation peaks.); fits of the complex permittivity using Havriliak–Negami functions for DMAZn_E (b) for one-relaxation processes in the high-temperature phase; and (c) for two relaxation processes in the high-temperature phase. (d) Fits for three relaxation processes in the pretransitional period. (e) Fitting for two relaxation processes in the low-temperature phase. Dashed lines and areas represent the relaxation peaks while the solid line represents the sum of the responses from all relaxation processes. The arrows represent the combination of Havriliak–Negami functions. | ||
 | ||
| Fig. 6 Relaxation map as a function of 1000/T for DMAZn_E (the linear fits represent Arrhenius processes). | ||
| Compound | E a (eV) | |||
|---|---|---|---|---|
| I | II | III | IV | |
| DMAZn (single crystal sample) | 0.2821 | — | 0.2025 | 0.2925 |
| DMAZn measurement 52 V mm−1 (pellet sample) | 0.26 ± 0.01 | 0.25 ± 0.01 | 0.23 ± 0.01 | 0.16 ± 0.01 |
| DMAZn_E (pellet sample) | 0.27 ± 0.01 | 0.25 ± 0.01 | 0.24 ± 0.01 | 0.20 ± 0.01 |
| DMAZn_E measurement 52 V mm−1 (pellet sample) | 0.28 ± 0.01 | 0.25 ± 0.01 | 0.23 ± 0.01 | 0.20 ± 0.01 |
3.5 DFT
Electronic structure calculations of DMAZn in different phases were begun from the optimization of crystal structures. Atomic coordinates obtained from diffraction data reported here and elsewhere44 have been used as starting points. It should be pointed out that for structures 1–4 tabulated in Table S4 (ESI†) the crystallographic data contained fractional occupancies, i.e., there were multiple equivalent positions of the DMA+ cation. Accordingly, to make possible electronic structure computations the fractional occupancies were brought to unity for each individual amine molecule. The obtained set of unique, optimized structures can be divided into two distinct subgroups differing in their relative internal energies, with the most stable structure being used as the reference: (1, 3, 4) and (2, 5, 6) – approx. 4 and 0 kJ mol−1 per 2 Zn atoms, respectively. A fundamental feature of the former, less stable subgroup is that its members preserve their original crystal symmetries after optimization. Conversely, the latter subgroup is more stable, but the symmetry change takes place in each case. Specifically, hexagonal structure 2 and monoclinic structure 6 both converged to the triclinic P1 symmetry, while the triclinic structure 5 showed no symmetry experimentally. Structure 5 has also been indicated as the most probable model based on the XRD data and this is confirmed by its low relative energy. Structure 6 was constructed based on the diffraction data reported for the low-temperature analogue compound with manganese. One way to assess whether the given structures are approximately the same is to compare their powder XRD (PXRD) diffractograms. Indeed, it turns out that PXRD traces (Fig. S7, ESI†) for structures 2, 5 and 6 are almost undistinguishable. This observation, together with the analysis of the geometry of the system (both of the framework and alignment of organic cation) and relative energies brought us to the conclusion that geometry optimization converges all three distinct experimental structures to almost the same system. What is more, when the precision of determination of unit cell parameters is reduced to 0.04 Å all three optimized structures are essentially indistinguishable. With a set of optimized structures in hand we have moved to the computational explanation of two (high and low frequency) relaxation processes observed in the dielectric spectra of low-temperature phase of DMAZn. To this end we explored few possible scenarios. We attribute the high-frequency process to tilting of the DMA+ cations being the residual or the rotation around an axis passing through two carbon atoms, as observed in the high-temperature phase (T > Tc). This specific process will be fully addressed in the further part of this section, describing the energetics of the cation rotation. First, we focus the discussion of the origin of the low-frequency process. We have analysed rotation of the methyl groups in DMA+ following some previous literature reports.59 For the analysis of the energy of this process, we performed SS-NEB calculations of the stepwise rotation (20°) of the methyl group up to 120°. It is important to note that calculations were performed for the cation located inside the DMAZn voids, to fully account for the impact of the real chemical environment. Fig. 7 (top) presents the MEP for the rotation of the methyl group. The energy required to cross the rotation barrier for one methyl group is equal to approx. 11.5 kJ mol−1 (i.e., 0.12 eV). This low energy suggests that methyl group rotation could be thermally excited; however, free energy analysis is required to evaluate the exact temperature of activation. Fig. 7 (bottom) shows the change of the directional components of dipole moment and its magnitude as a function of the angle of rotation. In terms of absolute values, the dipole moment change is basically insignificant (less than 0.02 a.u.). This low change of dipole moment value could not be seen in dielectric spectra. Based on this, we rejected the hypothesis that methyl group rotation is responsible for the low-frequency relaxation process. Another considered scenario that could be behind the two relaxation processes in the low-temperature dielectric spectra is the presence of a clustering effect. The clustering effect in this system could be observed after the high-temperature to the low-temperature phase transition, in which DMA+ cations are frozen in two different orientations which are not symmetrically equivalent (structure has no symmetry – P1 space group). Consequently, these two DMA+ may experience disparate local chemical environments. To check whether this scenario is plausible, we have analyzed the chemical environment of both cations and its energetic response to small distortions (rotation). Fig. 8a and c present the distorted (rotated) cations.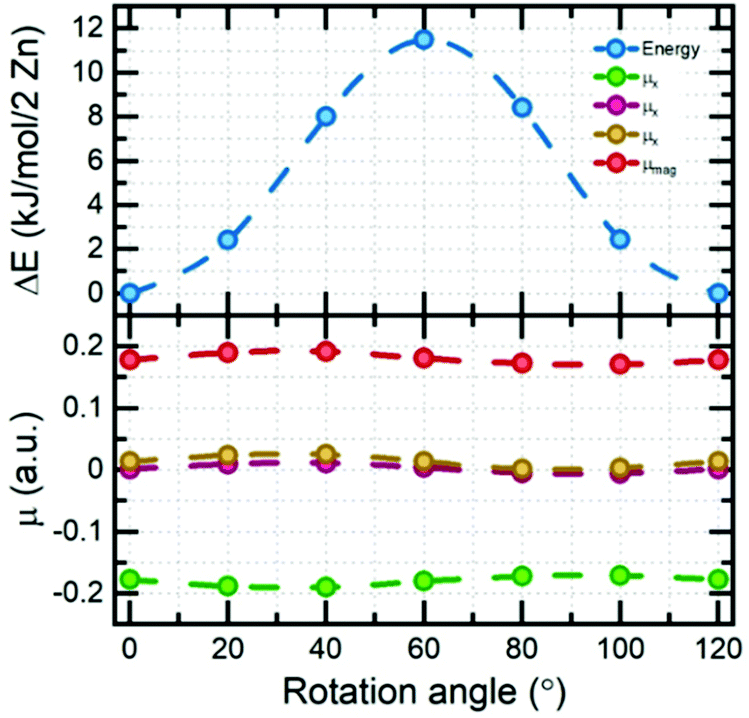 | ||
| Fig. 7 Energy and dipole moment as a function of the rotation angle of the methyl group. | ||
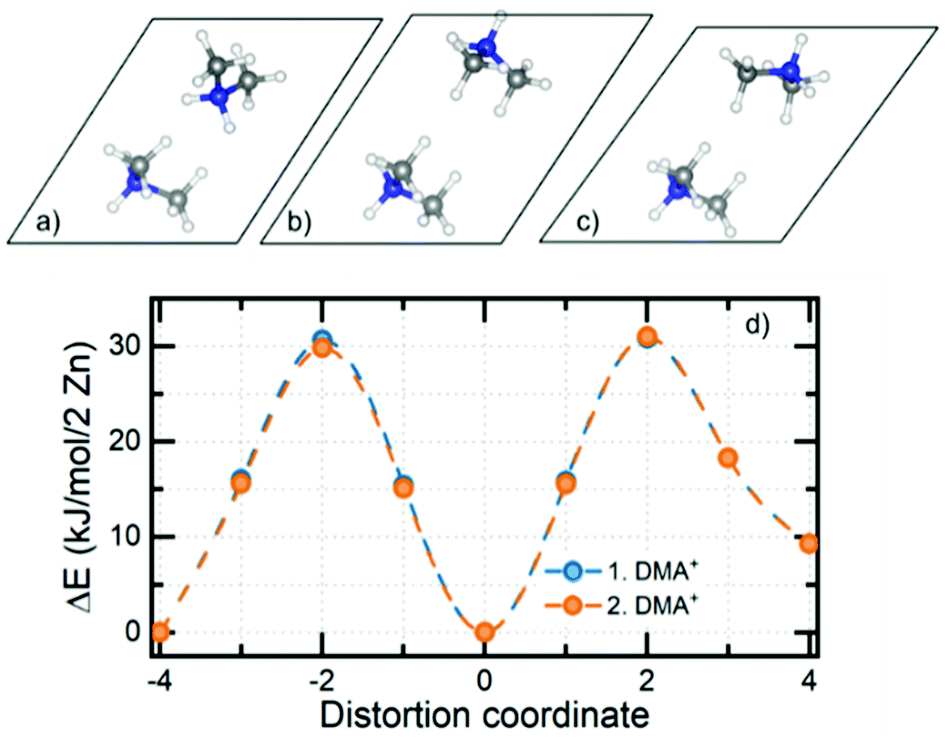 | ||
| Fig. 8 Distorted orientation of the DMA+ cation (top) and MEP of the rotation obtained from SS-NEB calculations (bottom). Distortion coordinate is assumed to go from structure a to b and from b to c. | ||
Both cations were rotated in the same manner; however, for clarity of presentation we show the visualization of rotation for one, selected (upper face of unit cell) cation. In Fig. 8b one can see the crystallographic orientation of both DMA+ cations. Again, we performed SS-NEB calculations with a, b and c optimized structures as endpoints. From Fig. 8d it is obvious that individual dynamics of a given DMA+ cation does not influence the rotation energetics of an adjacent cation. It is thus evident that both cations are equivalent in terms of chemical environment and response to rotation, and minimum energy paths between phases considered stable (or metastable) are almost indistinguishable. This brings us to the conclusion that that the low-frequency relaxation process cannot be explained by a clustering effect of any kind.
To check other possible scenarios, we performed full analysis of the rotation of arbitrarily selected DMA+ cations (one of the two present in the unit cell). To this end, we prepared eight different structures by rotating the cation around the axis passing through carbon atoms with increments of 45° (eight rotations – 360°). We label the structures with numbers from 0 to 7, as listed in Table S5 (ESI†). After geometry optimization we obtained 5 distinguishable structures – S0, S2, S3, S4 and S5 (Fig. 9, right panel). Structure S0 is the same as the experimental one, and is the most stable (based on relative energy – 0 kJ mol−1 per 2 Zn). Structures S1 and S7 are converged to the same geometry as S0 whereas S6 is equivalent to S5, hence we withdraw them from further discussion. Interestingly, structure S3 resembles a previously discussed phase in which both cations are aligned in an antiparallel fashion. It is a metastable structure, and its relative energy is equal to 3.84 kJ mol−1 per 2 Zn (comparable to structure 3 from Table S4, ESI†). The other three structures were not observed experimentally; however, it is possible for them to be excited by an electric field since the rotation of the cation changes the direction of its dipole moment. S2, S3 and S4 are distinguishable in simulated PXRDs (Fig. 9, bottom left); nevertheless, the differences do not seem to be significant. Interestingly, structures S1 and S5 exhibit a similar PXRD pattern, as compared to structures 2, 5 and 6 from Table S4 (ESI†). To fully confirm the existence of particular phases, one would have to perform experimental PXRD analysis with an applied electric field; however, this is beyond the scope of the present study. The high-frequency relaxation process is attributed to the residual tilts associated with the high-temperature process, i.e., cations are probing the potential energy landscape as shown in Fig. 8. In turn, the low-frequency process is attributed to the hopping between stable and metastable positions, as presented in Fig. 9, top left. Rotations of the DMA+ cation naturally change the dipole moment, and hence the process provides a dielectric response. Significantly lower frequency results from the existence of energy barriers between minima which must be crossed during rotations which simply requires more time. We emphasize that there can be many more minima which exist on the potential energy surface of cation rotation; nonetheless, it would be almost impossible to analyze all of them from the computational perspective since structures like MOFs and other framework materials in general show a very flat potential energy surface with many possible metastable phases.
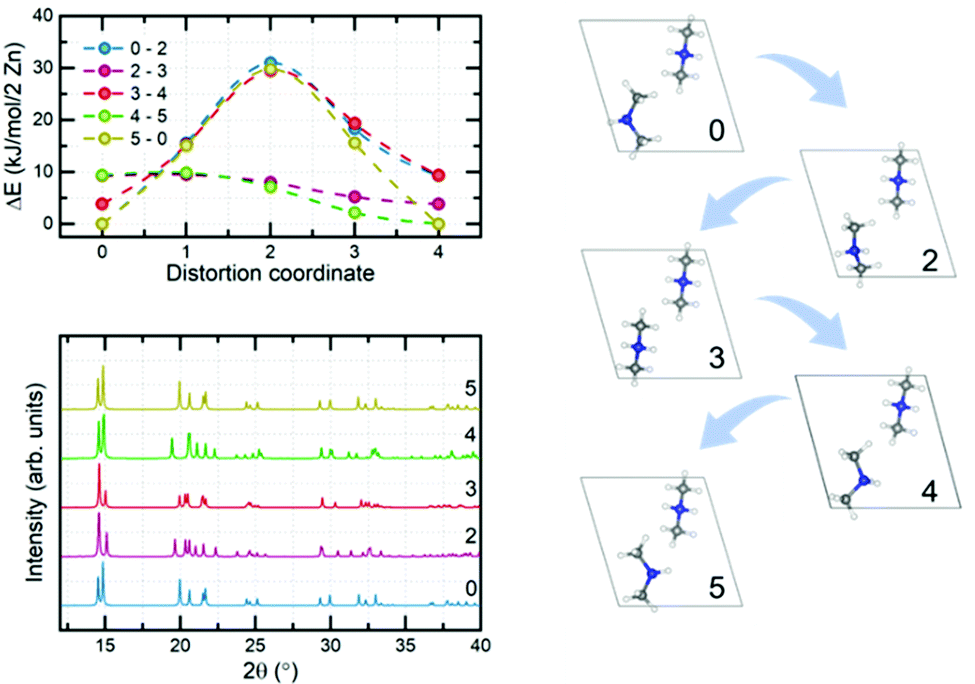 | ||
| Fig. 9 MEP of transition between stable and metastable DMA+ configurations obtained from geometry optimization (top left), corresponding PXRD diffractograms (bottom left) and visualization of their structure (right). | ||
Conclusions
Based on these studies, we report a much more complex behavior of DMAZn than that originally thought.Firstly, calorimetric studies of the non-deuterated DMAZn sample revealed a first-order phase transition with the onset at 167 K on heating and 156 K on cooling, in line with previous studies. However, the related change of entropy (∼4 J mol−1 K−1) is lower than the theoretical value (∼9.1 J mol−1 K−1). This means that the phase transition cannot be regarded as a simple order–disorder transformation. The calorimetric studies proved also that field-assisted crystallization does not change the properties of the synthesized compound and does not lead to the formation of other polymorphic forms. Similarly, deuteration of the formate linkers does not affect the nature of mechanism of the phase transition, leading only to a slight increase in the Tc value.
Secondly, XRD measurements on the deuterated analogue (DMAZnD) allowed observation of the splitting of Bragg peaks in its low-temperature phase. This feature can only be explained by symmetry reduction to the triclinic system in the low-temperature phase, not monoclinic as previously reported. A non-zero signal during second harmonic generation measurements allowed us to assign the crystal structure of the low-temperature phase of DMAZnD to triclinic P1 with two symmetry independent DMA+ cations. Such a structure of low-temperature phase is also expected in non-deuterated DMAZn due to previously-reported isomorphism with the DMAZnD analogue. The presence of two independent dimethylammonium ions is in line with molecular interpretation of cation dynamics inferred from DFT results. It is worth emphasizing that the triclinic symmetry of the low-temperature phase of DMAZnD contrasts with what has been determined for its manganese analogue [(CH3)2NH2][Mn(HCOO)3], whose low-temperature phase was modelled in the monoclinic Cc space group. The high-temperature symmetry of DMAZn is determined as Rc, in agreement with previous reports.
Temperature-resolved SHG measurements provided unequivocal proof of an acentric structure of the DMAZn low temperature phase. Hysteretic behavior of the temperature plot of SHG intensity supported the first-order nature of phase transition. From the application standpoint the capability of DMAZn to perform as a nonlinear optical switch of the SHG-on–SHG-off kind has been explored. Indeed, temperature cycling experiments demonstrated that the intensity of SHG for the low-temperature acentric phase is fully restored after one cooling–heating cycle as well as being stable across 8 consecutive cycles.
Finally, the dielectric response of DMAZn was revealed to be much more complex than originally thought. Namely, two dipolar relaxations are detectable in the high-temperature phase below 188 K. Their loss peaks lie close to each other and thus are partially overlapping. The dominant dielectric anomaly originates from reorientations of DMA+ cations between three symmetrically equivalent positions. Its activation energy is roughly 0.28 eV. In turn, the second, faster relaxation process of Ea = 0.25 eV has a continuation in the low-temperature phase. Most likely, it appears due to the formation of pretransitional clusters above the phase transition temperature. This feature confirms the complex character of the phase transition in DMAZn and may explain the discrepancy between theoretical and experimental values of entropy change around Tc. We also report two relaxation anomalies in the low-temperature phase. The low-frequency relaxation was previously attributed to methyl group rotations, but our DFT results demonstrate that methyl group rotation does not induce a change of the dipole moment high enough to be reflected in the dielectric response. Analysis of various scenarios, such as the clustering effect, have finally led us to the conclusion that the high-frequency relaxation process can be attributed to the residual tilts associated with the well-known high-temperature process i.e. rotation of 120 degrees between three equivalent positions, whereas the low-frequency process can be attributed to the hopping between stable and metastable positions of the DMA+ cation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are deeply grateful for financial support by the National Science Centre within the framework of the Opus13 project (Grant No. DEC-2017/25/B/ST3/02321). JKZ acknowledges Academia Iuvenum, Politechnika Wroclawska for support.Notes and references
- R. Shang, S. Chen, K.-L. L. Hu, B.-W. W. Wang, Z.-M. M. Wang and S. Gao, Chem. – Eur. J., 2016, 22, 6199–6203 CrossRef CAS PubMed.
- P. Jain, V. Ramachandran, R. J. Clark, D. Z. Hai, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS PubMed.
- P. Jain, N. S. Dalal, B. H. Toby, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2008, 130, 10450–10451 CrossRef CAS PubMed.
- M. Ptak, A. Sieradzki, M. Šimėnas and M. Maczka, Coord. Chem. Rev., 2021, 448, 214180 CrossRef CAS.
- X. Y. Wang, L. Gan, S. W. Zhang and S. Gao, Inorg. Chem., 2004, 43, 4615–4625 CrossRef CAS PubMed.
- M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) czka, A. Ggor, B. Macalik, A. Pikul, M. Ptak and J. Hanuza, Inorg. Chem., 2014, 53, 457–467 CrossRef PubMed.
czka, A. Ggor, B. Macalik, A. Pikul, M. Ptak and J. Hanuza, Inorg. Chem., 2014, 53, 457–467 CrossRef PubMed. - M. Maczka, A. Sieradzki, B. Bondzior, P. Dereń, J. Hanuza and K. Hermanowicz, J. Mater. Chem. C, 2015, 3, 9337–9345 RSC.
- Z. Zhang, H. Tang, D. Cheng, J. Zhang, Y. Chen, X. Shen and H. Yu, Results Phys., 2019, 12, 2183–2188 CrossRef.
- M. Sánchez-Andújar, L. C. Gómez-Aguirre, B. Pato Doldán, S. Yáñez-Vilar, R. Artiaga, A. L. Llamas-Saiz, R. S. Manna, F. Schnelle, M. Lang, F. Ritter, A. A. Haghighirad and M. A. Senaris-Rodriguez, CrystEngComm, 2014, 16, 3558–3566 RSC.
- M. Šimenas, M. Ptak, A. H. Khan, L. Dagys, V. Balevičius, M. Bertmer, G. Völkel, M. Maczka, A. Pöppl and J. Banys, J. Phys. Chem. C, 2018, 122, 10284–10292 CrossRef.
- P. Jain, A. Stroppa, D. Nabok, A. Marino, A. Rubano, D. Paparo, M. Matsubara, H. Nakotte, M. Fiebig, S. Picozzi, E. S. Choi, A. K. Cheetham, C. Draxl, N. S. Dalal and V. S. Zapf, npj Quantum Mater., 2016, 1, 1–6 Search PubMed.
- M. Mączka, A. Pietraszko, B. Macalik and K. Hermanowicz, Inorg. Chem., 2014, 53, 787–794 CrossRef PubMed.
- T. Asaji and K. Ashitomi, J. Phys. Chem. C, 2013, 117, 10185–10190 CrossRef CAS.
- S. K. Ramakrishna, K. Kundu, J. K. Bindra, S. A. Locicero, D. R. Talham, A. P. Reyes, R. Fu and N. S. Dalal, J. Phys. Chem. C, 2021, 125, 3441–3450 CrossRef CAS.
- W. Zhang, H.-Y. Y. Ye, R. Graf, H. W. Spiess, Y.-F. F. Yao, R.-Q. Q. Zhu and R.-G. G. Xiong, J. Am. Chem. Soc., 2013, 135, 5230–5233 CrossRef CAS PubMed.
- S. Bertaina, N. Abhyankar, M. Orio and N. S. Dalal, J. Phys. Chem. C, 2018, 122, 16431–16436 CrossRef CAS.
- P. Peksa, J. Trzmiel, M. Ptak, A. Ciupa-Litwa and A. Sieradzki, Materials, 2021, 14, 6150 CrossRef CAS PubMed.
- Z. Y. Du, T. T. Xu, B. Huang, Y. J. Su, W. Xue, C. T. He, W. X. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2015, 54, 914–918 CrossRef CAS PubMed.
- N. Abhyankar, J. J. Kweon, M. Orio, S. Bertaina, M. Lee, E. S. Choi, R. Fu and N. S. Dalal, J. Phys. Chem. C, 2017, 121, 6314–6322 CrossRef CAS.
- M. Mączka, A. Pietraszko, L. Macalik, A. Sieradzki, J. Trzmiel and A. Pikul, Dalton Trans., 2014, 43, 17075–17084 RSC.
- P. Peksa, J. Trzmiel, K. Fedoruk, A. Gagor, M. Šimenas, A. Ciupa, S. Pawlus, J. Banys, M. Maczka and A. Sieradzki, J. Phys. Chem. C, 2019, 123, 23594–23603 CrossRef CAS.
- T. Besara, P. Jain, N. S. Dalal, P. L. Kuhns, A. P. Reyes, H. W. Kroto and A. K. Cheetham, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6828–6832 CrossRef.
- B. Pato-Doldán, M. Sánchez-Andújar, L. C. Gómez-Aguirre, S. Yáñez-Vilar, J. López-Beceiro, C. Gracia-Fernández, A. A. Haghighirad, F. Ritter, S. Castro-García and M. A. Senaris-Rodriguez, Phys. Chem. Chem. Phys., 2012, 14, 8498–8501 RSC.
- M. Šimenas, S. Balčiunas, M. Maczka, J. Banys and E. E. Tornau, Phys. Chem. Chem. Phys., 2016, 18, 18528–18535 RSC.
- M. Šimenas, S. Balčiunas, A. Ciupa, L. Vilčiauskas, D. Jablonskas, M. Kinka, A. Sieradzki, V. Samulionis, M. Maczka and J. Banys, J. Mater. Chem. C, 2019, 7, 6779–6785 RSC.
- M. Šimenas, A. Ciupa, G. Usevičius, K. K. Aidas, D. Klose, G. Jeschke, M. Mczka, G. Völkel, A. Pöppl and J. J. Banys, Phys. Chem. Chem. Phys., 2018, 20, 12097–12105 RSC.
- M. Šimenas, A. Ciupa, M. Maczka, A. Pöppl and J. Banys, J. Phys. Chem. C, 2015, 119, 24522–24528 CrossRef.
- M. Mczka, M. Ptak and L. Macalik, Vib. Spectrosc., 2014, 71, 98–104 CrossRef.
- K. Asadi and M. A. van der Veen, Eur. J. Inorg. Chem., 2016, 4332–4344 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Formalik, M. Fischer, J. Rogacka, L. Firlej and B. Kuchta, J. Chem. Phys., 2018, 149, 064110 CrossRef PubMed.
- T. A. Manz and N. G. Limas, RSC Adv., 2016, 6, 47771–47801 RSC.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- E. Bitzek, P. Koskinen, F. Gähler, M. Moseler and P. Gumbsch, Phys. Rev. Lett., 2006, 97, 170201 CrossRef PubMed.
- J.-X. Gao, X.-N. Hua, X.-G. Chen and W.-Q. Liao, J. Mater. Chem. C, 2019, 7, 5348–5352 RSC.
- J. Zhang, Y. Zeng, H. Lu, X. Chen, X. Yuan and Z. Fu, Cryst. Growth Des., 2020, 20, 2617–2622 CrossRef CAS.
- H. Peng, Y. H. Liu, X. Q. Huang, Q. Liu, Z. H. Yu, Z. X. Wang and W. Q. Liao, Mater. Chem. Front., 2021, 5, 4756–4763 RSC.
- M. Śanchez-Andújar, S. Presedo, S. Ýã Nez-Vilar, S. Castro-García, J. Shamir and M. A. Senaris-Rodriguez, Inorg. Chem., 2010, 49, 1510–1516 CrossRef PubMed.
- A. Sieradzki, M. Maczka, M. Simenas, J. K. Zarȩba, A. Gagor, S. Balciunas, M. Kinka, A. Ciupa, M. Nyk, V. Samulionis, J. Banys, M. Paluch and S. Pawlus, J. Mater. Chem. C, 2018, 6, 9420–9429 RSC.
- I. Tables, Int. Tables Crystallogr. Vol. A Space-gr. symmetry, 2006, 1, 342–343 Search PubMed.
- M. Mączka, A. Gągor, N. L. M. Costa, W. Paraguassu, A. Sieradzki and A. Pikul, J. Mater. Chem. C, 2016, 4, 3185–3194 RSC.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195–1215 CrossRef CAS.
- K. Robinson, G. V. Gibbs and P. H. Ribbe, Science, 1971, 172, 567–570 CrossRef CAS PubMed.
- M. Rok, G. Bator, B. Zarychta, B. Dziuk, J. Repeć, W. Medycki, M. Zamponi, G. Usevičius, M. Šimėnas and J. Banys, Dalton Trans., 2019, 48, 4190–4202 RSC.
- J. K. Zaręba, M. Nyk and M. Samoć, Adv. Opt. Mater., 2021, 9, 2100216 CrossRef.
- L. He, P. P. Shi, M. M. Zhao, C. M. Liu, W. Zhang and Q. Ye, Chem. Mater., 2021, 33, 799–805 CrossRef CAS.
- M. Rok, B. Zarychta, J. Trojan-Piegza, A. Bil, A. Piecha-Bisiorek, J. K. Zaręba, W. Medycki and R. Jakubas, J. Mater. Chem. C, 2022, 10, 3036–3047 RSC.
- L. Xu, J.-X. Gao, X.-G. Chen, X.-N. Hua and W.-Q. Liao, Dalton Trans., 2018, 47, 16995–17003 RSC.
- H.-Y. Zhang, S.-Q. Lu, X. Chen, R.-G. Xiong and Y.-Y. Tang, Chem. Commun., 2019, 55, 11571–11574 RSC.
- X. G. Chen, Y. Z. Zhang, D. S. Sun, J. X. Gao, X. N. Hua and W. Q. Liao, Dalton Trans., 2019, 48, 11292–11297 RSC.
- M. Rok, A. Ciżman, B. Zarychta, J. K. Zarȩba, M. Trzebiatowska, M. Mączka, A. Stroppa, S. Yuan, A. E. Phillips and G. Bator, J. Mater. Chem. C, 2020, 8, 17491–17501 RSC.
- R. A. Cowley, S. N. Gvasaliya, S. G. Lushnikov, B. Roessli and G. M. Rotaru, Adv. Phys., 2011, 60, 229–327 CrossRef CAS.
- A. Bussmann-Holder and N. Dalal, Struct. Bonding, 2006, 124, 1–21 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc00089j |
| This journal is © The Royal Society of Chemistry 2022 |