
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Implications of gas-barrier properties in realizing the self-recovery of photodegraded CsPbBr3 perovskite nanocrystals†
Ikumi
Enomoto
,
Yoshiki
Iso
 * and
Tetsuhiko
Isobe
*
* and
Tetsuhiko
Isobe
*
Department of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: iso@applc.keio.ac.jp; isobe@applc.keio.ac.jp; Fax: +81 45 566 1551; Tel: +81 45 566 1558 Tel: +81 45 566 1554
First published on 10th December 2021
Abstract
Fluorescent CsPbBr3 perovskite nanocrystals (NCs) degraded by photoinduced desorption of surface ligands can exhibit self-recovery; however, irreversible oxidative damage from oxygen in the atmosphere must be avoided. In this work, gas-barrier polymer films made of ethylene vinyl alcohol (EVOH) and ethylene vinyl acetate (EVA) copolymers were investigated as sealing materials for CsPbBr3 NC films. The NC film covered with EVOH showed photodegradation with blackening and photoluminescence quenching under light excitation, while self-recovery of the optical properties was observed during dark storage. These phenomena were attributed to the photoinduced desorption of surface ligands (and thus the formation of surface defects) and their re-adsorption, respectively. In contrast, self-recovery did not occur when an EVA film was used. X-ray photoelectron spectroscopy indicated the formation of Pb–O bonds for the EVA-coated film. Thus, EVOH, which has a lower oxygen permeability than EVA, successfully prevented irreversible damage by permeated oxygen. This is an important finding for the reuse or lifetime-extension of CsPbBr3 NCs.
All-inorganic CsPbX3 (X = Cl, Br, or I) perovskite nanocrystals (NCs) exhibit remarkable properties, such as high photoluminescence (PL) quantum yields, narrow emission peaks, tunable emission wavelengths, and higher stabilities against heat and moisture, compared with organic–inorganic APbX3 (A: cation) perovskites.1,2 Accordingly, CsPbX3 NCs are promising materials for optoelectronic applications such as light-emitting diodes (LEDs),3–9 wide color gamut displays,10–12 solar devices,13–17 lasers,18–20 photodetectors,21–23 anti-counterfeiting agents,24–26 and photocatalysts.27,28 Although the toxic lead in CsPbX3 NCs is a concern in terms of commercialization, the current limit for lead (1000 ppm by weight) in the most recent Restriction of Hazardous Substances (RoHS) guidelines is not as severe as that for cadmium (100 ppm by weight), which is widely used in CdSe quantum dots.29
However, the application scope of CsPbX3 NCs is restricted owing to their instability under light irradiation.30,31 The photodegradation of the NCs is caused by changes in the adsorption states of surface ligands.32 The interactions between surface ligands and the NC surface are weak and dynamic,33 leading to their frequent desorption and the formation of surface defects and NC aggregation. When the repulsive forces between dispersed particles due to steric hindrance by surface ligands are lost, the van der Waals forces between the NCs cause aggregation.34 Furthermore, NC surfaces exposed to the atmosphere are degraded by oxygen as follows:35
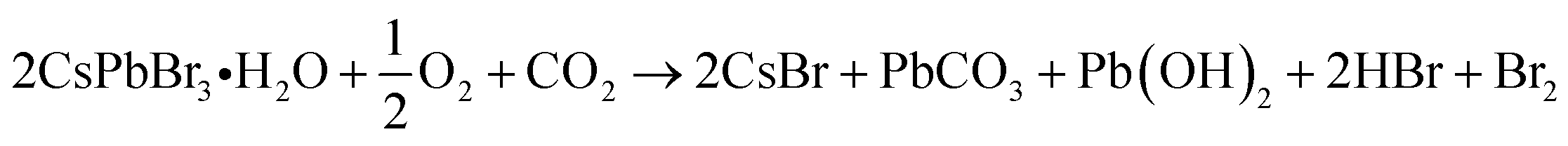 | (1) |
| Pb(OH)2 → PbO + H2O | (2) |
In recent years, there have been numerous studies on improving the stability of such NCs under light irradiation by various strategies,36 such as B-site doping (for ABX3 perovskites),37,38 repairing surface traps,39,40 forming core–shell structures,41 and matrix encapsulation.24
Although significant improvements in photostability have been reported, the PL intensity is not necessarily maintained, particularly when the irradiation intensity is enhanced or prolonged. As a novel strategy, the PL recovery of CsPbBr3 NCs to their initial state after photodegradation should enable their long-term use. The present authors previously reported that, after significant photodegradation by light excitation, solid CsPbBr3 NCs sealed with a glass plate exhibit complete self-recovery of PL properties.42 In that study, the PL intensity of the NCs decreased to ∼20% of the initial intensity under 72 h blue LED irradiation, but then spontaneously increased to 108% upon subsequent dark storage for 2400 h. The solid sample changed from yellow to black during irradiation owing to photoinduced desorption of the surface ligands, but the yellow color was restored after dark storage. We concluded that self-recovery occurs when the desorbed surface ligands are re-adsorbed during dark storage.
Since the self-recovery phenomenon of CsPbBr3 NCs did not occur when the NCs were exposed to air, it was necessary to seal the NCs with a sufficiently thick glass plate. However, in terms of device application scope, using a resin that is easier to mold and more flexible than glass is highly desirable.
In this work, the ethylene vinyl acetate (EVA) copolymer, which is widely used as an encapsulant in solar devices owing to its excellent weather resistance, and the ethylene vinyl alcohol (EVOH) copolymer, which has excellent gas-barrier properties, were used as sealing materials to evaluate their effects on the photodegradation and self-recovery of CsPbBr3 NC films. Film samples of CsPbBr3 NCs with oleic acid and oleylamine as surface ligands were prepared and the effects of the different sealing materials were evaluated by investigating the changes in the film's crystal structure, particle morphology, and optical properties upon degradation by light excitation and subsequent dark storage at 25 °C and 60% relative humidity.
The details of the experimental methods and conditions are given in the ESI.† X-ray diffraction (XRD) analysis revealed that the sample is obtained as cubic CsPbBr3 NCs (Fig. S3, ESI†). From the transmission electron microscopy (TEM) images (Fig. S4, ESI†), it can be observed that the average particle size of the NCs is 8.8 ± 1.4 nm. The UV-vis absorption and PL spectra of the as-prepared NCs dispersed in toluene show an absorption edge at ∼520 nm and an emission peak at 514.4 nm (Fig. S5(a), ESI†). The bandgap estimated from the Tauc plot is 2.39 eV (Fig. S5(b), ESI†), which is larger than the bandgap of cubic CsPbBr3 (∼2.3 eV) owing to the quantum size effect.1
The properties of the prepared CsPbBr3 NC films as illustrated in Fig. 1 were evaluated during continuous irradiation with 468 nm blue LED light for 72 h and subsequent 168 h dark storage. Fig. 2 shows the appearance of the CsPbBr3 NC films before and after irradiation and storage in the dark. The glass-coated film changes from yellow to black during 72 h irradiation. Correspondingly, its UV-vis absorbance (Fig. 3) increases over the entire wavelength range measured. However, the film regains its yellow color and initial absorbance profile almost completely upon subsequent 168 h dark storage. Similar changes are also observed for the EVOH-coated film. These color changes are attributed to the photoinduced desorption and readsorption of surface ligands on the NCs.42
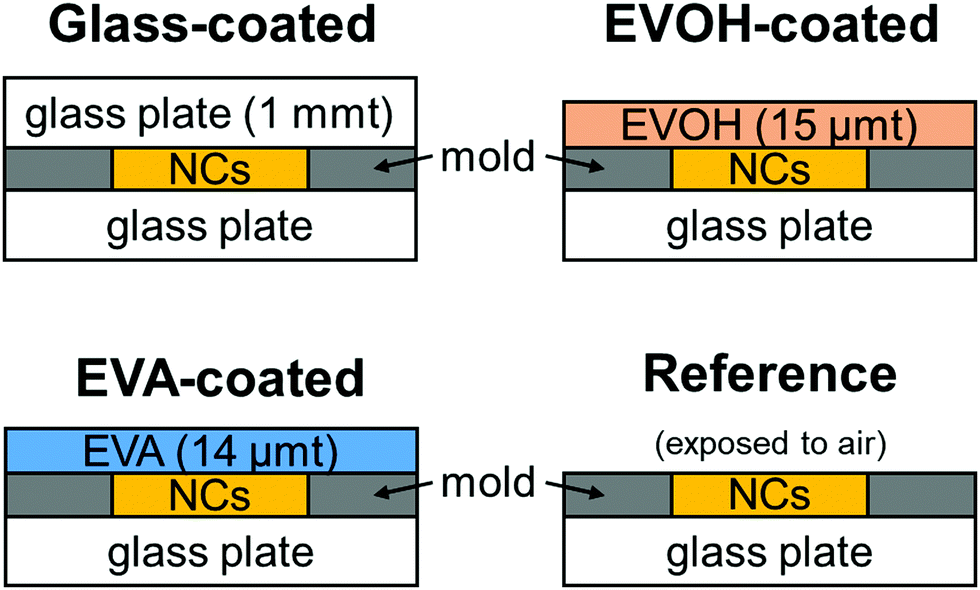 | ||
| Fig. 1 Schematics of the cross-sectional structure of the CsPbBr3 NC films prepared in this study. | ||
 | ||
| Fig. 2 Images of the CsPbBr3 NC films before and after 72 h irradiation and after subsequent 168 h dark storage. Captured under (left) white light and (right) 365 nm UV light. | ||
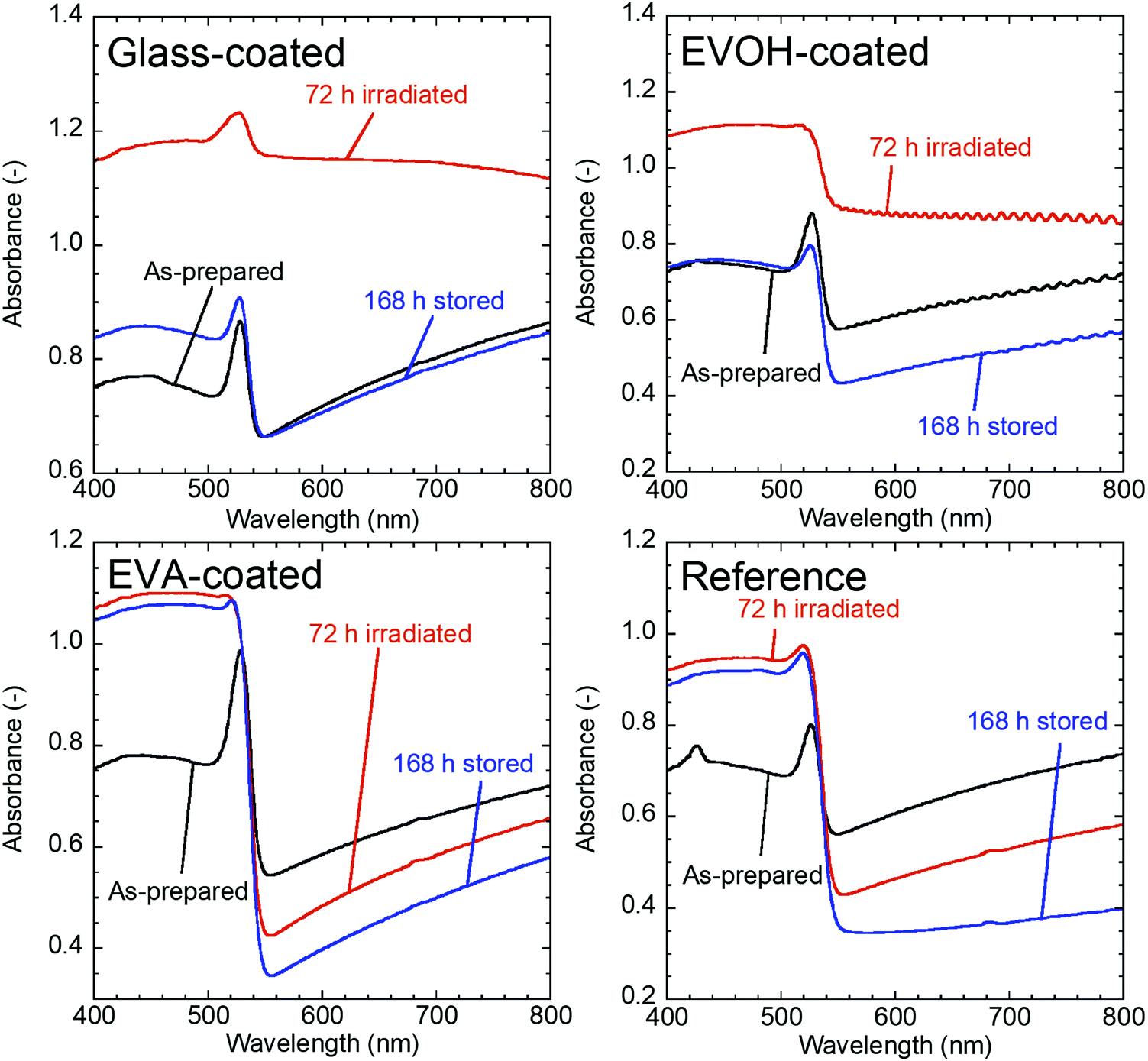 | ||
| Fig. 3 UV-vis spectra of CsPbBr3 NC films before and after 72 h irradiation and after subsequent 168 h dark storage. | ||
Conversely, the EVA-coated film changes from yellow to orange, as also observed for the reference film, which is exposed to ambient air. The film cannot recover its color, even upon subsequent dark storage for 168 h. The UV-vis spectra of the reference and EVA-coated films also exhibit similar changes during irradiation and dark storage.
Fig. 4 shows the changes in the PL spectra and corresponding PL peak intensities for the films. The intensities for all the samples decrease to 25–50% of the initial values during 72 h irradiation. However, upon 168 h dark storage, the glass- and EVOH-coated films recover their PL intensities to 102% and 77%, respectively. In contrast, the EVA-coated film and reference sample do not exhibit PL self-recovery. It should be noted that the shoulder peak at a longer wavelength could be explained by the photon recycling effect.43 Such a shoulder peak was not observed for the blackened samples by irradiation for 72 h because the photon recycling effect seems to be reduced by the increased nonradiative relaxation.44
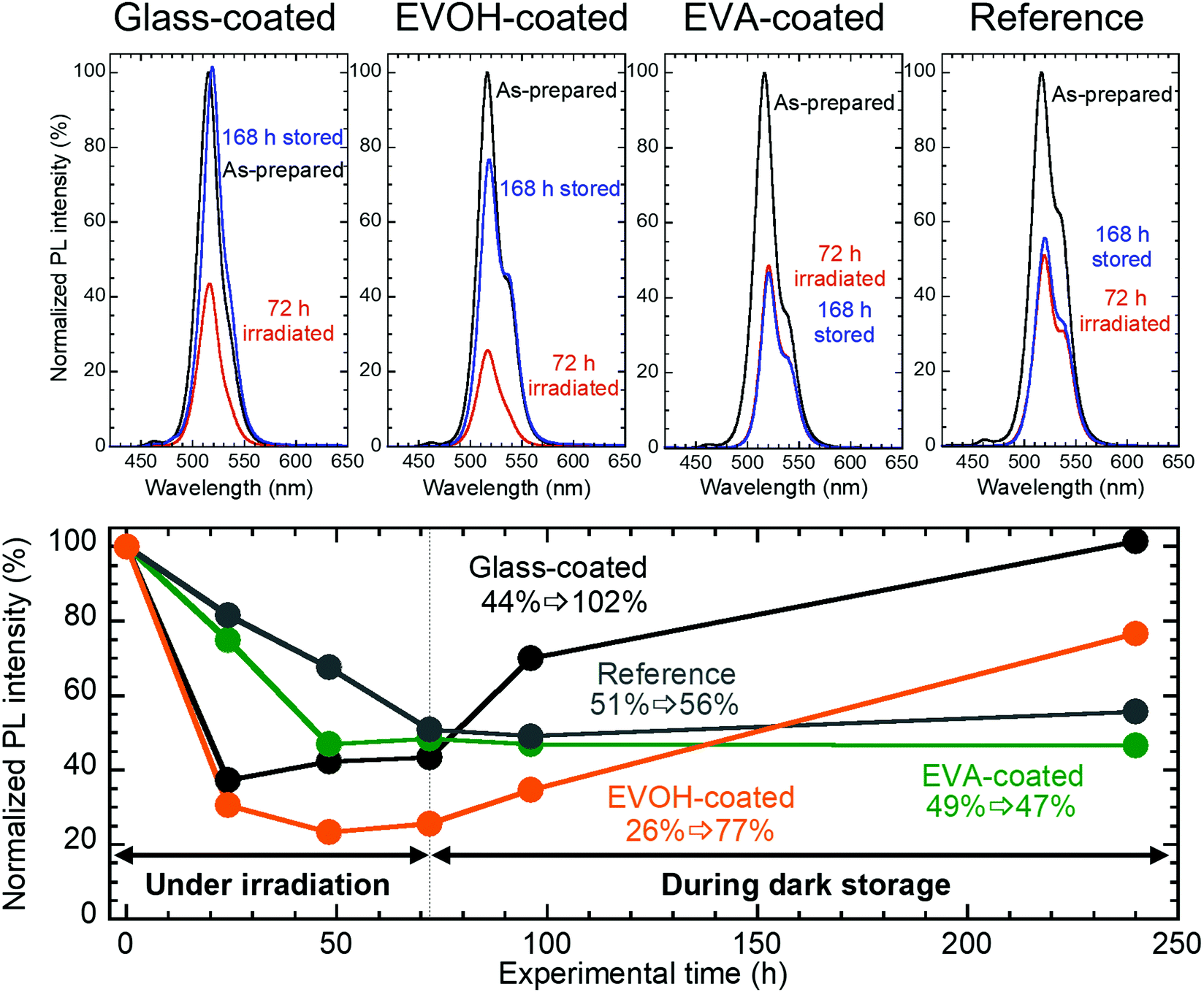 | ||
| Fig. 4 PL spectra and the corresponding PL peak intensities for CsPbBr3 NC films before and after 72 h irradiation and after subsequent 168 h dark storage. λex = 400 nm. | ||
PL decay curves were measured to determine PL lifetimes (see Fig. S6 and Table S1, ESI†). The average PL lifetime for the EVOH-coated film decreases from 27.8 to 11.1 ns upon 72 h irradiation, indicating PL quenching by non-radiative transitions through defects formed under irradiation. According to our previous study, the surface defects presumed to be the cause of sample blackening are generated by the photoinduced desorption of surface ligands adsorbed on the NCs.42 Upon subsequent 168 h storage in the dark, the average PL lifetime of the EVOH-coated film increases to 29.9 ns, indicating a decrease in the non-radiative transition rate. Therefore, the recovery of absorbance and PL intensity mentioned above is attributed to the reduction of surface defects by readsorption of the desorbed surface ligands. However, the PL decay curves and the corresponding PL lifetimes of the EVA-coated film do not exhibit similar trends. This cannot be explained in terms of the photoinduced desorption and readsorption of surface ligands. Accordingly, other degradation mechanisms should be investigated in future studies.
The TEM observation (Fig. S7, ESI†) revealed that the average particle size for the glass-coated film increases from 8.8 ± 1.4 nm to 10.0 ± 1.9 nm upon 72 h irradiation and remains at 10.3 ± 2.6 nm upon subsequent 168 h dark storage. Similar increases in particle size are observed for the other samples.
These increases in primary particle size can be explained by the following two mechanisms: (i) agglomeration of NCs through contact between the crystal surfaces exposed by desorption of the surface ligands and (ii) crystal growth caused by ion supply to the exposed crystal surface, probably through ionic transport in residual oleic acid and oleylamine molecules that chelate to free metal cations and halide anions in the NC material collected by centrifugation. After light irradiation, the average particle size does not increase because of readsorption of the surface ligands.
XRD analysis was performed to evaluate changes in the crystal structure for the CsPbBr3 NC films upon continuous irradiation (Fig. S8, ESI†). The XRD patterns of all the samples do not change upon 72 h irradiation, revealing that the cubic CsPbBr3 structure is maintained. According to the TEM and XRD analyses, morphology and crystal structure are not related to photodegradation and self-recovery. Therefore, the observed blackening and self-recovery are attributed to reversible photoinduced desorption and readsorption of surface ligands.42 However, the irreversible degradation without blackening observed for the EVA-coated film and reference sample require further explanation.
X-ray photoelectron spectroscopy (XPS) analysis was performed to investigate the oxidation state of the NCs. The wide-scan XPS spectrum of the as-prepared NCs shown in Fig. 5(a) reveals the presence of Cs, Pb, and Br.45 In our previous work, the Pb 4f5/2 and 4f7/2 peaks for CsPbBr3 NCs exposed to ambient air under blue LED irradiation shifted to lower energies due to oxidation by oxygen, whereas the NCs sealed by a glass plate maintained the peak positions after the same irradiation.44 The peak shifts were attributed to the appearance of the Pb–O peaks at lower energies compared to the Pb–Br peaks; therefore, the changes in the Pb 4f peaks for the EVOH- and EVA-coated films upon 72 h irradiation and subsequent 168 h storage in the dark were investigated (Fig. 5(b)). The Pb 4f5/2 and 4f7/2 peaks for the EVOH-coated film appear at 143.1 and 138.3 eV, respectively. These binding energies are attributed to Pb–Br bonds.34,44 Conversely, both Pb 4f peaks for the EVA-coated film shift to lower energies at 142.9 and 138.0 eV, respectively, after 72 h irradiation. This shift was also observed after subsequent 168 h storage in the dark. According to the peak deconvolutions (see Fig. S9, ESI†), Pb–O peaks appeared at lower binding energies of 142.8 and 137.9 eV than the Pb–Br peaks at 143.1 and 138.3 eV,35,44 indicating oxidation of CsPbBr3 by oxygen permeated through the EVA film during irradiation. It should be noted that oxidation should occur only near the surface of the NCs because no byproduct is detected by XRD analysis. The irreversible degradation of the EVA-coated film can be explained by oxidation of the exposed surface of NCs. Oxygen that permeated through the EVA film oxidizes the NC surface owing to the loss of surface ligands by photoinduced desorption. This irreversible chemical reaction prevents the recovery of the UV-vis absorption and PL properties for the EVA-coated film and reference sample. The oxidized surface phase might prevent significant decrease in the PL lifetime under irradiation (Fig. S6 and Table S1, ESI†) by passivating the inner fluorescent CsPbBr3 phase, whereas its absorption of excitation light causes the deterioration of PL intensity. In contrast, the EVOH-coated film is not oxidized because of the excellent gas-barrier ability of the EVOH film. The oxygen permeability coefficient of the EVOH film at a relative humidity of 60% and 1 atm is estimated to be ∼0.008 mL mm (m2 day atm)−1,46 and this value is much lower than that of the EVA film (145.8 mL mm (m2 day atm)−1).47 Therefore, the self-recovery of CsPbBr3 NCs can be realized by a suitable resin film instead of a glass plate.
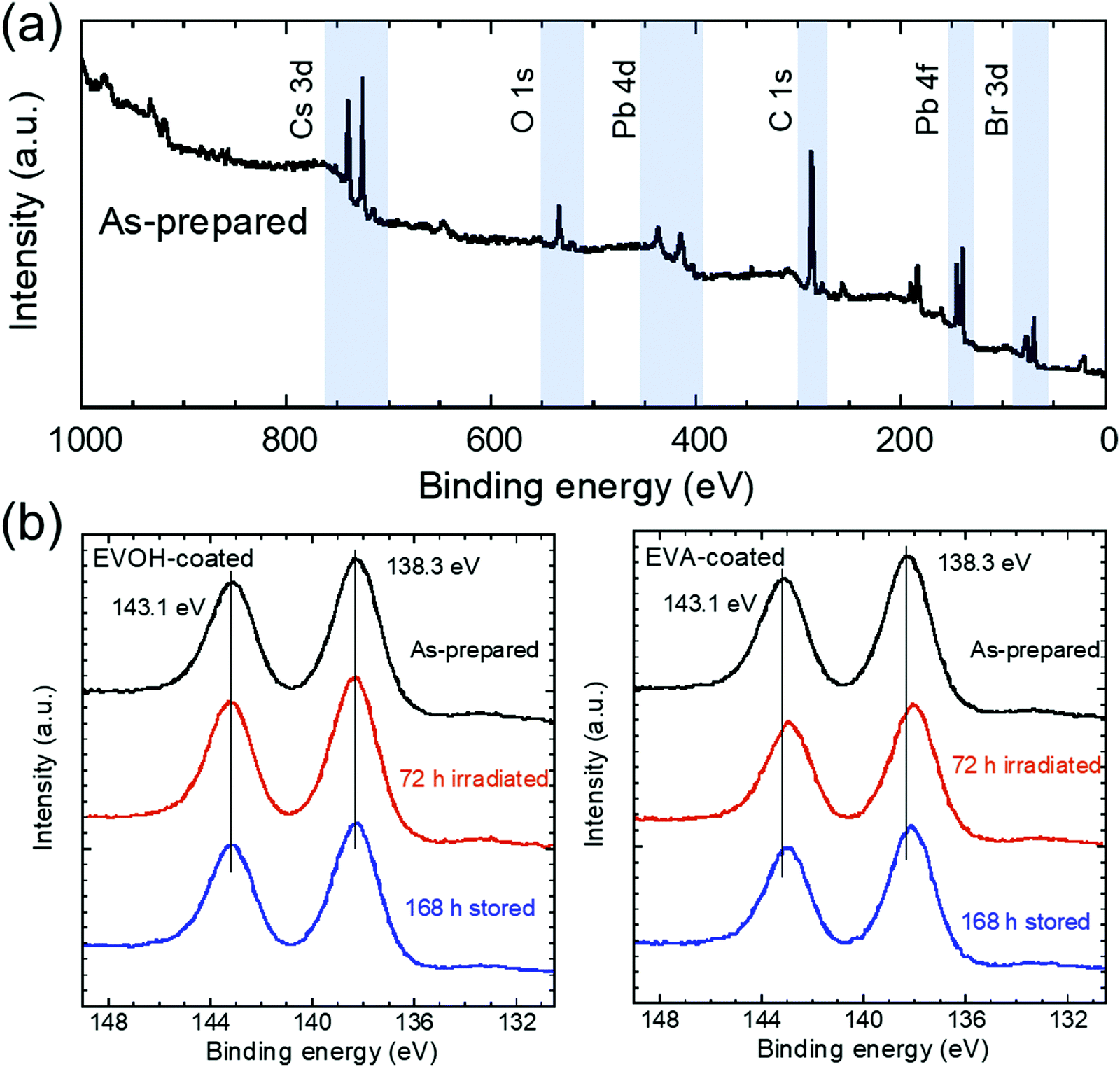 | ||
| Fig. 5 (a) XPS spectrum of the as-prepared CsPbBr3 NC film. (b) Pb 4f peaks after 72 h irradiation and subsequent 168 h dark storage. | ||
In this work, the effects of coating with EVA and EVOH films on the photodegradation and self-recovery of CsPbBr3 NC films were evaluated. The EVOH-coated film shows blackening and a decrease in PL intensity under light excitation, followed by recovery upon subsequent dark storage, similar to the glass-coated film. These phenomena are attributed to the photoinduced desorption of surface ligands, which induces the formation of surface defects and their readsorption after irradiation. Conversely, the EVA-coated film shows irreversible photodegradation similar to that observed for the reference sample, which is exposed to ambient air. This irreversible degradation cannot be explained in terms of the reversible desorption and readsorption of surface ligands. In the XPS spectra of the EVOH-coated film, Pb 4f peaks assigned to Pb–Br bonding are observed. Conversely, peaks attributed to Pb–O bonding are observed for the EVA-coated film, revealing that oxygen permeates through the EVA film. The EVOH film has a much lower oxygen permeability coefficient than the EVA film, preventing irreversible oxidation. Therefore, the self-recovery of photodegraded CsPbBr3 NCs can be realized by the use of a suitable resin film material, such as EVOH. These are important findings and will have far-reaching implications for the application of CsPbBr3 self-recovery as a means to realize its reuse or lifetime-extension for next-generation optoelectronic devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Hosokawa Powder Technology Foundation, the Nippon Sheet Glass Foundation for Materials Science and Engineering, the Mizuho Foundation for the Promotion of Sciences, and JSPS KAKENHI Grant Number JP20K15131.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- C. Chen, D. Li, Y. Wu, C. Chen, Z. G. Zhu, W. Y. Shih and W. H. Shih, Nanotechnology, 2020, 31, 225602 CrossRef CAS PubMed.
- M. Cao, Y. Xu, P. Li, Q. Zhong, D. Yang and Q. Zhang, J. Mater. Chem. C, 2019, 7, 14412–14440 RSC.
- H. Wang, X. Zhang, Q. Wu, F. Cao, D. Yang, Y. Shang, Z. Ning, W. Zhang, W. Zheng, Y. Yan, S. V. Kershaw, L. Zhang, A. L. Rogach and X. Yang, Nat. Commun., 2019, 10, 665 CrossRef CAS PubMed.
- J. S. Yao, J. Ge, K. H. Wang, G. Zhang, B. S. Zhu, C. Chen, Q. Zhang, Y. Luo, S. H. Yu and H. Bin Yao, J. Am. Chem. Soc., 2019, 141, 2069–2079 CrossRef CAS PubMed.
- J. H. Park, A. Y. Lee, J. C. Yu, Y. S. Nam, Y. Choi, J. Park and M. H. Song, ACS Appl. Mater. Interfaces, 2019, 11, 8428–8435 CrossRef CAS PubMed.
- W. Yang, F. Gao, Y. Qiu, W. Liu, H. Xu, L. Yang and Y. Liu, Adv. Opt. Mater., 2019, 7, 1900546 CrossRef.
- P. Cao, B. Yang, F. Zheng, L. Wang and J. Zou, Ceram. Int., 2020, 46, 3882–3888 CrossRef CAS.
- J. Tong, J. Wu, W. Shen, Y. Zhang, Y. Liu, T. Zhang, S. Nie and Z. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 9317–9325 CrossRef CAS PubMed.
- Y. Yin, Z. Hu, M. U. Ali, M. Duan, L. Gao, M. Liu, W. Peng, J. Geng, S. Pan, Y. Wu, J. Hou, J. Fan, D. Li, X. Zhang and H. Meng, Adv. Mater. Technol., 2020, 5, 2000251 CrossRef CAS.
- S. Mei, B. Yang, X. Wei, H. Dai, Z. Chen, Z. Cui, G. Zhang, F. Xie, W. Zhang and R. Guo, Nanomaterials, 2019, 9, 832 CrossRef CAS PubMed.
- F. Wang, M. Yang, S. Ji, L. Yang, J. Zhao, H. Liu, Y. Sui, Y. Sun, J. Yang and X. Zhang, J. Power Sources, 2018, 395, 85–91 CrossRef CAS.
- J. Duan, Y. Wang, X. Yang and Q. Tang, Angew. Chem., Int. Ed., 2020, 59, 4391–4395 CrossRef CAS PubMed.
- W. Zhang, X. Liu, B. He, Z. Gong, J. Zhu, Y. Ding, H. Chen and Q. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 4540–4548 CrossRef CAS PubMed.
- X. Liu, X. Tan, Z. Liu, H. Ye, B. Sun, T. Shi, Z. Tang and G. Liao, Nano Energy, 2019, 56, 184–195 CrossRef CAS.
- Y. Gao, Y. Wu, H. Lu, C. Chen, Y. Liu, X. Bai, L. Yang, W. W. Yu, Q. Dai and Y. Zhang, Nano Energy, 2019, 59, 517–526 CrossRef CAS.
- Q. Zhang, R. Su, X. Liu, J. Xing, T. C. Sum and Q. Xiong, Adv. Funct. Mater., 2016, 26, 6238–6245 CrossRef CAS.
- Z. Liu, Q. Shang, C. Li, L. Zhao, Y. Gao, Q. Li, J. Chen, S. Zhang, X. Liu, Y. Fu and Q. Zhang, Appl. Phys. Lett., 2019, 114, 101902 CrossRef.
- S. Yuan, D. Chen, X. Li, J. Zhong and X. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 18918–18926 CrossRef CAS PubMed.
- P. Ramasamy, D. H. Lim, B. Kim, S. H. Lee, M. S. Lee and J. S. Lee, Chem. Commun., 2016, 52, 2067–2070 RSC.
- J. Zeng, H. Zhou, R. Liu and H. Wang, Sci. China Mater., 2019, 62, 65–73 CrossRef CAS.
- J. Zeng, X. Li, Y. Wu, D. Yang, Z. Sun, Z. Song, H. Wang and H. Zeng, Adv. Funct. Mater., 2018, 28, 1804394 CrossRef.
- F. Zhang, Z. Shi, S. Li, Z. Ma, Y. Li, L. Wang, D. Wu, Y. Tian, G. Du, X. Li and C. Shan, ACS Appl. Mater. Interfaces, 2019, 11, 28013–28022 CrossRef CAS PubMed.
- X. Yu, L. Wu, D. Yang, M. Cao, X. Fan, H. Lin, Q. Zhong, Y. Xu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14527–14532 CrossRef CAS PubMed.
- Y. Liu, F. Li, L. Qiu, K. Yang, Q. Li, X. Zheng, H. Hu, T. Guo, C. Wu and T. W. Kim, ACS Nano, 2019, 13, 2042–2049 CAS.
- Y. W. Liu, S. H. Guo, S. Q. You, C. Y. Sun, X. L. Wang, L. Zhao and Z. M. Su, Nanotechnology, 2020, 31, 215605 CrossRef CAS PubMed.
- Y. F. Xu, M. Z. Yang, B. X. Chen, X. D. Wang, H. Y. Chen, D. Bin Kuang and C. Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- J. Chen, D. Liu, M. J. Al-Marri, L. Nuuttila, H. Lehtivuori and K. Zheng, Sci. China Mater., 2016, 59, 719–727 CrossRef CAS.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- T. Kosugi, Y. Iso and T. Isobe, Chem. Lett., 2019, 48, 349–352 CrossRef CAS.
- Y. Wang, X. Li, S. Sreejith, F. Cao, Z. Wang, M. C. Stuparu, H. Zeng and H. Sun, Adv. Mater., 2016, 28, 10637–10643 CrossRef CAS PubMed.
- S. Huang, Z. Li, B. Wang, N. Zhu, C. Zhang, L. Kong, Q. Zhang, A. Shan and L. Li, ACS Appl. Mater. Interfaces, 2017, 9, 7249–7258 CrossRef CAS PubMed.
- J. Li, L. Wang, X. Yuan, B. Bo, H. Li, J. Zhao and X. Gao, Mater. Res. Bull., 2018, 102, 86–91 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- S. Zou, Y. Liu, J. Li, C. Liu, R. Feng, F. Jiang, Y. Li, J. Song, H. Zeng, M. Hong and X. Chen, J. Am. Chem. Soc., 2017, 139, 11443–11450 CrossRef CAS PubMed.
- H. Liu, Z. Wu, J. Shao, D. Yao, H. Gao, Y. Liu, W. Yu, H. Zhang and B. Yang, ACS Nano, 2017, 11, 2239–2247 CrossRef CAS PubMed.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- H. Li, Y. Qian, X. Xing, J. Zhu, X. Huang, Q. Jing, W. Zhang, C. Zhang and Z. Lu, J. Phys. Chem. C, 2018, 122, 12994–13000 CrossRef CAS.
- Z. J. Li, E. Hofman, J. Li, A. H. Davis, C. H. Tung, L. Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- K. Kidokoro, Y. Iso and T. Isobe, J. Mater. Chem. C, 2019, 7, 8546–8550 RSC.
- Z. Gan, X. Wen, W. Chen, C. Zhou, S. Yang, G. Cao, K. P. Ghiggino, H. Zhang and B. Jia, Adv. Energy Mater., 2019, 9, 1900185 CrossRef.
- K. Miyashita, K. Kidokoro, Y. Iso and T. Isobe, ACS Appl. Nano Mater., 2021, 4, 12600–12608 CrossRef CAS.
- K. Xu, E. T. Vickers, B. Luo, Q. Wang, A. C. Allen, H. Wang, V. Cherrette, X. Li and J. Z. Zhang, Sol. Energy Mater. Sol. Cells, 2020, 208, 110341 CrossRef CAS.
- Z. Zhang, I. J. Britt and M. A. Tung, J. Appl. Polym. Sci., 2001, 82, 1866–1872 CrossRef CAS.
- Y. Zhong, D. Janes, Y. Zheng, M. Hetzer and D. De Kee, Polym. Eng. Sci., 2007, 47, 1101–1107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc05077j |
| This journal is © The Royal Society of Chemistry 2022 |