
Boosting organic afterglow efficiency via triplet–triplet annihilation and thermally-activated delayed fluorescence†
Jiulong
Zhang‡
,
Jiuyang
Li‡
,
Xun
Li
,
Shou
Yuan
,
Yan
Sun
,
Yunlong
Zou
,
Yingtong
Pan
and
Kaka
Zhang
 *
*
Key Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, People's Republic of China. E-mail: zhangkaka@sioc.ac.cn
First published on 18th January 2022
Abstract
Due to the spin-forbidden nature of organic phosphorescence, it is challenging to achieve high afterglow efficiency in room-temperature organic afterglow systems, especially for those with long afterglow emission wavelengths. Here we report the incorporation of triplet–triplet annihilation (TTA) and thermally-activated delayed fluorescence (TADF) mechanisms to significantly enhance the organic afterglow efficiency in dopant–matrix systems under ambient conditions. Difluoroboron β-diketonate (BF2bdk) compounds are selected as luminescent dopants and designed with naphthalene functional groups to show a relatively strong tendency of intersystem crossing. 4-Methoxybenzophenone matrices are employed to facilitate intersystem crossing of BF2bdk excited states via dipole–dipole interactions and meanwhile suppress nonradiative decay and quenching of BF2bdk triplet excited states. Besides phosphorescence decay, the dopant–matrix systems exhibit additional pathways to harvest triplet energies via TTA and TADF, leading to the significant improvement of the organic afterglow efficiency. The afterglow materials can be melt-cast into desired shapes, can function as multicolor-encoded anti-counterfeiting objects, and can be processed into aqueous dispersions, which display background-free bioimaging properties.
Introduction
Manipulation of triplet excited states is of vital importance in diverse aspects of photofunctional systems such as room-temperature phosphorescence (RTP),1–5 aggregation-induced emission,6 triplet–triplet annihilation (TTA),7–15 thermally activated delayed fluorescence (TADF),16–23 and organic afterglow materials.24–32 Recent years have witnessed very exciting advancements of room-temperature organic phosphorescence and afterglow materials, because these materials exhibit intriguing applications in microenvironment oxygen sensing, anti-counterfeiting, optical storage, bioimaging and sensors.33–38 However, due to the spin-forbidden nature of phosphorescence, it remains a formidable task to achieve high afterglow efficiency (emission lifetime >0.1 s) in purely organic systems.Pioneering studies have shown that judicious molecular design, aggregation state control, and supramolecular assembly can be used to control the properties of triplet excited states and improve the organic afterglow efficiency.39–47 Recently, two-component dopant–matrix design strategies have been reported by our research group and others to exhibit strong capability in devising high-performance organic afterglow materials.48–58 Through the manipulation of triplet excited state properties in the dopant–matrix systems, organic materials with high afterglow efficiency, ultralong afterglow lifetimes, and intense afterglow brightness have been achieved.48–52 The dopant–matrix strategy also allows a very flexible choice of both luminescent dopants and organic matrices, giving rise to diverse organic afterglow materials with different compositions and intriguing properties.48–52 Besides, two-component donor–acceptor systems based on retarded charge recombination have been reported to exhibit surprisingly long afterglow durations of up to hours.53–56 These afterglow systems showed decreased performance when exposed to ambient conditions because of the instability of photo-generated radical cations and radical anions. The donor–acceptor systems have also been extended to the fabrication of aqueous afterglow materials and efficient afterglow materials with long emission wavelengths.57,58
Despite the above success, it is still challenging to fabricate highly efficient organic afterglow materials under ambient conditions, especially for those with long afterglow emission wavelengths.24–38 Fig. S1 (ESI†) illustrates the current status of the research of organic afterglow materials where only a very small percentage of organic materials show both high afterglow efficiency and long emission lifetimes. Fig. S2 (ESI†) exhibits the chemical structures of selected high-performance organic afterglow materials, among which the afterglow emission wavelengths have been found to be mostly restricted to the range of 400 nm to 550 nm. To date, highly efficient afterglow materials with long emission wavelengths, for example, larger than 550 nm or 600 nm, remain rarely reported (Fig. S3 and S4, ESI†).
Besides the population of triplet excited states, harvesting triplet energies is another key to achieve high afterglow efficiency. Inspired by the fantastic achievements of TTA and TADF in the area of metal-complex-based systems and OLED research areas,7–23 we conceive that, if the triplet excited states of organic afterglow systems can be harvested by TTA and TADF pathways, a drastic enhancement of the organic afterglow efficiency would result (Scheme 1). Different from conventional TTA and TADF material design,7–23,59,60 moderate or modest rate constants of TTA and TADF (kTTA and kTADF) are required to maintain the afterglow emission lifetimes > 0.1 s; large kTTA and kTADF lead to drastic shortage of emission lifetimes. Since organic systems without the presence of heavy atoms typically possess phosphorescence rate constants (kP) on the order of 10−2 to 103 s−1, moderate or modest values of kTTA and kTADF in the range of 10−1 to 101 s−1 would be sufficient to open additional pathways to significantly harvest triplet energies and improve the afterglow efficiency (Scheme 1).
 | ||
| Scheme 1 Enhancing organic afterglow efficiency in dopant–matrix systems via TTA and TADF mechanisms. | ||
On the basis of the above consideration, here we design naphthalene-containing BF2bdk luminescent dopants with relatively strong tendency of intersystem crossing and relatively low energy levels of excited states (Scheme 2). 4-Methoxybenzophenone (MeOBP) matrices are used to stabilize the singlet excited states of BF2bdk via dipole–dipole interactions, reduce ΔEST given that the energy level of BF2bdk triplets is less affected by organic matrices, and consequently facilitate both forward and reverse intersystem crossing of BF2bdk excited states. At the same time, the organic matrices can suppress nonradiative decay of BF2bdk triplets by their rigid microenvironment and protect BF2bdk triplets from oxygen quenching by encapsulation. Because of the relatively large population of triplet excited states, TTA processes in NPhBF2–MeOBP, NαNBF2–MeOBP and NβNBF2–MeOBP afterglow systems can be switched on to enhance the afterglow efficiency. It has been found that NPhRedBF2–MeOBP afterglow materials exhibit a TADF-type organic afterglow mechanism with high efficiency and an emission wavelength of >600 nm. The organic afterglow materials have been found to display excellent processability into desired shapes and aqueous dispersions, which exhibit promising characteristics for advanced anti-counterfeiting and high-contrast bioimaging applications.
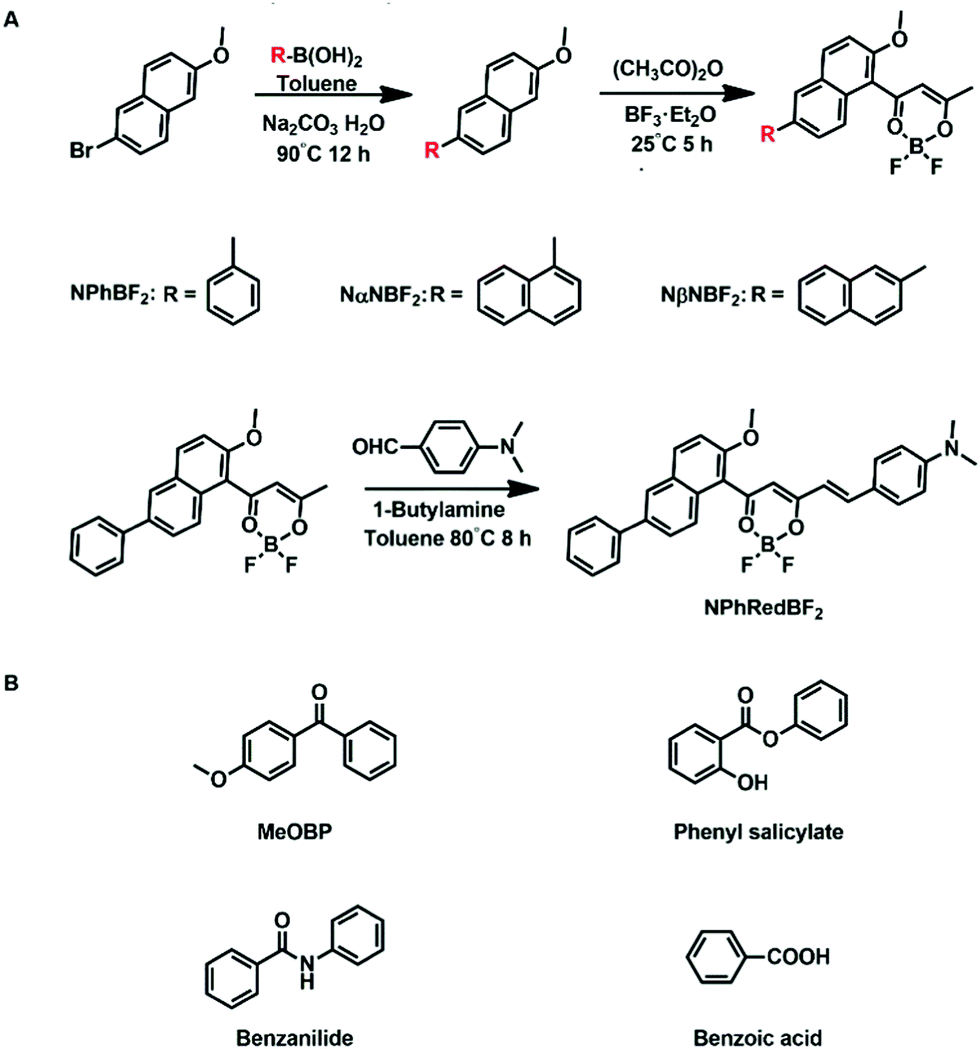 | ||
| Scheme 2 Synthesis of naphthalene-containing BF2bdk luminescent compounds via a cascade reaction and further modification. | ||
Results and discussion
Design and synthesis of naphthalene-containing BF2bdk luminescent compounds
The naphthalene system is known to possess high quantum yields of intersystem crossing (ΦISC) but relatively small molar absorption coefficient. In view of these, we decorate naphthalene substrates with BF2bdk functional groups by the cascade reaction developed in our recent studies (Scheme 2)61,62 to enhance their absorption in visible regions and maintain their high ΦISC. The cascade reaction of aromatic substrates in acetic anhydride and boron trifluoride diethyl etherate is very simple and straightforward, which can be successfully performed even without the use of Schlenk techniques. The naphthalene-containing BF2bdk luminescent compounds have been obtained with reasonable isolation yields. NPhBF2 compounds were further allowed to react with 4-dimethylaminobenzaldehyde to form boron difluoride hemicurcuminoid compounds with larger aromatic conjugation lengths (Scheme 2) and consequently with lower singlet excited state energy levels for red afterglow materials (vide infra). The naphthalene-containing BF2bdk luminescent compounds were structurally characterized by 1H NMR, 13C NMR, 19F NMR, 11B NMR, LRMS, HRMS, and single crystal X-ray diffraction (Fig. 1A), as well as UV-vis and steady-state emission studies (see ESI†). | ||
| Fig. 1 (A) Single crystal structure of NPhBF2. (B) UV-vis spectra of naphthalene-containing BF2bdk in dichloromethane solutions. (C) TD-DFT calculated results of NβNBF2 and NPhRedBF2 S1 states. Blue and green isosurfaces correspond to hole and electron distributions, respectively. TD-DFT calculations were performed in the ORCA 4.2.1 program with the B3LYP functional and def2-TZVP(-f) basis set. (D) Steady-state emission spectra of naphthalene-containing BF2bdk in dichloromethane solutions. | ||
Table 1 summarizes the photophysical properties of the naphthalene-containing BF2bdk compounds in dilute solutions; UV-vis, emission spectra and excited state decay profiles are shown in Fig. S5–S7 (ESI†). NPhBF2, NαNBF2 and NβNBF2 in dichloromethane solutions show UV-vis absorption bands in the range of 360 nm to 460 nm (Fig. 1B), which can be assigned to intramolecular charge transfer (ICT) transition from aromatic groups to the dioxaborine moieties.63,64 TD-DFT calculations, which show typical ICT characteristics in S1 states, support these assignments (Fig. 1C). Given that the LUMO levels of the naphthalene-containing BF2bdk compounds are fixed by dioxaborine functional groups, the absorption of naphthalene-containing BF2bdk compounds in the visible regions can be attributed to the electron-rich nature of naphthalene functional groups. By extending the conjugation lengths of the naphthalene-containing electron-donating functional groups, NPhRedBF2 compounds have been found to exhibit intense absorption bands ranging from 400 nm to 600 nm (Fig. 1B and C). Steady-state emission studies show that NPhBF2, NαNBF2 and NβNBF2 in dichloromethane solutions possess broad emission bands in the range of 425 nm to 650 nm, while NPhRedBF2 display 550–750 nm emission bands with maxima at 606 nm (Fig. 1D and Fig. S6, ESI†).
| Entry | λ abs (nm) | λ em (nm) | τ (ns) | Φ(%) |
|---|---|---|---|---|
| NPhBF2 in dichloromethane | 400 | 518 | 5.5 | 9.4 |
| NPhBF2 in tetrahydrofuran | 391 | 493 | 8.6 | 35.1 |
| NαNBF2 in dichloromethane | 396 | 524 | 6.5 | 8.4 |
| NαNBF2 in tetrahydrofuran | 386 | 494 | 8.5 | 33.9 |
| NβNBF2 in dichloromethane | 402 | 529 | 5.7 | 5.9 |
| NβNBF2 in tetrahydrofuran | 394 | 500 | 9.2 | 30.4 |
| NPhRedBF2 in dichloromethane | 528 | 618 | 0.9 | 15.5 |
| NPhRedBF2 in tetrahydrofuran | 516 | 606 | 1.1 | 21.4 |
Afterglow material fabrication via a dopant–matrix design strategy
The naphthalene-containing BF2bdk powders in the present study exhibit bright fluorescence upon excitation but show no room-temperature afterglow after removal of the excitation source (Fig. S8, ESI†). A dopant–matrix design strategy has been used for the fabrication of organic afterglow materials. Diverse organic matrices have been tested (Scheme 2B). The materials obtained by doping NβNBF2 into MeOBP show the most significant afterglow properties with relatively high afterglow brightness and long afterglow durations (Fig. S9, ESI†); MeOBP matrices alone show insignificant room-temperature afterglow (Fig. S10, ESI†). Therefore, the organic matrices were fixed as MeOBP for the preparation of afterglow materials in the following studies. It is found that afterglow materials prepared by melt casting exhibit better afterglow performance than those obtained by a grinding method (Fig. 2, Fig. S11–S14, ESI†). The melt-cast NPhBF2–MeOBP, NαNBF2–MeOBP, and NβNBF2–MeOBP materials exhibit orange afterglow, while NPhRedBF2–MeOBP materials display orange red afterglow under ambient conditions (Fig. S14, ESI†). At 77 K, the naphthalene-containing BF2bdk powders show weak afterglow (Fig. S15, ESI†), whereas the BF2bdk–MeOBP samples at 77 K exhibit significant organic afterglow (Fig. S16, ESI†). These observations indicate the successful fabrication of afterglow materials by the dopant–matrix design strategy. | ||
| Fig. 2 Photographs of (A) NPhBF2–MeOBP-0.5%, (B) NαNBF2–MeOBP-0.5% and (C) NβNBF2–MeOBP-0.5% afterglow materials under 365 nm UV light and after removal of the UV light. | ||
Photophysical studies of the BF2bdk–MeOBP materials have been performed (Table 2 and Table S1, ESI†). NPhBF2–MeOBP, NαNBF2–MeOBP, and NβNBF2–MeOBP materials display 450–650 nm emission bands in the steady-state fluorescence spectra. The delayed emsision spectra show 550–750 nm phosphorescence bands and 450–530 nm delayed fluorescence bands with identical emission maxima to the fluorescence peak (Fig. 3A–C); dual emission has been observed.65–67 Besides, it is found that the steady-state fluorescence spectra of NPhBF2–MeOBP, NαNBF2–MeOBP, and NβNBF2–MeOBP materials display red-shifted fluorescence maxima by increasing BF2bdk doping concentrations (Fig. 3A–C), which suggest molecular aggregation of BF2bdk in MeOBP matrices via π–π interactions. The phosphorescence maxima show insignificant changes upon the variation of the doping concentrations, which agrees well with the observations in phosphorescence spectra of other reported systems; the phosphorescence spectra of many organic compounds are insensitive to the states and the microenvironments of these organic compounds.68
| Entry | λ em (nm) | τ (ms) (DF) | τ (ms) (RTP) | Φ (%) |
|---|---|---|---|---|
| NPhBF2-0.5% melt-cast sample | 505(TTA) | 106(21.2%) | 261 | 60.0 |
| 585(RTP) | 286(78.5%) | |||
| NαNBF2-0.5% melt-cast sample | 510(TTA) | 181(21.8%) | 275 | 42.2 |
| 590(RTP) | 352(68.2%) | |||
| NβNBF2-0.5% melt-cast sample | 515(TTA) | 166(31.8%) | 250 | 40.1 |
| 595(RTP) | 327(68.2%) |
 | ||
| Fig. 3 (A–C) The room-temperature emission spectra collected in fluorescence mode and phosphorescence mode (1 ms delay) of (A) NPhBF2–MeOBP, (B) NαNBF2–MeOBP and (C) NβNBF2–MeOBP afterglow materials with weight ratios fixed at 0.1%, 0.5% and 1%, respectively. The spectra collected in fluorescence mode denote steady-state emission spectra. The spectra collected in phosphorescence mode represent delayed emission spectra that contain both phosphorescence and delayed fluoresence. (D) The room-temperature emission decay of NβNBF2–MeOBP-1% afterglow materials monitored at 520 nm and 595 nm, respectively. (E) The isosurface maps of NβNBF2 singlet and triplet excited states based on the TD-DFT calculated results. Blue and green isosurfaces correspond to hole and electron distributions, respectively. (F) TD-DFT calculated energy levels of NβNBF2 singlet and triplet excited states and their spin–orbit coupling matrix elements (SOCME). (G) The double logarithmic plot of the room-temperature emission decay profile of NβNBF2–MeOBP-0.5% afterglow materials monitored at 515 nm. (H) Schematic illustration of the TTA mechanism. | ||
The significant room-temperature phosphorescence can be attributed to the relatively strong tendency of intersystem crossing of the naphthalene-containing BF2bdk luminescent compounds. TD-DFT calculations exhibit rich channels of S1-to-Tn intersystem crossing with relatively large spin–orbit coupling matrix elements (Fig. 3E, F, Fig. S17, ESI†), some of which possess relatively small ΔEST values. Because of the relatively large population of BF2bdk triplet excited states in the dopant–matrix systems, it is understandable that the delayed emission spectra of NPhBF2–MeOBP, NαNBF2–MeOBP, and NβNBF2–MeOBP materials exhibit 450–530 nm delayed fluorescence bands (Fig. 3C); NβNBF2–MeOBP materials show the most significant delayed fluorescence bands (Fig. S18, ESI†). The relative intensity of the delayed fluorescence bands is found to increase with the BF2bdk doping concentration, which suggests the formation of triplet–triplet annihilation in the dopant–matrix systems. TTA is a bimolecular process and increases with triplet concentrations. The double logarithmic plot of the emission decay profile of the NβNBF2–MeOBP-0.5% sample monitored at 515 nm obeys a power law with a slope of −1.77 (Fig. 3G). With reference to the reported studies, these observations support that the 515 nm delayed emission bands are originated from bimolecular TTA processes (Fig. 3H).7–15 It is also found that the delayed emission spectra of NβNBF2 solution in dichloromethane at 77 K show weak delayed fluorescence signals in the range of 500 nm to 570 nm (Fig. S19, ESI†). These observations further support the involvement of TTA in the NβNBF2 system.
The total emission of the samples, for example, the NβNBF2–MeOBP-0.5% afterglow materials, consists of prompt fluorescence, delayed fluorescence and room-temperature phosphorescence. Although the fluorescence components are much larger than the phosphorescence components in the steady-state emission spectra such as the spectra of NβNBF2–MeOBP afterglow materials as shown in Fig. 3C, the delayed fluorescence plus phosphorescence constitute a large percent of the total emission as estimated from the intensity integral of both fluorescence and phosphorescence decay (Fig. S20, ESI†). The emission decay profiles of NβNBF2–MeOBP-0.5% afterglow materials monitored at 515 nm and 595 nm have been collected by TCSPC techniques (Fig. S20, ESI†). The emission decay of NβNBF2–MeOBP-0.5% afterglow materials monitored at 515 nm can be fit into a triple-exponential decay with τ1 of 30 ms (16.5%), τ2 of 203 ms (52.5%) and τ3 of 681 ms (31.0%) (Fig. S20, ESI†). Similar to the reported studies,40,62 the prompt fluorescence lifetimes (the τ1 part) measured using a microsecond flash lamp are in millisecond regimes. The accurate lifetimes of prompt fluorescence of NβNBF2–MeOBP-0.5% afterglow materials have been measured using a picosecond pulsed diode laser to be τ1 = 1.9 ns (29.5%) and τ2 = 6.5 ns (70.5%) (Fig. S20, ESI†). The τ2 and τ3 parts can be assigned to delayed fluorescent components, which constitute 83.5% of the total fluorescence. The emission decay monitored at 595 nm can be fit into a triple-exponential decay with τ1 of 3.7 ms (1.5%), τ2 of 77.9 ms (22.3%) and τ3 of 289 ms (76.2%) (Fig. S20, ESI†). The τ2 and τ3 parts contribute to afterglow behaviors, which constitute 98.5% of the total phosphorescence (Text S2, ESI†).
Other BF2bdk–MeOBP materials have also been found to show long-lived emission characteristics when monitored at RTP and TTA regions (Fig. S21–S24, ESI†). The photoluminescence quantum yields (PLQYs) of BF2bdk–MeOBP materials have been measured to be in the range of 40%–60% using a Hamamatsu absolute PLQY measurement system based on a standard protocol (Table 2). Fig. S1–S4 (ESI†) show that it is challenging to achieve highly efficient organic afterglow materials with long emission wavelengths. The PLQYs and afterglow lifetimes of the present long-emission-wavelength systems (Table 2) are among the highest values in the reported studies (Fig. S1–S4, ESI†). The TTA mechanism that can harvest triplet energies is of crucial importance to enhance organic afterglow efficiency.7–15 The kTTA values can be estimated from the delayed fluorescence lifetimes to be on the order of 100–101 s−1, similar to the rate constants of phosphorescence decay. These moderate kTTA values are necessary to improve afterglow efficiency and simultaneously maintain afterglow lifetimes.
Enhancing red organic afterglow efficiency via TADF
For the fabrication of red organic afterglow materials, molecular systems with low energy levels of excited states are required. However, due to the energy gap law, all the related photophysical processes such as radiative decay and nonradiative decay would be increased in the systems with low energy levels of excited states.69–71 As a result, the increased nonradiative decay would compete with radiative decay, and meanwhile the increase of radiative decay and nonradiative decay would shorten the emission lifetimes of the organic systems, both of which are detrimental to the fabrication of highly efficient red afterglow materials.In the present study, it is found that NPhRedBF2–MeOBP-0.01% materials exhibit orange red afterglow under ambient conditions upon ceasing the excitation source (Fig. S14, ESI†). The steady-state emission spectra of NPhRedBF2–MeOBP materials display a fluorescence band in the range of 525 nm–725 nm with emission maxima at around 600 nm (Fig. 4A). Interestingly, the room-temperature phosphorescence spectra show nearly identical delayed fluorescence bands to those of the steady-state fluorescence spectra, which suggests a TADF mechanism in the dopant–matrix system. The emission decay profile as shown in Fig. 4B can be fit into τ1 = 33 ms (69.3%), τ2 = 117 ms (19.2%), and τ3 = 313 ms (11.5%). Afterglow emission of NPhRedBF2–MeOBP materials can be obtained by visible light excitation (Fig. S25, ESI†) and 530 nm laser excitation (Fig. S26, ESI†). This indicates that the contribution of energy transfer from MeOBP triplets to NPhRedBF2 dopants is not necessary for the emergence of organic afterglow in the NPhRedBF2–MeoBP system,72,73 because MeOBP possesses insignificant absorption in the visible region (Fig. S27, ESI†). Besides, at such a low doping concentration, delayed fluorescence from TTA should not have such a large contribution to dominate the whole delayed fluorescence spectra. It is noted that boron difluoride curcuminoid and hemicurcuminoid compounds have been found to show TADF behaviours in several reported studies.74–77 In addition, due to the presence of two or multiple electron-donating functional groups, the types of ICT excited states of NPhRedBF2 can be enriched. The nature of some triplet states may be different from that of the S1 state, so ISC and RISC can be accelerated to some extent according to the El-Sayed rule. Moreover, TD-DFT calculations, where multiple ISC channels show relatively larger SOCME values and some of these channels possess relatively small ΔEST values, support that the organic systems possess a moderate tendency of RISC (Fig. 4C, Fig. S28, ESI†). It has been found that the afterglow colour of NPhRedBF2–MeOBP-0.01% materials at 77 K (red afterglow from phosphorescence decay of triplet states, Fig. S16D, ESI†) shows red-shifted behaviours when compared to that at room temperature (orange red, Fig. S14, ESI†), which suggests that the TADF mechanism is frozen at 77 K. Similar to the NPhRedBF2–MeOBP materials, NPhRedBF2–benzophenone materials also exhibit TADF-type organic afterglow properties under ambient conditions (Fig. S29, ESI†). The MeOBP matrices showed strong phosphorescence from 77 K to 240 K and thus largely interfered with the temperature-dependent phosphorescence measurements as reported in our recent study;78 so benzophenone (BP) matrices were used in the place of MeOBP. At 77 K, the phosphorescence spectra of NPhRedBF2–benzophenone materials have been found to exhibit a structured emission band ranging from 630 nm to 850 nm with emission maxima at 658 nm (Fig. 5). This 630–850 nm emission band can be attributed to phosphorescence decay of NPhRedBF2 triplets within the benzophenone matrices. Upon increasing the temperature, the delayed fluorescence bands at approximately 580 nm gradually increase and dominate the whole delayed emission spectra at 300 K (Fig. 5). These studies further support the TADF mechanism of NPhRedBF2–matrix afterglow materials, since the RISC process that is the rate-determining step for TADF is frozen at 77 K and can be activated at room temperature. All the above observations and analysis support our proposed TADF mechanism in the dopant–matrix systems. The triplet excited states can undergo reverse intersystem crossing to reach singlet excited states and subsequently emit delayed fluorescence, which can significantly enhance the organic afterglow efficiency of the red afterglow systems (Fig. 4D). The kTADF values can be estimated from the delayed fluorescence lifetimes to be on the order of 100–102 s−1. When compared to the TADF emitters with large kTADF values of 103–106 s−1,16–23,59,60 the moderate kTADF values are a distinct feature of the present study different from those of the TADF emitters for efficient OLEDs. Organic afterglow systems with moderate kTADF values can simultaneously harvest triplet energies by the TADF mechanism and maintain afterglow lifetimes. The PLQYs of NPhRedBF2–MeOBP materials at 0.005%, 0.05% and 0.5% doping concentrations have been determined to be 56.3%, 18.6% and 6.5%, respectively; the decrease of PLQY upon increasing the doping concentration can be caused by aggregation of NPhRedBF2 molecules in MeOBP matrices. NPhRedBF2–MeOBP-0.005% afterglow materials with such high PLQYs have been rarely achieved in the reported studies (Fig. S1–S4, ESI†).
 | ||
| Fig. 4 (A) Room-temperature steady-state emission and delayed emission spectra of NPhRedBF2–MeOBP afterglow materials at different doping concentrations. (B) Room-temperature emission decay of NPhRedBF2–MeOBP-0.01% afterglow materials excited at 365 nm and monitored at 599 nm. (C) and (D) Schematic representations of (C) energy levels and spin orbital coupling matrix elements and (D) the proposed mechanism of organic afterglow in the NPhRedBF2–MeOBP system. | ||
 | ||
| Fig. 5 Temperature-dependent delayed emission spectra of NPhRedBF2–benzophenone samples. | ||
By increasing the doping concentration, the fluorescence spectra of the NPhRedBF2–MeOBP materials show the emergence of an even lower-energy emission signal at 631 nm (Fig. 4A and 6), which can be attributed to molecular aggregation in the dopant–matrix systems; most BF2bdk systems show red-shifted emission upon molecular aggregation.61–64 In the delayed fluorescence spectra, the lower-energy emission bands at 631 nm persist (Fig. 4A and 5), indicating that the delayed fluorescence bands can be readily adjusted by increasing the doping concentration to red regions.
 | ||
| Fig. 6 Photographs of afterglow objects with desired shapes prepared by the melt casting technique of (A) NPhRedBF2–MeOBP-0.01%, (B) NPhRedBF2–MeOBP-0.1% and (C) (left–right) NPhRedBF2–MeOBP-0.01%, NαN–MeOBP-0.5% and NβNBF2–MeOBP-0.5% under 365 nm UV light and after removal of the UV light. | ||
The role of organic matrices in dopant–matrix afterglow systems
The role of organic matrices for the emergence of organic afterglow under ambient conditions is an open question in the research fields of room-temperature phosphorescence and organic afterglow systems. It has been reported that triplet-to-triplet excited state energy transfers from organic matrices to luminescent dopants can give rise to organic afterglow.72,73 In the present study, the BF2bdk–MeOBP materials can be excited by 420 nm visible light to exhibit organic afterglow (Fig. 3A–C). In particular, in the case of the NPhRedBF2–MeOBP system, afterglow can be observed by 530 nm excitation (Fig. S26, ESI†). Since MeOBP possesses insignificant absorption in the visible region (Fig. S27, ESI†), we argue that triplet-to-triplet excited state energy transfer is not necessary for the formation of dopant–matrix afterglow materials in the present systems.Several recently reported studies proposed that organic matrices with T1 levels sandwiched between S1 and T1 levels of luminescent dopants can serve as bridges for the singlet-to-triplet intersystem crossing of the luminescent dopants, giving rise to room-temperature organic afterglow.38,51 This is not the case in the present study, because the naphthalene-containing BF2bdk luminescent dopants, especially in the NPhRedBF2 system, possess lower S1 and T1 levels than the T1 level of MeOBP matrices. With reference to the studies of Adachi's group and our research group, a high T1 level of organic matrices is necessary to avoid afterglow quenching caused by triplet-to-triplet energy transfer from luminescent dopants to organic matrices.62,79
On the basis of our previous study52,61,62 and the current findings, we propose that the MeOBP matrices with large dipole moments can interact with and perturb the excited states of naphthalene-containing BF2bdk luminescent dopants via dipole–dipole interactions. These interactions can stabilize the singlet excited states of the luminescent dopants and consequently reduce ΔEST given that the energy level of BF2bdk triplets is less affected by organic matrices, leading to the enhancement of intersystem crossing of BF2bdk excited states; in the case of NPhRedBF2, reverse intersystem crossing can also be enhanced. Meanwhile, the MeOBP matrices can inhibit nonradiative decay of BF2bdk triplets by their rigid microenvironments and protect BF2bdk triplets from oxygen quenching by encapsulation. This is our understanding on the role of organic matrices for the fabrication of dopant–matrix afterglow materials. In control experiments without the presence of MeOBP matrices, the TADF property of NPhRedBF2 has not been observed in the solution state (Fig. S30, ESI†).
Functionalities of the dopant–matrix afterglow materials
Because of the relatively low melting points of MeOBP matrices, the afterglow materials can be processed into objects with desired shapes by the melt casting technique with the aid of silicone moulds (Fig. 2, 6 and Fig. S31, ESI†). Fig. 6C shows the combination of afterglow numbers with different emission colours prepared from NPhRedBF2–MeOBP-0.01%, NαN–MeOBP-0.5% and NβNBF2–MeOBP-0.5% materials, where NαN represents 6-methoxy-1,2′-binaphthalene. By taking advantage of the different shapes, different afterglow colours and different afterglow duration times, the combination of these materials shows the potential for the fabrication of multicolour-encoded anti-counterfeiting labels or devices.24–38For most biomedical applications, the afterglow materials should be processed into aqueous dispersions. However, it is found that, in many reported studies, these processing procedures can lead to a significant decrease or complete loss of the afterglow properties in the material systems. In the present study, we were inspired by the widely used technique for the preparation of emulsions and suspensions. Pluronic F-127 surfactants have been used to disperse hydrophobic NβNBF2–MeOBP materials. Since the NβNBF2–MeOBP materials are in solid states at room temperature, we first melt the materials by heating to 65 °C and then inject the molten samples into a hot aqueous solution (80 °C) of Pluronic F-127 surfactants under sonication. It is found that the sonication can efficiently disperse the molten samples into very small droplets that can be stabilized by Pluronic F-127 surfactants to form a milky dispersion. The hot milky dipsersion exhibits fluorescence properties under 365 nm UV light but shows no afterglow upon swithching off the UV light; in the molten state, the molecular motion of NβNBF2 should be very active, so nonradiative decay can quench the triplet states of NβNBF2 molecules. After rapid cooling, the aqueous dispersion displays orange afterglow with a duration of 1.6 s as observed by human eyes in a dark room (Fig. 7A). These observations indicate that, by the processing methods in the present study, an aqueous disperison of room-temperature organic afterglow materials can be readily obtained without the use of organic solvents or expensive equipment.
 | ||
| Fig. 7 (A) and (B) Photographs of (A) NβNBF2–MeOBP-0.5% and (B) NβNBF2–MeOBP-0.5% + R6G afterglow dispersion under 365 nm UV light and after removal of the UV light. (C) and (D) Preliminary bioimaging studies of the NβNBF2–MeOBP-0.5% afterglow dispersion in living (C) fish and (D) mice. | ||
Due to their long-lived emission characteristics, the capability of NβNBF2–MeOBP afterglow dispersions in avoiding the interference of background fluorescence has been tested. Rhodamine 6G has been added to the afterglow dispersions of NβNBF2–MeOBP materials. In the UV-on state, only yellow fluorescence from rhodamine 6G has been observed. In the UV-off state, the yellow fluorescence disappears immediately, whereas the aqueous dispersion exhibits orange afterglow that is originated from NβNBF2–MeOBP materials (Fig. 7B). These observations indicate that the aqueous afterglow materials possess intriguing properties to eliminate background interference via a time-gated imaging mode. Furthermore, preliminary in vivo bioimaging studies have been performed. The aqueous afterglow dispersion can be readily drawn by syringes and injected into animal tissues. After injecting into fish tails, the bioimaging is found to be interfered with by the fluorescence from the anal fin in the UV-on mode, while this interference can be completely removed in the UV-off mode where only NβNBF2–MeOBP materials show afterglow emission (Fig. 7C). The preliminary bioimaging studies on mice also show very clean background in the UV-off state (Fig. 7D). These studies suggest that, because of their long-lived excited state nature, the aqueous afterglow materials show promising applications in biomedical fields.
Conclusions
In conclusion, the present study demonstrates that triplet–triplet annihilation and thermally-activated delayed fluorescence mechanisms can be incorporated into organic room-temperature phosphorescence systems to remarkably enhance the afterglow quantum efficiency of the material systems under ambient conditions. A distinct feature of the present study is the moderate or modest rate constants of triplet–triplet annihilation and thermally-activated delayed fluorescence, which can significantly improve the afterglow efficiency and simultaneously maintain long afterglow lifetimes.The molecular design of luminescent dopants is very simple; naphthalene-containing BF2bdk compounds are selected to increase the quantum yields of intersystem crossing and molar absorption coefficients for the significant population of triplet excited states. The organic matrices with large molecular dipole moments are also readily available and very cheap. The organic matrices not only suppress nonradiative decay and quenching but also facilitate forward and reverse intersystem crossing via dipole–dipole interactions. These endow great opportunities to the dopant–matrix design strategy for the fabrication of high-performance organic afterglow materials.
Because of the flexibility of component selection of the dopant–matrix design strategy, it is anticipated that diverse organic afterglow materials with intriguing photophysical properties can be obtained. We also envisage that these afterglow materials can be widely used in fields such as data storage and encryption, as well as high-contrast bioimaging and highly sensitive optical analyses.
Experimental
Preparation of organic afterglow materials
For the preparation of the BF2bdk–MeOBP materials, naphthalene-containing BF2bdk in dichloromethane solution and MeOBP in dichloromethane solution were first added into an agate mortar. After grinding and solvent evaporation, BF2bdk–MeOBP afterglow powders were obtained. Other afterglow materials with different doping concentrations, different BF2bdk dopants and different organic matrices were prepared through similar processes. For the preparation of melt-cast afterglow objects with desired shapes, the BF2bdk–MeOBP powders were first heated to form molten mixtures, and then poured into silicone moulds with desired shapes. After cooling to room temperature, afterglow objects with various shapes were obtained.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (22175194), Shanghai Scientific and Technological Innovation Project (20QA1411600, 20ZR1469200), and Hundred Talents Program from Shanghai Institute of Organic Chemistry (Y121078).Notes and references
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang and Q. Zheng, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- X. Qiao and D. Ma, Mater. Sci. Eng., R, 2020, 139, 100519 CrossRef.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- R. Ieuji, K. Goushi and C. Adachi, Nat. Commun., 2019, 10, 5283 CrossRef PubMed.
- T.-A. Lin, C. F. Perkinson and M. A. Baldo, Adv. Mater., 2020, 32, 1908175 CrossRef CAS PubMed.
- B. Joarder, N. Yanai and N. Kimizuka, J. Phys. Chem. Lett., 2018, 9, 4613–4624 CrossRef CAS PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- K. J. Fallon, E. M. Churchill, S. N. Sanders, J. Shee, J. L. Weber, R. Meir, S. Jockusch, D. R. Reichman, M. Y. Sfeir, D. N. Congreve and L. M. Campos, J. Am. Chem. Soc., 2020, 142, 19917–19925 CrossRef CAS PubMed.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- D. Yang, J. Han, M. Liu and P. Duan, Adv. Mater., 2019, 31, 1805683 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32, 2003911 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed.
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef.
- Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed.
- Y. Yu, M. S. Kwon, J. Jung, Y. Zeng, M. Kim, K. Chung, J. Gierschner, J. H. Youk, S. M. Borisov and J. Kim, Angew. Chem., Int. Ed., 2017, 56, 16207–16211 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed.
- Z. Xie, X. Zhang, H. Wang, C. Huang, H. Sun, M. Dong, L. Ji, Z. An, T. Yu and W. Huang, Nat. Commun., 2021, 12, 3522 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai, Q. Wu, N. Gan, X. Xu, Z. An and W. Huang, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- P. Wei, X. Zhang, J. Liu, G.-G. Shan, H. Zhang, J. Qi, W. Zhao, H. H.-Y. Sung, I. D. Williams, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9293–9298 CrossRef CAS PubMed.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 32, 2001348 CrossRef CAS PubMed.
- H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Adv. Funct. Mater., 2019, 29, 1807243 CrossRef.
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- Y. Sun, G. Wang, X. Li, B. Zhou and K. Zhang, Adv. Opt. Mater., 2021, 9, 2100353 CrossRef CAS.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS PubMed.
- P. Alam, N. L. C. Leung, J. Liu, T. S. Cheung, X. Zhang, Z. He, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, C. C. S. Chan, K. S. Wong, Q. Peng and B. Z. Tang, Adv. Mater., 2020, 32, 2001026 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- Y. Lei, W. Dai, Y. Tian, J. Yang, P. Li, J. Shi, B. Tong, Z. Cai and Y. Dong, J. Phys. Chem. Lett., 2019, 10, 6019–6025 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- B. Zhou, G. Wang, X. Wang, W. Guo, J. Li and K. Zhang, J. Mater. Chem. C, 2021, 9, 3939–3947 RSC.
- X. Wang, Y. Sun, G. Wang, J. Li, X. Li and K. Zhang, Angew. Chem., Int. Ed., 2021, 60, 17138–17147 CrossRef CAS PubMed.
- P.-Z. Chen, L.-Y. Niu, Y.-Z. Chen and Q.-Z. Yang, Coord. Chem. Rev., 2017, 350, 196–216 CrossRef CAS.
- C.-T. Poon, W. H. Lam, H.-L. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 13992–13993 CrossRef CAS PubMed.
- S. K. Behera, S. Y. Park and J. Gierschner, Angew. Chem., Int. Ed., 2020, 60, 22624–22638 CrossRef PubMed.
- N. A. Kukhta and M. R. Bryce, Mater. Horiz., 2021, 8, 33–55 RSC.
- J. Chen, X. Chen, Y. Liu, J. Zhao, Z. Yang, Y. Zhang and Z. Chi, Chem. Sci., 2021, 12, 9201–9206 RSC.
- G. N. Lewis and M. Kasha, J. Am. Chem. Soc., 1944, 66, 2100–2116 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- R. Englman and J. Jortner, J. Lumin., 1970, 1, 134–142 CrossRef.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- J. Wang, H. Zhang, L.-Y. Niu, X. Zhu, Y.-F. Kang, R. Boulatov and Q.-Z. Yang, CCS Chem., 2020, 2, 1391 CrossRef CAS.
- K. Jinnai, R. Kabe and C. Adachi, Adv. Mater., 2018, 30, 1800365 CrossRef PubMed.
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. D. S. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- N. R. Paisley, S. V. Halldorson, M. V. Tran, R. Gupta, S. Kamal, W. R. Algar and Z. M. Hudson, Angew. Chem., Int. Ed., 2021, 60, 18630–18638 CrossRef CAS PubMed.
- A. Shahalizad, A. Malinge, L. Hu, G. Laflamme, L. Haeberlé, D. M. Myers, J. Mao, W. G. Skene and S. Kéna-Cohen, Adv. Funct. Mater., 2020, 31, 2007119 CrossRef.
- A. D’Aléo, M. H. Sazzad, D. H. Kim, E. Y. Choi, J. W. Wu, G. Canard, F. Fages, J.-C. Ribierre and C. Adachi, Chem. Commun., 2017, 53, 7003–7006 RSC.
- Y. Sun, J. Liu, J. Li, X. Li, X. Wang, G. Wang and K. Zhang, Adv. Opt. Mater., 2021, 2101909 CrossRef.
- N. Notsuka, R. Kabe, K. Goushi and C. Adachi, Adv. Funct. Mater., 2017, 27, 1703902 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, supporting figures. CCDC 2115255. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc04903h |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |