
Revisiting syntheses of Fe3O4 nanoparticles in water and lower alcohols and their resistive switching properties†
Mariia A.
Mikhailova
a,
Ivan Yu.
Chernyshov
ab,
Georgii A.
Illarionov
a,
Denis S.
Kolchanov
 a,
Oleg A.
Kuchur
a,
Alexandr V.
Vinogradov
a,
Sofia M.
Morozova
ac and
Maxim I.
Morozov
*a
a,
Oleg A.
Kuchur
a,
Alexandr V.
Vinogradov
a,
Sofia M.
Morozova
ac and
Maxim I.
Morozov
*a
aLaboratory of Solution Chemistry of Advanced Materials and Technologies, ITMO University, Lomonosova str. 9, St.Petersburg, 191002, Russian Federation. E-mail: morozov@scamt-itmo.ru
bInorganic Systems Engineering Group, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands
cN.E. Bauman Moscow State Technical University, 5 Baumanskaya 2-ya str., Moscow, 105005, Russian Federation
First published on 24th November 2021
Abstract
Magnetite (Fe3O4) nanoparticles have found numerous applications due to the ease of fabrication, favourable combination of physical and chemical properties, as well as environmental and biological safety. At the same time, their functional applications in memristive devices remain underexplored, especially with regard to nanocrystalline phases that can be obtained by various means of solution chemistry. In this study, we examine the physical properties, morphology, and biocompatibility of magnetite nanoparticles obtained by hydrolytic and non-hydrolytic synthesis routes. For this purpose, we have revisited two solution chemistry methods for obtaining magnetite nanoparticles in water and lower alcohols. Notably, magnetite nanoparticles obtained via the hydrolytic route have demonstrated an appreciably high value of the resistive switching ratio ROFF/RON ∼ 103 which is comparable to the highest values ever reported for iron oxide memristors. The advantageous suitability of the hydrolytically synthesized magnetite nanoparticles for resistive switching applications is rationalized by their higher purity and crystallinity, as well as by the plausible activity of residual water molecules and hydroxy groups on facilitating the topotactic redox reaction associated with the resistive switching phenomenon.
Introduction
Transition metal oxide nanoparticles (NPs) and nanostructures are extensively used in various functional applications including in electronics,1 photonics,1,2 catalysis,3,4 biosensing5,6 and thermoelectronic7 devices. The current trend of technological convergence makes such materials very attractive for various multifunctional applications. It particularly concerns memristive electronics, one of the promising areas of rapidly developing technologies with yet unexplored multifunctionality.8,9Memristive electronics is based on a concept of bioinspired computing that simulates the activity of biological neural networks. In turn, a memristor is an electrical prototype of a synapse that functions as a passive element of an electric circuit whose resistance depends on the previously passed electric current.10–12 The electrical relation of a memristive element to a biological neuron is usually implemented11,13,14 in terms of the Hodgkin–Huxley model.15 Thus, memristive devices are feasible for applications in biohybrid circuits and interfaces.16–18 Technological implementation of such devices may thus require biocompatibility of both materials in use and the fabrication methods.19 The state of the art and future expectations in this field have been outlined in recent topical reviews.20,21
Magnetite NPs (Fe3O4) are characterized by low toxicity and high biocompatibility,22 and thus have been proposed for various medical23 and biomedical24,25 applications, though the in vivo applications usually require specific biocompatible polymer coatings.26,27
The resistive switching properties of magnetite thin films were thoroughly studied in 2007, and the primary electroforming mechanism was argued to be a redox transformation between Fe3O4 and γ-Fe2O3.28 Since 2008, this class of materials has been classified as memristors.1,11,12
Most of the reported examples of iron oxide memristors have been fabricated by physical methods such as atomic layer deposition,29,30 pulsed laser deposition,31 epitaxial growth32 or various sputtering techniques.28,33,34 Meanwhile, in contrast to other memristive oxides such as TiO2,21 the solution chemistry approach to fabrication of Fe3O4 memristors is still poorly explored. There are almost no examples of memristive devices based on magnetite nanoparticles obtained by classical co-precipitation and sol–gel synthesis routes. An exception was an early study by Kim et al.,35 but the results of the functional characterization were seemingly misinterpreted, as was notified later in the correction report.36 Nonetheless, the solution chemistry approaches to fabrication of oxide-based electronic devices may generally be well competitive with the physical deposition techniques,21 especially regarding the production cost and the technological flexibility, typically allowing controllable purity, crystallinity, and morphology of the synthesized materials.37–41
Fe3O4 nanocrystals can be obtained by means of solution chemistry using hydrolytic or non-hydrolytic processes. The main synthetic strategies and growth mechanisms have been addressed in several comprehensive reviews.41–45
The hydrolytic synthesis of magnetite NPs is usually performed by adding a base to an aqueous mixture of ferric and ferrous salts.46 Then the product can be converted into a stable hydrosol using an acidic or alkaline peptizing agent47 or various organic surfactants.48,49 The use of stabilizing agents apparently affects the acidity and purity of the hydrosol product. More recently, a method of obtaining a pure and pH-neutral hydrosol of magnetite NPs has been proposed based on tailoring of the initial non-stochiometric ratio of Fe2+/Fe3+ ions with its further adjustment via the Schikorr reaction50 assisted by ultrasound.51 Thus, phase purity, pH-neutrality and environmental safety are the factors that certainly merit consideration of the hydrolytic synthesis for designing biocompatible electronic devices.
The non-hydrolytic synthesis of magnetite NPs is usually carried out using solvents with a relatively high boiling temperature, typically above 200 °C. Examples include benzyl alcohol52 and 2-pyrrolidone.53 Surfactants, such as oleic acid or oleates, are typically used for reducing the particle size and preventing their aggregation.35,54,55 Implementation of these strategies may be inappropriate for memristive films, as their properties are generally sensitive to any structural and phase defects.1
An alternative reasonable approach may concern synthesis in low boiling point and less toxic solvents, such as lower aliphatic alcohols. Synthesis of magnetite particles in isopropyl alcohol was first studied by Dubois and Demazeau.56 In that study, the reported particle sizes fell beyond the range of nanomaterials and varied from 100 nm to 1.2 μm. Later, the nanocrystalline phases of magnetite (7–35 nm) were obtained in the solvothermal systems at temperatures in the range of 200–350 °C and pressures in the range of 1.8–20 MPa.57,58 The influence of the solvent nature on the size and composition of nanoparticles was comprehensively investigated for the synthesis of magnetite and hematite phases from iron acetylacetonate in lower alcohols, cyclohexane, and toluene.57 Additionally, the presence of a small amount of water for the synthesis of iron oxides in n-propanol was reported to drastically affect the reaction mechanism, favouring the formation of hematite nanocrystals.57,58 Despite a few systematic studies,57,59 the non-hydrolytic synthesis of iron oxides in lower aliphatic alcohols is still much less investigated compared to the cases of zirconium and titanium oxides.60 Particularly, the lower temperature limit and the possibility of obtaining stable sols have not been carefully explored for this route, yet. Furthermore, the mechanisms of nanoparticle formation have mainly been studied for the cases of non-hydrolytic sol–gel synthesis, with most examples involving transformations based on chlorides, isopropoxides and acetylacetonates of titanium and zirconium,60 while the formation of Fe3O4 nanoparticles involving the redox mechanism is less understood.
In this study, we revisit the key solution chemistry methods of obtaining Fe3O4 nanoparticles based on syntheses in water,51 benzyl alcohol,52 and lower aliphatic alcohols57 with respect to their potential for application in biocompatible and environmentally sustainable memristive devices. For the case of synthesis in lower aliphatic alcohols, we address a detailed study of iron oxide phase formation from iron acetylacetonate by a solvothermal process at 175 °C, which is at the low temperature margin reported for the synthesis of Fe3O4 in 1-propanol57 and yet has not been investigated for other lower aliphatic alcohols. In addition, this is the only known temperature previously reported for the synthesis of single-phase magnetite NPs in benzyl alcohol.52 On top of that, it is just a few degrees above the decomposition temperature of iron acetylacetonate.61,62
Materials and methods
Chemicals
Iron(III) acetylacetonate, Fe(AcAc)3 (≥97%, Sigma-Aldrich), was dried at 50 °C and 45 mbar to remove moisture. Methanol, MeOH (≥99.8%, Fisher Scientific); ethanol, EtOH (95%); 1-propanol, n-PrOH (≥99.9%, Ekos-1); isopropanol, i-PrOH (≥99.9%, Vekton); 1-butanol, n-BuOH (≥99%, Ekos-1); isobutanol, i-BuOH (≥99%, Chimmed); and benzyl alcohol, BnOH (≥99%, Ekos-1), were dehydrated by distillation over anhydrous CuSO4. The precursors for hydrolytic synthesis, FeCl2·4H2O (≥98%, Sigma-Aldrich), FeCl3·6H2O (≥97%, Sigma-Aldrich), and NH4OH (water solution, ∼25% NH3 basis, puriss. p.a., Sigma-Tec), were used without any additional treatment.Hydrolytic synthesis
The hydrolytic synthesis of pure magnetite hydrosol and nanoparticles (NPs) was performed by fully adopting a previously reported method without any modification.51Non-hydrolytic synthesis
Iron oxide NPs were prepared by the solvothermal process in a series of different lower alcohols. 15 mL of a respective dehydrated alcohol was added to 0.2 g of Fe(AcAc)3 powder under an inert atmosphere ([Fe3+] ≈ 0.04 M). The reaction mixture was transferred into a 50 mL Teflon liner, placed into a stainless-steel autoclave, and carefully sealed. The autoclave was placed in an oven for heat treatment at selected temperatures. The heating rate was set at 1 °C min−1, and the dwell time was 24 h. The resulting reaction mixture was cooled down to ambient temperature, and the iron oxide containing product dried out. The processing scheme is illustrated in Fig. S1 (ESI†).Fabrication of functional inks and deposition of thin films
Two types of the functional inks were prepared. For the case of the hydrolytically synthesized magnetite, an aqueous sol with 4 wt% Fe3O4 was mixed with methanol at a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, while for the case of magnetite synthesized in i-PrOH, the obtained powder was redispersed in MeOH at a concentration of 0.04 wt%. Thus, the first type of ink contained 1.18 wt% Fe3O4, 28.44 wt% H2O, and 70.38 wt% MeOH, while the second type consisted of 0.04 wt% Fe3O4 and 99.96 wt% MeOH. Fig. S2a (ESI†) illustrates the variation of the rheological properties in the colloidal system consisting of 4 wt% Fe3O4 hydrosol and various amounts of methanol. For the functional inks, the optimal concentrations of the components were chosen and experimentally verified in the regions of lower surface tension and higher colloidal stability, as illustrated in Fig. S2a and b (ESI†).
:3, while for the case of magnetite synthesized in i-PrOH, the obtained powder was redispersed in MeOH at a concentration of 0.04 wt%. Thus, the first type of ink contained 1.18 wt% Fe3O4, 28.44 wt% H2O, and 70.38 wt% MeOH, while the second type consisted of 0.04 wt% Fe3O4 and 99.96 wt% MeOH. Fig. S2a (ESI†) illustrates the variation of the rheological properties in the colloidal system consisting of 4 wt% Fe3O4 hydrosol and various amounts of methanol. For the functional inks, the optimal concentrations of the components were chosen and experimentally verified in the regions of lower surface tension and higher colloidal stability, as illustrated in Fig. S2a and b (ESI†).
Fluorine doped tin oxide (FTO) glass substrates (25 × 25 × 2 mm3, <10 Ohm cm−2, FTO thickness: 950 nm) were cleaned in an ultrasound bath. The surface hydrophilization was performed at air plasma for 10 minutes using a Femto Low Pressure Plasma System (Diener Electronic GmbH, Germany). Thin films of Fe3O4 nanoparticles were obtained by deposition of the obtained inks onto FTO glass substrates using an APT Spin – 150i NPP spin coater (SPS Europe, France). The coating regime was set as follows: 2 layers of functional inks were deposited onto an FTO glass substrate at a coating speed of 1000 rpm (1800 rpm for the second layer), 10 s per layer. For each layer, a drop of 700 μL of the functional inks was taken. The prepared thin films were dried in a vacuum chamber at 100 °C for 24 hours. The thickness of the obtained Fe3O4 layers was evaluated using a Solver NexT atomic force microscope (NT-MDT, Russia) to be ca. 80 nm and 35 nm for the films with the hydrolytically and non-hydrolytically obtained Fe3O4 nanoparticles, respectively. The corresponding AFM images are shown in Fig. S3a and b (ESI†).
Materials characterization
The phase composition of the obtained samples was primarily determined by the X-ray diffraction (XRD) method using an X-ray diffractometer (Rigaku SmartLab 3, Japan). The microscopic study was implemented with a Merlin high-resolution scanning electron microscope (SEM) (Zeiss, Germany). The Raman spectra were obtained using a LabRAM HR800 spectrometer (Horiba Jobin Yvon, Japan) equipped with a He–Ne laser (532 nm) and a 100× Olympus lens. The measurements were conducted at laser powers of 50 μW and 500 μW. The mean values of the hydrodynamic radii and ζ-potential of the particles in the synthesized sols were determined using a Photocor Compact-Z particle size analyser (Photocor Ltd., Estonia). The surface area, pore volume, and pore size distribution were evaluated by measuring N2 adsorption–desorption isotherms at 77 K using a NOVA 1200e gas adsorption analyser (Quantachrome, USA). The samples were degassed at 120 °C for 8 h prior to characterization. The obtained data were processed using BET and BJH models.Cytotoxicity assay
The biocompatibility of the obtained iron oxide NPs was assessed using a human dermal fibroblast (HDF) cell line. A standard methylthiazol tetrazolium (MTT) assay63 was adopted as follows: the samples were placed into sterile 6-well flat-bottom plates. An additional empty plate was reserved for the control test. Cells of the human dermal fibroblast (HDF) line were taken each in a quantity of ∼5 × 103 and seeded in the 6-well plates with the samples, as well as in the empty (control) one. The cells were incubated at 37 °C for 72 h (5% CO2). The incubated product was tested by adding 0.2 mL of MTT (5 mg mL−1) for 2 h, then the MTT–formazan product was dissolved in 0.2 mL of dimethyl sulfoxide (DMSO), and the optical absorption was measured at 570 nm using an Infinite F50 plate reader (Tecan, Austria). The experimental reproducibility was provided with three isolated batches prepared for each of the experimental tests.Functional characterization
The magnetization hysteresis curves for the prepared iron oxide samples were obtained at room temperature using a domestically assembled vibration magnetometer. The thin film samples of glass/FTO/Fe3O4 were subjected to electrical characterization. The bottom electrode was connected to the FTO layer, while an external copper tip was used as the top electrode for probing the voltage–current hysteresis in the magnetite thin layer. The measurements were performed using a Keithley 6430 Sub-Femtoamp Remote SourceMeter operating in the standard two-point regime and KickStart Instrument Control Software Version 2.0.6 (Tektronix).Theoretical modelling
Quantum chemical calculations were used for modelling Fe3+/Fe2+ reduction and alcohol dehydration processes. The Fe3+/Fe2+ reduction was modelled as an alcohol's homolytic α-H migration to the Fe3+–O–Fe3+ moiety, which was previously shown to be an active centre in methane activation processes.64,65 The alcohol dehydration process was modelled as an alcohol-to-ether SN2 reaction. Geometry optimization and frequency calculations were performed using Gaussian 16 software at the PBE0–D3/Def2–SVP level of theory.66 The solvent medium was modelled as ethanol using the PCM model due to the absence of parametrization for all alcohols used in the study. The effect of different solvents was calculated using the COSMO-RS solvation model67 as implemented in the COSMOtherm package.68 All iron ions (Fe3+ and Fe2+) were considered at the high spin state. Activation energies were calculated using electronic energies with zero-point energy correction.Results and discussion
Effect of solvents in non-hydrolytic synthesis
First, we address the synthesis of iron oxide NPs in a series of aliphatic alcohols, namely, MeOH, EtOH, n-PrOH, i-PrOH, n-BuOH, and i-BuOH, at 175 °C. A reference synthesis of magnetite NPs in aromatic benzyl alcohol (BnOH) was performed under the same conditions as a well-known example.52 The optimal temperature and time profile (175 °C, 1 °C min−1, 24 h) was first experimentally determined for the synthesis in i-PrOH (Fig. S1, Table S1, ESI†) and then remained unchanged for other alcohols considered in this section.The XRD patterns of the obtained materials are shown in Fig. 1. The determined phase ratios and particle sizes are listed in Table 1. All the reactions resulted in the formation of iron oxide NPs. The single-phase products were obtained by the syntheses in EtOH (α-Fe2O3), n,i-PrOH (Fe3O4), n-BuOH (Fe3O4), and BnOH (Fe3O4). Mt and Ht phases have also manifested themselves with black and red brown colours, as illustrated in Fig. S4a and b (ESI†), correspondingly. The synthesis in i-BuOH resulted in a major product of Fe3O4 with a minor quantity of α-Fe2O3 (∼7%), while a notable amount of unidentified by-product has been observed for the case of synthesis in MeOH (see Fig. S5 and Table S2 for details, ESI†).
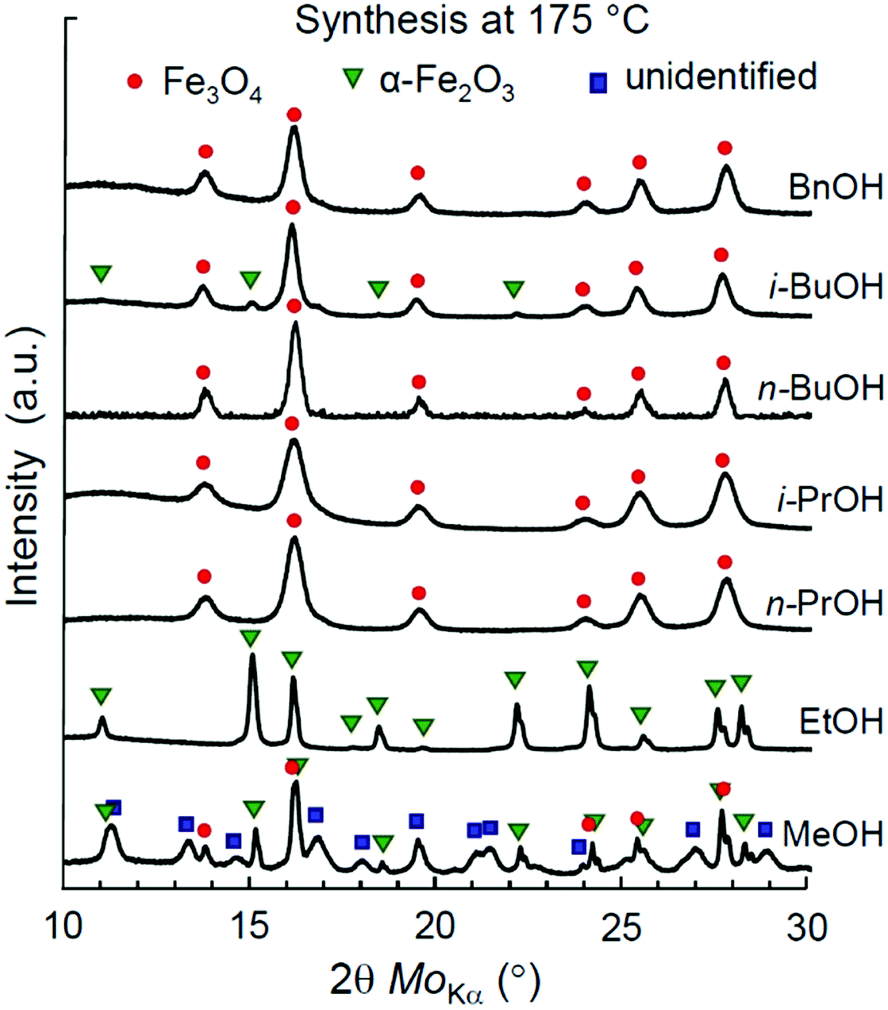 | ||
| Fig. 1 XRD patterns of iron oxide nanoparticles synthesized in lower alcohols at 175 °C. | ||
| Alcohol | Phase ratioa (%) | Crystallite sizeb (nm) | ζ-potentialc (mV) |
|---|---|---|---|
| a Mt – Fe3O4 (magnetite), Ht – α-Fe2O3 (hematite), UP – unidentified phases (see Fig. S5 and Table S2 for details, ESI). b The mean value (by Scherrer). c N/S – not stable in solvents as used for synthesis; N/D – no data. | |||
| MeOH | Mt:Ht (70:30) + UP |
10.5 | N/S |
| EtOH | Ht | 15.4 | N/S |
| n-PrOH | Mt | 7.3 | N/S |
| i-PrOH | Mt | 6.7 | N/S |
| n-BuOH | Mt | 9.2 | N/S |
| i-BuOH | Mt:Ht (93:7) |
9.1 | N/S |
| BnOH | Mt | 8.8 | 28 |
| BnOH,52 | Mt | 10.5 | N/D |
The identified iron oxide phases in all syntheses have been attributed to either α-Fe2O3 (hematite, Ht) or Fe3O4 (magnetite, Mt). The presence of γ-Fe2O3 (maghemite, Mht) cannot be fully excluded based on XRD characterization, as both Mt and Mht possess similar crystal structures of an inverse spinel and thus cannot be clearly distinguished by XRD.69 Nonetheless, we do not expect the presence of any maghemite product in the samples obtained by the non-hydrolytic synthesis due to the following reasons: (1) all the samples identified as Mt were black in colour, as illustrated in Fig. S4a (ESI†), while Mht would clearly manifest itself by a brown tinge. (2) Mht is known to be relatively metastable and typically originates from Fe3O4 as a result of topotactic transformation under oxidizing conditions requiring temperatures above 200 °C, especially in the presence of water.70–72 None of these conditions has been fulfilled at the solvothermal process. (3) No presence of Mht was reported in the previous studies addressing the synthesis with the same precursors.52,57 Particularly, no Mht was detected by Mössbauer spectroscopy.52 The possibility of Mht phase appearance in other cases will be discussed later in this study.
The hematite nanoparticles synthesized in ethanol showed a relatively high mean crystallite size of 15.4 nm, while all the Mt nanoparticles were characterized by smaller sizes ranging from 6.7 to 9.2 nm. The smallest crystallite sizes of pure magnetite NPs were obtained in i-PrOH (6.7 nm) and in BnOH (8.8 nm), in line with the previously reported data for the syntheses in n-PrOH processed at 200 °C (7 nm)57 and in BnOH at 175 °C (10.5 nm).52
Redispersion in MeOH
Obtaining a stable sol is one of the key features advancing the possibility of thin film fabrication by cost efficient and flexible methods such as tape casting, spin coating, and ink-jet printing.73 In this regard, we analysed whether the products synthesized in each alcohol are redispersible in the same medium. As summarized in Table 1, only benzyl alcohol may serve as both reaction and redispersion media. Mt NPs synthesized in BnOH were found to be redispersible in BnOH and resulted in a stable sol with a ζ-potential of 28 mV. In contrast, none of the examined aliphatic alcohols could serve simultaneously as both reaction and dispersion media for obtaining the colloidal ink of iron oxide nanoparticles despite their very small sizes.Next, we addressed the possibility of obtaining a stable colloid by redispersing magnetite NPs in MeOH, an alcohol solvent with a relatively high polarity. For a case study, we have chosen the fine-grade Fe3O4 nanoparticles obtained in i-PrOH and subjected them to double centrifugation at 104 rpm for 10 min and subsequent replacement of the solvent with pure MeOH.
The optimal concentration of Mt NPs in MeOH was experimentally determined to be 0.04 wt% with respect to the highest colloidal stability, as illustrated in Fig. S2b (ESI†). This dispersion demonstrated a smooth dependence of the mean hydrodynamic radius (Rh) and ζ-potential on the duration of the ultrasonic treatment, as shown in Fig. 2. The gradual decrease of the mean hydrodynamic radius along with an increase of ζ-potential was observed during the first 90 min of sonication. Further elongation of the ultrasound treatment resulted in reaching a plateau, followed by a sharp growth of particle agglomerates. Thus, the optimal sonication time for redispersion of magnetite NPs in MeOH was 1.5 h. The obtained Mt sol with Rh = 34 nm and a ζ-potential of 32 mV has demonstrated high colloidal stability that lasted for at least 30 days.
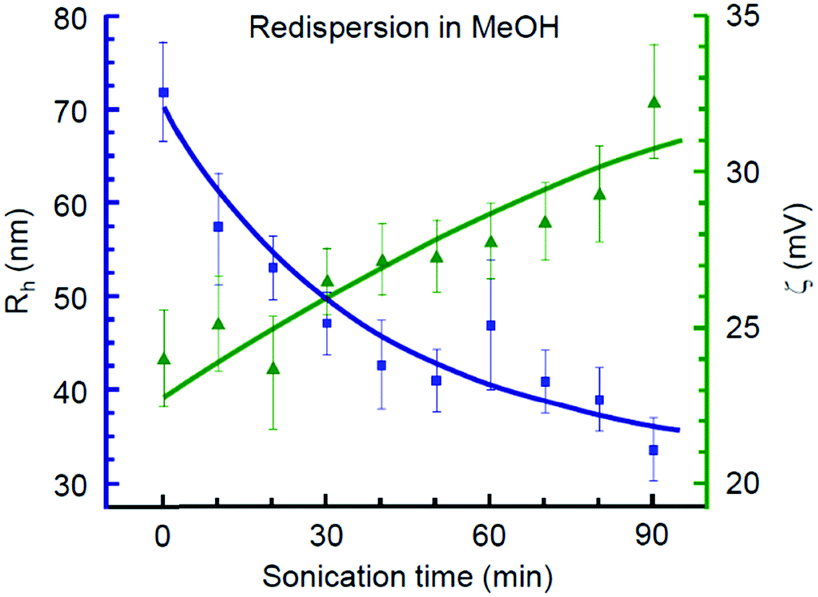 | ||
| Fig. 2 DLS mean hydrodynamic radius and ζ-potential of Fe3O4 nanoparticles synthesized in i-PrOH and redispersed in MeOH at various stages of ultrasound treatment. | ||
Influence of water presence in i-PrOH
Next, we experimentally address the effect of water presence in i-PrOH on the phase equilibrium of the iron oxide products. The influence of water can be manifold. As mentioned previously, it advances the formation of Mht over Ht at an early stage of Mt oxidation.70–72 This, however, may occur only in the case in which the prior Fe3O4 phase has been synthesized. Since the formation of Fe3O4 from Fe(AcAc)3 requires reduction of iron(III), the above-mentioned effect of water is very unlikely. Alternatively, a small amount of water may significantly accelerate the reaction of Ht formation from Fe(AcAc)3, thus skipping or essentially surpassing the reaction of Fe3+ reduction. The latter effect was carefully investigated by Kominami et al. for the synthesis of Mt NPs in mixtures of Fe(AcAc)3, n-PrOH, and 0.5, 1, or 3 vol% of water at 300 °C.57Here, we investigate the same effect for the synthesis in i-PrOH at 175 °C with a higher resolution of the variable water content. The XRD patterns of the iron oxide products synthesized with water added in quantities of 0.3–0.8 wt% are shown in Fig. S6 (ESI†). The calculated Mt:Ht phase ratios and the mean crystallite sizes of the obtained products are summarized in Table 2.
A drastic transition between the Mt and Ht phase ratio occurred at 0.5 wt% of water, while only trace quantities of Ht was observed at a lower water content. The mean size of the crystallites steadily increases with an increase of the water content in the reaction mixture (Table 2). Thus, addition of a small amount of water to i-PrOH significantly affects the crystallinity and the oxidation state of iron oxide products. These conclusions are consistent with the previously reported results by Kominami et al.57 and provide them an extension for the case of a similar reaction system at a lower temperature and a lower water content.
Mechanism of iron oxide synthesis in lower alcohols
In this section we discuss possible reaction pathways for iron oxide formation in the reactions of Fe(AcAc)3 with aliphatic and benzyl alcohols. An intuitive model of magnetite formation in such conditions presumes two basic processes: (1) a chemical interaction resulting in an oxide compound and (2) reduction of iron (Fe3+/Fe2+) favouring magnetite over other forms of iron oxides. An interplay between the kinetics of the above processes should supposedly determine the final product of interaction between the initial precursors under any given synthesis conditions.The most probable processes leading to the formation of iron oxide are shown in Scheme 1. These are two known alcohol dehydration routes producing either alkenes (Scheme 1a) or ethers (Scheme 1b). The occurrence of such processes in the presence of a transition metal oxide catalyst has been demonstrated in several studies.74,75 Hypothetically, other possible routes leading to iron oxide formation may involve alkylation of acetylacetonate and thermal decomposition of Fe(AcAc)3. While experimental evidence of the former one is questionable,75 the latter is known to pass via intermediate formation of iron carbonate, Fe2(CO3)3,76 whose decomposition occurs at temperatures above 450 °C,61 and thus can be neglected.
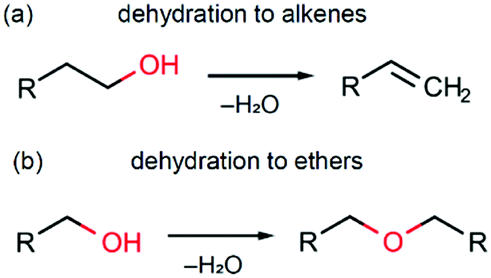 | ||
| Scheme 1 Alcohol dehydration pathways. | ||
The key possible reactions leading to Fe3+/Fe2+ reduction are summarized in Scheme 2. An acid–base catalysed reaction at the cleavage of the acetylacetone molecule leads to formation of acetate and acetone, as expanded in Scheme 2a. This process was justified in several studies by detection of ester products.75,77 In other studies, this mechanism was suggested to be the primary reaction pathway for the cases of magnetite synthesis in dibenzyl ether in the presence of oleic acid.78,79 Particularly, the acetate anion can act as a reducing agent, as shown in Scheme 2b. The other iron reduction pathway is a redox interaction between Fe(AcAc)3 and an alcohol molecule, as illustrated in Scheme 2c. This interaction has previously been investigated as an alcohol's homolytic α-H migration to the Fe3+–O–Fe3+ moiety that behaves as an active centre in methane activation processes.64,65 Finally, one more route of a direct Fe3+/Fe2+ reduction by the acetylacetonate anion is illustrated in Scheme 2d. In the literature, it was first proposed as the formation of the acetylacetone radical.80 Later, the process was shown to result in radical polymerization, in which the acetylacetonate radical was captured by the polymerizing molecule.62 Therefore, the amount of the acetylacetone radicals in this process shall be extremely small and thus can be neglected.
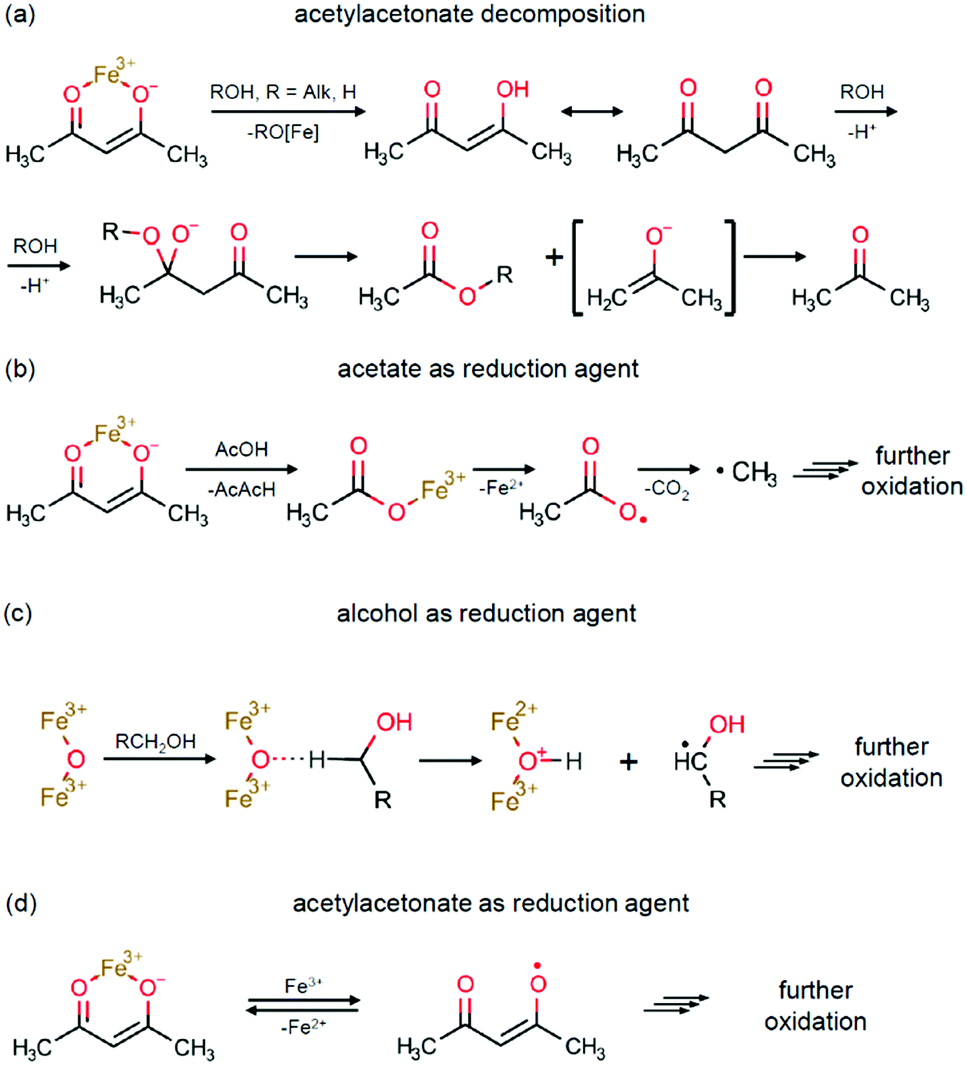 | ||
| Scheme 2 Fe3+/Fe2+ reduction pathways: (a) acetylacetonate decomposition; (b) acetate as a reduction agent; (c) alcohol as a reduction agent; and (d) acetylacetonate as a reduction agent. | ||
In order to assess the effect of different alcohols on the reaction products upon a solvothermal interaction with Fe(AcAc)3, the relative reactivity of alcohols has been evaluated for the key stages of alcohol dehydration (Scheme 1b) and iron reduction (Scheme 2c) using quantum chemical calculations (see Fig. S7 and Table S3 for details, ESI†). The results are summarized in Table 3. The calculated values of the activation energies (Ea) for the reduction mechanism suggest that almost all aliphatic alcohols addressed in the study possess nearly the same reduction ability. The difference in Ea varies within the error limit of DFT. A minor exception is isopropanol, which shows slightly lower activation energies for both mechanisms.
| Alcohol | Activation energy, Ea (kJ mol−1) | Iron oxide products at 175 °Ca | |
|---|---|---|---|
| Fe3+/Fe2+ reduction | Dehydration | ||
| a Mt – Fe3O4 (magnetite), Ht – α-Fe2O3 (hematite). | |||
| MeOH | 106.9 | 34.5 | Mt, Ht |
| EtOH | 102.7 | 34.3 | Ht |
| n-PrOH | 99.6 | 32.0 | Mt |
| i-PrOH | 98.7 | 30.3 | Mt |
| n-BuOH | 100.8 | 31.4 | Mt |
| i-BuOH | 102.6 | 34.3 | Mt, Ht |
| BnOH | 78.2 | 14.1 | Mt |
In contrast, the activation energies for benzyl alcohol are significantly lower for both reduction and dehydration processes. This suggests that the reactions with benzyl alcohol must be capable of developing relatively high rates. The obtained deviations in the calculated Ea for the reduction reactivity of benzyl and isopropyl alcohols are likely associated with the high stability of benzyl and secondary carbocations and radicals compared to the primary ones. However, the reasons for the minor difference between the other alcohols are not obvious and seem to be due to more subtle effects.
Comparison of the calculated activation energies for the reduction effect of different alcohols with the oxidation state of iron in the corresponding reaction products obtained experimentally (Table 3) suggests that the reaction mechanism based on alcohols as the only reduction agents (Scheme 2c) is not fully sufficient for rationalizing the redox aspect of the solvothermal synthesis addressed in the study. Therefore, it seems that the processes of acetylacetonate decomposition and the reduction effect of acetate anions (Scheme 2a and b) are also involved in the redox interaction, while the complex interaction of both mechanisms and the apparent inhibition of the redox rate in the cases of syntheses in MeOH and EtOH still remain unclear. In a study by Kominami et al.,57 the syntheses performed at 300 °C in MeOH also resulted in a mixed phase product of Mt and Ht, while the product in EtOH was pure Mt.
To figure out the threshold temperature for the synthesis of Mt in EtOH, we have performed an additional series of syntheses at 250, 275, and 300 °C. A phase analysis of the reaction products by XRD is demonstrated in Fig. 3. Apparently, the redox effect for the synthesis in EtOH was achieved at higher temperatures compared to syntheses in higher aliphatic alcohols, while an almost pure Mt product was obtained only at 300 °C.
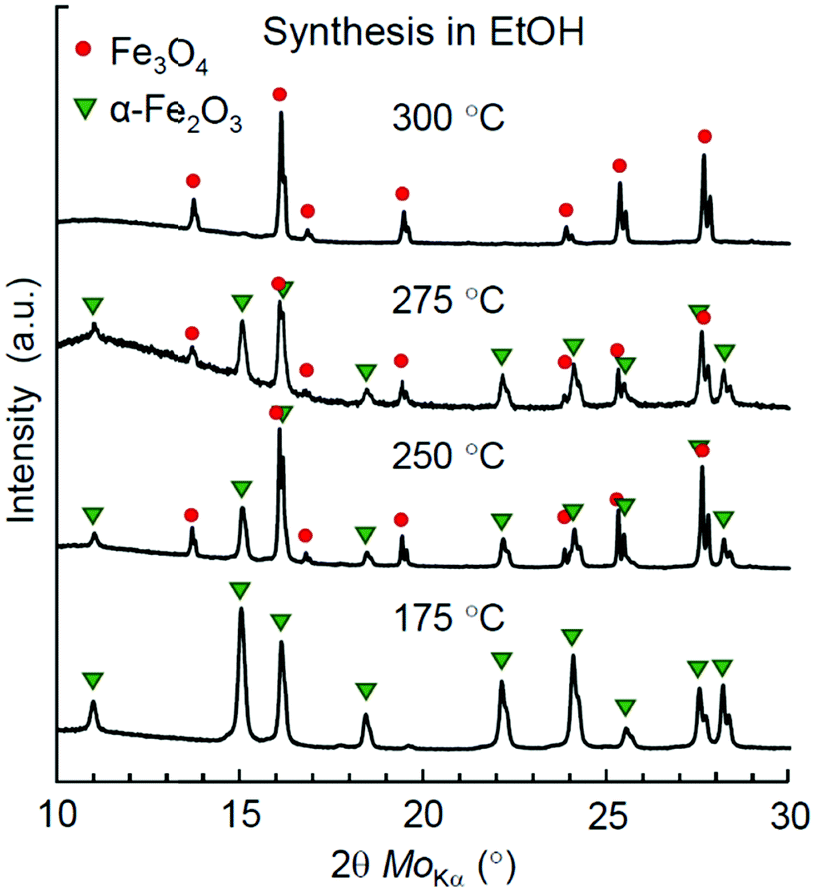 | ||
| Fig. 3 XRD patterns of iron oxide NPs synthesized in EtOH at 175–300 °C. | ||
Finally, to shed more light on the role of iron acetylacetonate decomposition in the overall redox interaction, we made an additional trial for synthesis in BnOH at 150 °C. The reduction ability of BnOH is the highest among the lower alcohols addressed in the study (see Table 3), while the chosen temperature is just a few degrees below the decomposition temperature of Fe(AcAc)3 (ca. 165 °C).61,62 The reaction product was a powder with well distinguishable dark-brown colour (Fig. S4c, ESI†), though an XRD analysis revealed only the peak series of Mt (Fig. S8, ESI†). The observed dark-brown colour is thus a strong indication of an incomplete iron reduction that may be attributed to the presence of either Mht or, more likely, amorphous Fe2O3 in the synthesized product.
Based on the analysis provided above and the data obtained in this and previous studies,52,57 the experimentally explored temperature ranges of Mt crystallization as the only single-phase precipitate in solvothermal processing of iron acetylacetonate in lower alcohols are depicted in Fig. 4.
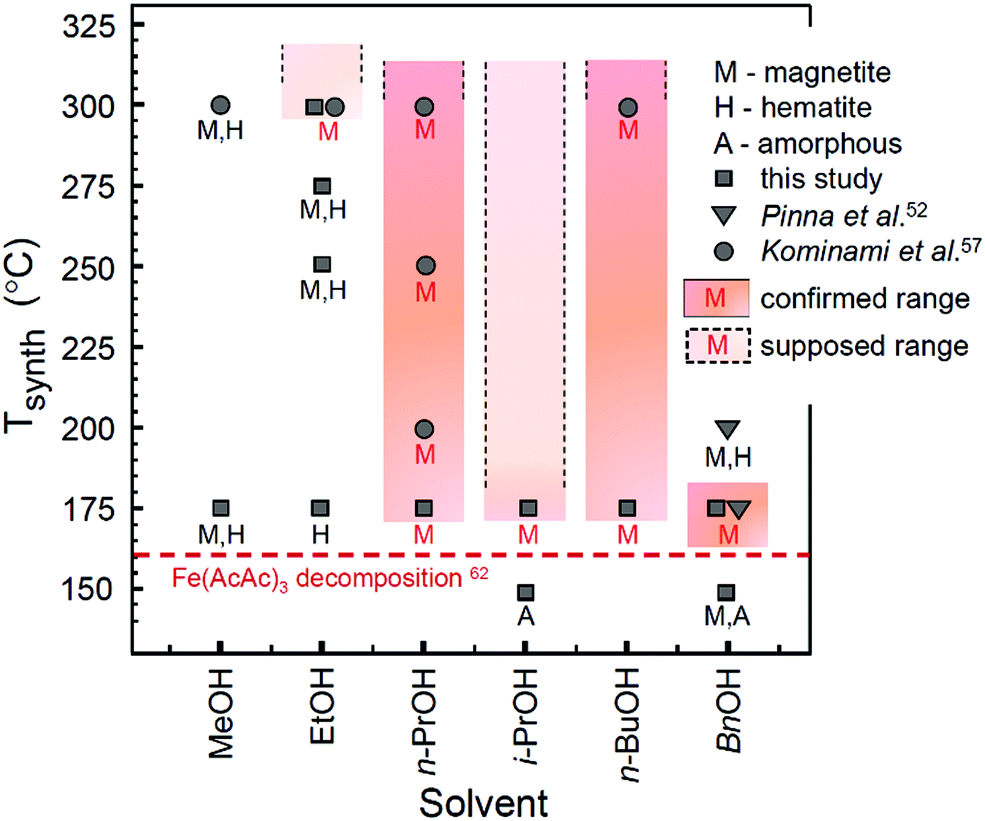 | ||
| Fig. 4 Diagram of iron oxide formation in a solvothermal system of Fe(AcAc)3 and lower alcohols. Filled colour areas designate the single-phase Mt product. The plotted data are taken from this study (synthesis time: 24 h) and adopted from ref. 52 (synthesis time: 48 h) and ref. 57 (synthesis time: 2 h). The initial decomposition temperature for Fe(AcAc)3 (horizontal dashed line) is taken from ref. 62. | ||
Notably, only a narrow range of temperatures is available for synthesis of single-phase Mt in BnOH. In view of the reaction model discussed above, this can be rationalized by a combination of two factors: (1) the relatively high rate of the dehydration reaction (Table 3) may result in the production of iron oxide faster than completion of the reduction processes, which seemingly limits the upper temperature range of Mt formation as the single phase product; and (2) inactivation of one of the reduction mechanisms, most likely the one associated with acetylacetonate (Scheme 2a and b), limits the formation of single phase Mt at lower temperatures.
Properties of Mt NPs obtained in water and lower alcohols
In this section we provide specific characterization of the obtained materials and discuss some of their key properties such as biocompatibility, magnetization, and resistive switching.The phase purity, crystallinity, and morphology of magnetite nanoparticles obtained via hydrolytic and non-hydrolytic (in i-PrOH) routes have been examined using XRD (Fig. S9, ESI†), SEM (Fig. S10, ESI†), and nitrogen adsorption–desorption (Fig. S11, ESI†).
The XRD analysis (Fig. S9, ESI†) showed the presence of only the magnetite phase in both samples, which visually appeared as black powders (Fig. S4a, ESI†) and apparently demonstrated magnetic behaviour (Fig. S1, ESI†). The mean crystallite sizes by Scherrer differed as 15 nm for Mt NPs derived by the hydrolytic process and 6.7 nm for the ones synthesized in i-PrOH.
Raman spectra for the same products are shown in Fig. 5. In both samples the structures of Mt and Mht bands have been observed with an initial laser power of 50 μW. The broad band features around 350, 500, and 700 cm−1 are usually referred as an identification of γ-Fe2O3 (Mht).81,82 However, these bands have a much stronger manifestation in the case of the hydrolytic sample. Appearance of these bands usually results from Mt oxidation induced by the heat of laser excitation, as was pointed in many studies.51,52,81,82 As such, they cannot serve as any proof for the purpose of phase analysis. Particularly, the appearance of Mht bands upon Raman characterization of Mt samples obtained by the hydrolytic route was reported for a laser power of 344 μW, while at a lower power (30 μW) these features were minor.51 Furthermore, Pinna et al. addressed this issue in detail and demonstrated that the presence of Mht features in the Raman spectra of Mt NPs synthesized in BnOH was eventually not confirmed by non-destructive Mössbauer spectroscopy.52
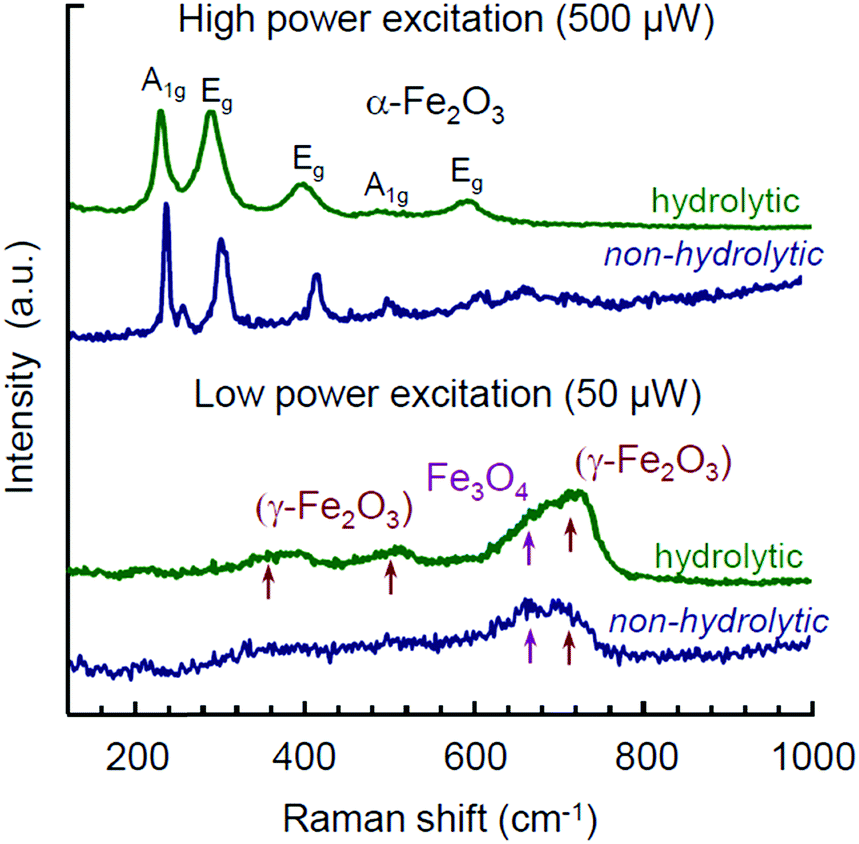 | ||
| Fig. 5 Raman spectra for Mt NPs synthesized by hydrolytic and non-hydrolytic (in i-PrOH) routes measured at laser powers of 50 and 500 μW. | ||
In view of the above and considering Mht to be an oxidation product of the initially formed Mt, the interpretation of Raman spectra obtained from different samples may rather shed light on the relative difference between the samples with respect to the degree of Mt to Mht transformation occurring under the same experimental conditions. The stronger manifestation of Mht features in the Raman spectrum by the hydrolytic sample compared to the one synthesized in i-PrOH points to one of the following two aspects: (1) in contrast to the reduction-type processing in lower alcohols, the hydrolytic synthesis is based on the non-stoichiometric excess of a ferrous precursor with subsequent adjustment of the oxidation process and colloidal stabilization.51 Thus, a possibility of Mht appearance as the minor by-product of such synthesis cannot be formally excluded. (2) The presence of water and hydroxy group residues in the product of the hydrolytic synthesis51 may enhance the topotactic transformation of Fe3O4 to γ-Fe2O3 upon heating.70–72 In any of these cases, we take up the effect of seemingly more facilitated Mt-to-Mht transformation in the hydrolytically obtained Mt NPs as their distinctive feature. It may be especially relevant for memristive applications, as the resistive switching in iron oxide memristors is associated with the same type of reversible redox transformations between Fe3O4 and γ-Fe2O3 phases.28
The SEM images of Mt NPs obtained by the hydrolytic and non-hydrolytic routes are shown in Fig. S10 (ESI†). In the case of the Mt sample obtained hydrolytically (Fig. S10a, ESI†), the mean particle size in the SEM images correlates well with the mean crystallite size determined by XRD: 11 and 10 nm, correspondingly. This indicates a high crystallinity of the hydrolytically synthesized Mt NPs. In contrast, the non-hydrolytic Mt nanoparticles appeared to be larger on a SEM image (Fig. S10b, ESI†) compared to the mean crystallite size characterized by XRD: 25 vs. 6.7 nm, correspondingly. This suggests that the synthesis product in i-PrOH contains a larger amount of amorphous phase and organic residues. The latter was pointed out and justified in a previous study by Kominami et al. based on measuring the weight loss by means of thermogravimetry.57
The magnetization hysteresis for Mt NPs obtained in i-PrOH is shown in Fig. S12 (ESI†) in comparison with the previously reported data for the hydrolytic counterpart.51 The specific magnetization of the non-hydrolytic sample reaches saturation at ca. 46 emu g−1 with a relatively low coercivity (∼12 Oe). The measured value of the saturated specific magnetization is nearly 50% of the corresponding bulk magnetization (92 emu g−1).83 In contrast, the specific magnetization of the hydrolytically synthesized Mt NPs was reported to be ca. 88 emu g−1,51 which is close to that of the bulk counterpart. The difference in specific magnetization demonstrated by the samples prepared via hydrolytic and non-hydrolytic routes is seemingly another manifestation of their different crystallinity and purity.
The possible presence of organic residues in samples of Mt NPs obtained by the non-hydrolytic method brings up an issue of their toxicity. To assess the biocompatibility of the obtained materials, an MTT assay has been performed with HDF cells. The assessment was made for xerogel samples. The samples obtained in i-PrOH, including the one previously redispersed in MeOH, were dried at 20 °C and 45 mbar for several hours to evaporate the alcohols. The xerogel samples obtained by the hydrolytic synthesis and by the synthesis in BnOH were dried at 80 °C and 45 mbar for several hours. The results on the viability of HDF cells incubated on the examined materials are shown in Fig. 6. All the samples, except the one synthesized in BnOH, have demonstrated a reasonably high biocompatibility within the range of the normalized HDF cell viability 95–97%. A sample synthesized in BnOH has demonstrated lower biocompatibility at a level of 71% of the control HDF cell viability. Thus, the by-products contaminating Mt NPs synthesized in BnOH are relatively more toxic and may require additional treatment and additional toxicity examination prior to biocompatible applications. The by-products may also include unevaporated residue of BnOH owing to its relatively high boiling point (205 °C).
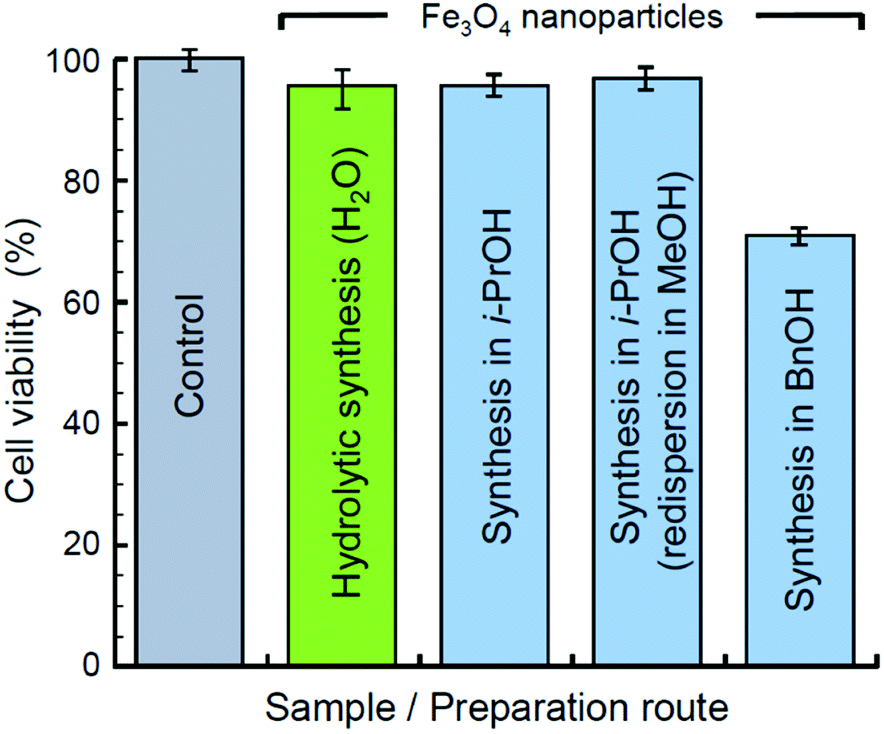 | ||
| Fig. 6 Viability of HDF cells incubated solely (control) and on substrates of Mt NPs synthesized in water, i-PrOH, and BnOH. | ||
The resistive switching properties of magnetite NPs synthesized by the hydrolytic and non-hydrolytic (i-PrOH) methods are shown in Fig. 7. Notably, the hydrolytically processed sample has demonstrated a very high resistive switching ratio ROFF/RON ∼ 103 (Fig. 7a), which is comparable to or higher than most of the values previously reported for iron oxide memristors.28–32,84 In contrast, the non-hydrolytic sample has demonstrated a much lower resistive switching ratio ROFF/RON ∼ 2 (Fig. 7b). Table 4 summarizes and compares the characterized properties of the two types of magnetite NPs obtained by the hydrolytic and non-hydrolytic syntheses.
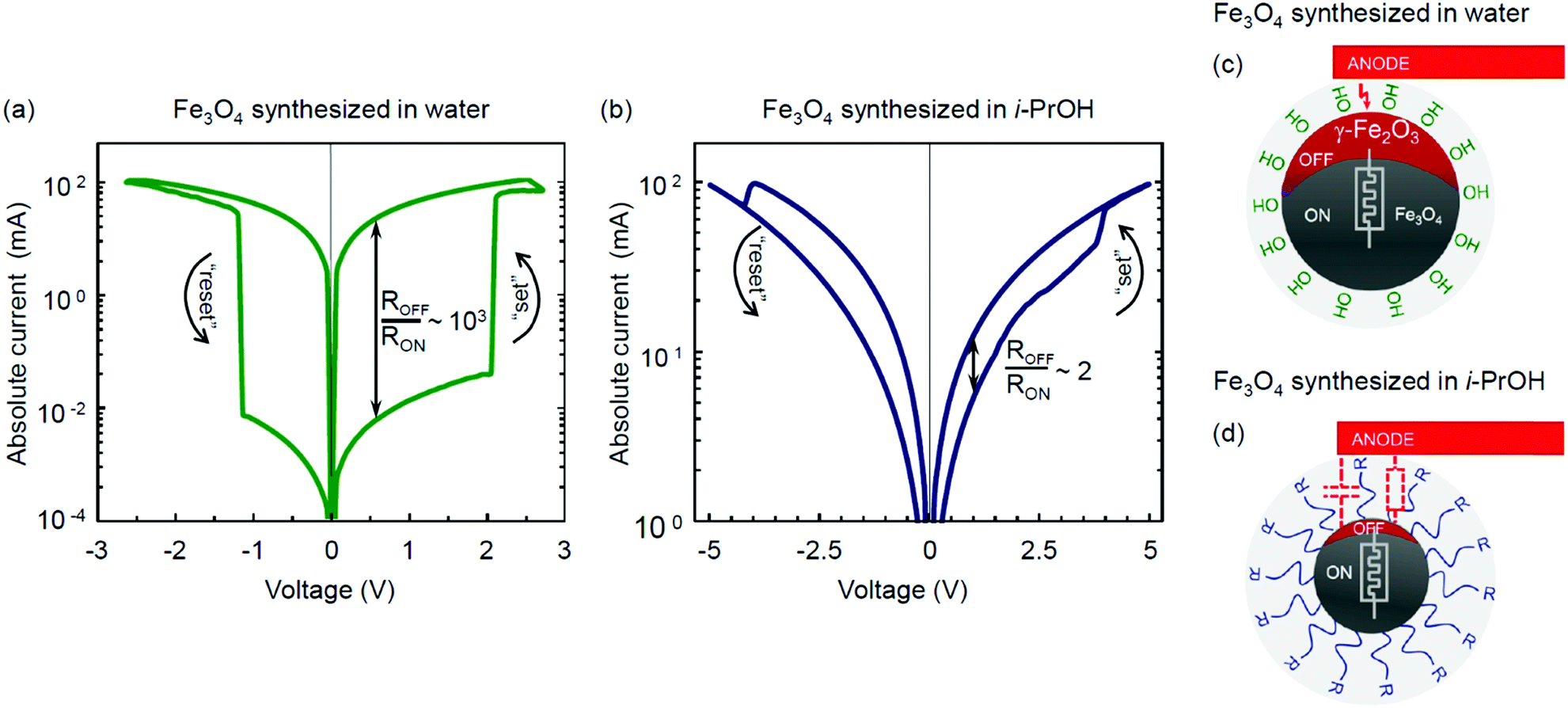 | ||
| Fig. 7 Bipolar resistive switching in Fe3O4 thin films obtained via (a) hydrolytic and (b) non-hydrolytic (i-PrOH) methods and schemes of the resistive switching redox interaction in iron oxide nanoparticles synthesized via (c) hydrolytic and (d) non-hydrolytic routes. | ||
| Properties \ Sample | Fe3O4 (hydrolytic) | Fe3O4 (i-PrOH) |
|---|---|---|
| a Based on N2 adsorption–desorption isotherms (Fig. S11, ESI). b Viability by the MTT assay with the HDF cell line. c Data from ref. 51. | ||
| Mean particle size by DLS | 60 | 68 |
| Mean particle size by SEM | 11 | 25 |
| Mean crystallite size by XRD | 10 | 6.7 |
| BET surface areaa (m2 g−1) | 122 | 87 |
| BJH pore volumea (cm3 g−1) | 0.29 | 0.23 |
| BJH pore diametera (nm) | 9.5 | 6.7 |
| Biocompatibilityb (%) | 97 | 96 |
| Magnetization (emu g−1) | 88c | 46 |
| Resistive switching ratio (ROFF/RON) | ∼103 | ∼2 |
Several mechanisms of the resistive switching phenomenon in transition metal oxides have been proposed and widely discussed in the literature. These include the formation and rupture of a conductive filament in an initial forming process,1,85,86 the electromigration processes at the metal–insulator interface,1,87 and the non-filamentary migration and polarization of ions.88–90 The case of electroforming in Fe3O4 was first investigated by Odagawa et al.,28 who argued its resistive switching properties to originate from the electrically induced redox reaction between the Fe3O4 and γ-Fe2O3 phases. The magnitude of the resistive ratio demonstrated by the hydrolytically synthesized magnetite (Fig. 7a) is quantitatively consistent with the values reported in that study.28
As discussed previously, the following two key factors may be beneficial for the effective charge transfer and resistive switching in Mt memristive thin films obtained by the hydrolytic route: (1) higher crystallinity and absence of organic contamination at the grain boundaries and (2) ease of transformation to γ-Fe2O3 upon oxidation.
The high purity and crystallinity of the hydrolytically derived magnetite is anticipated with the absence of any organic precursors, surfactants, or peptizing agents in the fabrication protocol. As a result, these NPs appeared to be of nearly the same size as the mean crystallite size characterized by XRD (Table 4). In contrast, the synthesis with organic precursors results in apparent particle agglomeration and contamination by organic combustion residues. The latter were evaluated to be in the range of 4–6 wt% according to the weight loss analysis.57 However, a series of experimental attempts to purify magnetite nanoparticles by thermal annealing in an Ar atmosphere at temperatures of 250, 350, and 500 °C have not succeeded in improving their resistive switching properties.
The only hypothetically possible residues from the hydrolytic process are water molecules, as well as hydroxy and amino groups. The presence of both adsorbed water molecules and hydroxy groups at the surface of Mt NPs obtained by the hydrolytic method was justified for temperatures up to 200 °C by means of infrared spectroscopy.51 At another point, water molecules are known to facilitate the topotactic transformation from Fe3O4 to γ-Fe2O3 upon oxidation.70–72 If this is the case, it may rationalize the higher level of Mht manifestation in the Raman spectra upon characterization with a laser power of 50 μW (Fig. 5), as well as the higher amplitude of the resistive switching based on the same type of transformation28 in the hydrolytically synthesized magnetite (Fig. 7a). The resistive switching is bipolar and reversible. The reverse transition from an oxidized metastable γ-Fe2O3 to Fe3O4 is energetically favourable and thus does not require any facilitation. The rationalized scenarios for different resistive switching efficiencies in the hydrolytically and non-hydrolytically synthesized Mt NPs are schematically illustrated in Fig. 7c and d, respectively.
The high resistive switching ratio observed in a thin film prepared with the hydrolytically synthesized Fe3O4 nanoparticles opens up prospects for their application in emerging memristive devices, including various biocompatible and environmentally sustainable cases. Fig. S13 (ESI†) expands the key features of the resistive switching hysteresis illustrated in Fig. 7a in the time (Fig. S13a–c, ESI†) and low-signal (Fig. S13d and e, ESI†) characterization domains.
The primary characterization of the resistive switching properties provided in this study for the hydrolytically and non-hydrolytically synthesized magnetite marks a substantial difference in the electrical functionality of Fe3O4 nanoparticle assemblies resulting from their different synthetic origins. Further analysis of other memristive functionalities such as switching stability, memory retention, synaptic potentiation and depression, as well as other related issues are beyond the scope of this study and will be addressed elsewhere.
Conclusions
In this study we have revisited two solution-based synthesis routes of obtaining magnetite nanoparticles in water and lower alcohols. In the first part of the study, we have explored the low-temperature range of the solvothermal formation of iron oxides in a series of lower alcohols in the vicinity of the decomposition temperature of Fe(AcAc)3 used as the primary source of iron.The experimental examination of the lower temperature regime has demonstrated that the solvothermal interaction results in a pure Fe3O4 product for the cases of synthesis in n-PrOH, i-PrOH, n-BuOH, and BnOH at 175 °C, which is just a few degrees above the decomposition point of iron acetylacetonate. Syntheses in other alcohols at this temperature resulted in the formation of Fe2O3 either as a single-phase product (in EtOH) or as a component of mixed-phase products (in MeOH and i-BuOH). By theoretical consideration, we have addressed the formation of Fe3O4 in lower alcohols as a result of kinetically competing processes, namely, dehydration and reduction.
The DFT calculations of the activation energies for the redox reaction with alcohol as the reduction agent did not indicate any notable difference in the relative reduction ability of lower aliphatic alcohols, thus suggesting that their key role in the solvothermal synthesis is rather dehydration that supplies water for further reactive formation of iron oxide. Seemingly, the Fe3+/Fe2+ transition in the solvothermal process involves an interaction with other reduction agents, of which the most plausible candidate is the acetate anion. None of the examined lower alcohols was sufficient to provide complete reduction of iron at a temperature of 150 °C, which is below the point of Fe(AcAc)3 thermal decomposition. The obtained results have been summarized along with previously reported data52,57 in a diagram illustrating the dependence of iron oxide phase products on the lower alcohol solvent and synthesis temperature (Fig. 4).
All the synthesis routes addressed in the study are compatible with obtaining stable colloids, either in the same dispersion medium as that used in the synthesis (H2O, BnOH) or in MeOH. This allows preparation of functional inks for thin film fabrication using spin-coating, tape casting, or inkjet printing techniques.
Characterization of magnetite nanoparticles obtained by the solvothermal synthesis in lower alcohols demonstrated them to be inferior for several applications with respect to the hydrolytically synthesized counterparts. Particularly, they possess lower purity, lower crystallinity, and as a result demonstrate a lower specific magnetization and a lower resistive switching ratio. Synthesis with BnOH is characterized by a relatively narrow temperature regime for obtaining single-phase Fe3O4 nanoparticles, and the resulting product has demonstrated a relatively higher toxicity compared to the other products examined in the study. In contrast, Fe3O4 nanoparticles produced by the hydrolytic route are free of organic residues and possess high crystallinity. They have demonstrated excellent resistive switching properties with an ROFF/RON ratio of ∼103 and a switching voltage of about ±2 V, which are comparable or superior to the previously reported values for iron oxide thin films. In contrast to various organic by-products contaminating nanoparticles obtained via the solvothermal process, the possible residues of the hydrolytic synthesis mainly include water and hydroxy groups, which at least do not interfere or at most assist the topotactic oxidizing transition from Fe3O4 to Fe2O3 associated with the resistive switching phenomenon. Thus, the hydrolytically synthesized magnetite nanoparticles can be prospective for fabrication of emerging resistive switching devices, especially regarding their possible biocompatible and environmentally sustainable applications.
Author contributions
A. V. V., S. M. M. and M. I. M. conceptualized the work; M. A. M. performed the synthesis and primary materials characterization; M. A. M., D. S. K. and G. A. I. carried out thin film fabrication and optimization of functional inks; O. A. K. carried out the cytotoxicity assay; G. A. I. performed the electrical characterization; I. Y. C. performed the DFT calculations; M. A. M., S. M. M. and M. I. M. prepared the original manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is fulfilled with the financial support from the Russian Science Foundation (Project No. 19-19-00433). X-ray studies were performed using the equipment of the Engineering Center of the St. Petersburg State Institute of Technology (Technical University). Dr I. S. Mukhin and V. A. Sharov (Academic University, St. Petersburg, Russia) are kindly acknowledged for the assistance with SEM and Raman characterization. Dr E. A. Pidko is greatly acknowledged for the access to the Gaussian 16 and COSMOtherm packages. IYC acknowledges NWO Domain Science for the use of the national computer facilities.References
- R. Waser, R. Dittmann, G. Staikov and K. Szot, Redox-Based Resistive Switching Memories - Nanoionic Mechanisms, Prospects, and Challenges, Adv. Mater., 2009, 21(25–26), 2632–2663, DOI:10.1002/adma.200900375.
- F. Diao and Y. Wang, Transition Metal Oxide Nanostructures: Premeditated Fabrication and Applications in Electronic and Photonic Devices, J. Mater. Sci., 2017, 53(6), 4334–4359, DOI:10.1007/s10853-017-1862-3.
- Y. Wang, J. Li and Z. Wei, Transition-Metal-Oxide-Based Catalysts for the Oxygen Reduction Reaction, J. Mater. Chem. A, 2018, 6(18), 8194–8209, 10.1039/c8ta01321g.
- J. Védrine, Heterogeneous Catalysis on Metal Oxides, Catalysts, 2017, 7(11), 341, DOI:10.3390/catal7110341.
- I. Şerban and A. Enesca, Metal Oxides-Based Semiconductors for Biosensors Applications, Front. Chem., 2020, 8, 354, DOI:10.3389/fchem.2020.00354.
- M. Zakharzhevskii, A. S. Drozdov, D. S. Kolchanov, L. Shkodenko and V. V. Vinogradov, Test-System for Bacteria Sensing Based on Peroxidase-Like Activity of Inkjet-Printed Magnetite Nanoparticles, Nanomaterials, 2020, 10(2), 313, DOI:10.3390/nano10020313.
- R. Schmidt and A. W. Brinkman, Studies of the Temperature and Frequency Dependent Impedance of an Electroceramic Functional Oxide NTC Thermistor, Adv. Funct. Mater., 2007, 17(16), 3170–3174, DOI:10.1002/adfm.200600878.
- M. A. Zidan, J. P. Strachan and W. D. Lu, The Future of Electronics Based on Memristive Systems, Nat. Electron., 2018, 1(1), 22–29, DOI:10.1038/s41928-017-0006-8.
- J. J. Yang, D. B. Strukov and D. R. Stewart, Memristive Devices for Computing, Nat. Nanotechnol., 2012, 8(1), 13–24, DOI:10.1038/nnano.2012.240.
- L. Chua, Memristor-The Missing Circuit Element, IEEE Trans. Circuit Theory, 1971, 18(5), 507–519, DOI:10.1109/tct.1971.1083337.
- D. B. Strukov, G. S. Snider, D. R. Stewart and R. S. Williams, The Missing Memristor Found, Nature, 2008, 453(7191), 80–83, DOI:10.1038/nature06932.
- L. Chua, Resistance Switching Memories Are Memristors, Appl. Phys. A: Mater. Sci. Process., 2011, 102(4), 765–783, DOI:10.1007/s00339-011-6264-9.
- M. D. Pickett, G. Medeiros-Ribeiro and R. S. Williams, A Scalable Neuristor Built with Mott Memristors, Nat. Mater., 2012, 12(2), 114–117, DOI:10.1038/nmat3510.
- C. Du, W. Ma, T. Chang, P. Sheridan and W. D. Lu, Biorealistic Implementation of Synaptic Functions with Oxide Memristors through Internal Ionic Dynamics, Adv. Funct. Mater., 2015, 25(27), 4290–4299, DOI:10.1002/adfm.201501427.
- A. L. Hodgkin and A. F. Huxley, A Quantitative Description of Membrane Current and Its Application to Conduction and Excitation in Nerve, J. Physiol., 1952, 117(4), 500–544, DOI:10.1113/jphysiol.1952.sp004764.
- D. Gater; A. Iqbal; J. Davey and E. GaleConnecting Spiking Neurons to a Spiking Memristor Network Changes the Memristor Dynamics. 2013 IEEE 20th International Conference on Electronics, Circuits, and Systems (ICECS). IEEE 2013. DOI:10.1109/icecs.2013.6815469.
- I. Gupta, A. Serb, A. Khiat, R. Zeitler, S. Vassanelli and T. Prodromakis, Real-Time Encoding and Compression of Neuronal Spikes by Metal-Oxide Memristors, Nat. Commun., 2016, 7, 12805, DOI:10.1038/ncomms12805.
- A. Serb, A. Corna, R. George, A. Khiat, F. Rocchi, M. Reato, M. Maschietto, C. Mayr, G. Indiveri, S. Vassanelli and T. Prodromakis, Memristive Synapses Connect Brain and Silicon Spiking Neurons, Sci. Rep., 2020, 10, 2590, DOI:10.1038/s41598-020-58831-9.
- G. A. Illarionov, D. S. Kolchanov, O. A. Kuchur, M. V. Zhukov, E. Sergeeva, V. V. Krishtop, A. V. Vinogradov and M. I. Morozov, Inkjet Assisted Fabrication of Planar Biocompatible Memristors, RSC Adv., 2019, 9(62), 35998–36004, 10.1039/C9RA08114C.
- A. Chiolerio, M. Chiappalone, P. Ariano and S. Bocchini, Coupling Resistive Switching Devices with Neurons: State of the Art and Perspectives, Front. Neurosci., 2017, 11, 70, DOI:10.3389/fnins.2017.00070.
- G. A. Illarionov, S. M. Morozova, V. V. Chrishtop, M.-A. Einarsrud and M. I. Morozov, Memristive TiO2: Synthesis, Technologies, and Applications, Front. Chem., 2020, 8, 724, DOI:10.3389/fchem.2020.00724.
- D. Chen, Q. Tang, X. Li, X. Zhou, J. Zang, W. Xue, J Xiang and C. Guo, Biocompatibility of Magnetic Fe3O4 Nanoparticles and Their Cytotoxic Effect on MCF-7 Cells, Int. J. Nanomed., 2012, 7, 4973–4982, DOI:10.2147/ijn.s35140.
- A. Ito, M. Shinkai, H. Honda and T. Kobayashi, Medical Application of Functionalized Magnetic Nanoparticles, J. Biosci. Bioeng., 2005, 100(1), 1–11, DOI:10.1263/jbb.100.1.
- J. L. Corchero and A. Villaverde, Biomedical Applications of Distally Controlled Magnetic Nanoparticles, Trends Biotechnol., 2009, 27(8), 468–476, DOI:10.1016/j.tibtech.2009.04.003.
- A. Ito and M. Kamihira, Tissue Engineering Using Magnetite Nanoparticles, Progress in Molecular Biology and Translational Science, Elsevier, 2011, pp. 355–395 DOI:10.1016/b978-0-12-416020-0.00009-7.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Magnetic Iron Oxide Nanoparticles: Synthesis, Stabilization, Vectorization, Physicochemical Characterizations, and Biological Applications, Chem. Rev., 2008, 108(6), 2064–2110, DOI:10.1021/cr068445e.
- P. Tartaj, M. a. d. P. Morales, S. Veintemillas-Verdaguer, T. Gonz lez-Carre o and C. J. Serna, The Preparation of Magnetic Nanoparticles for Applications in Biomedicine, J. Phys. D: Appl. Phys., 2003, 36(13), R182–R197, DOI:10.1088/0022-3727/36/13/202.
- A. Odagawa, Y. Katoh, Y. Kanzawa, Z. Wei, T. Mikawa, S. Muraoka and T. Takagi, Electroforming and Resistance-Switching Mechanism in a Magnetite Thin Film, Appl. Phys. Lett., 2007, 91(13), 133503, DOI:10.1063/1.2789178.
- S. Porro, K. Bejtka, A. Jasmin, M. Fontana, G. Milano, A. Chiolerio, C. F. Pirri and C. Ricciardi, A Multi-Level Memristor Based on Atomic Layer Deposition of Iron Oxide, Nanotechnology, 2018, 29(49), 495201, DOI:10.1088/1361-6528/aae2ff.
- X. Wan, F. Gao, X. Lian, X. Ji, E. Hu, L. He, Y. Tong and Y. Guo, Bio-Mimicked Atomic-Layer-Deposited Iron Oxide-Based Memristor with Synaptic Potentiation and Depression Functions, Jpn. J. Appl. Phys., 2018, 57(6), 60303, DOI:10.7567/jjap.57.060303.
- C. Yun, X. G. Chen, J. B. Fu, C. S. Wang, H. L. Du, G. C. Xiong, G. J. Lian, Y. C. Yang and J. B. Yang, Fabrication of Ferrimagnetic FeOx Thin Film and the Resistance Switching of Au/FeOx/Pt Heterostructure, J. Appl. Phys., 2013, 113(17), 17C303, DOI:10.1063/1.4794138.
- T. Ishibe, Y. Maeda, T. Terada, N. Naruse, Y. Mera, E. Kobayashi and Y. Nakamura, Resistive Switching Memory Performance in Oxide Hetero-Nanocrystals with Well-Controlled Interfaces, Sci. Technol. Adv. Mater., 2020, 21(1), 195–204, DOI:10.1080/14686996.2020.1736948.
- Y. Zhang, W. He, Y. Wu, K. Huang, Y. Shen, J. Su, Y. Wang, Z. Zhang, X. Ji, G. Li, H. Zhang, S. Song, H. Li, L. Sun, R. Zhao and L. Shi, Highly Compact Artificial Memristive Neuron with Low Energy Consumption, Small, 2018, 14(51), 1802188, DOI:10.1002/smll.201802188.
- C. Wang, W. He, Y. Tong and R. Zhao, Investigation and Manipulation of Different Analog Behaviors of Memristor as Electronic Synapse for Neuromorphic Applications, Sci. Rep., 2016, 6, 22970, DOI:10.1038/srep22970.
- T. H. Kim, E. Y. Jang, N. J. Lee, D. J. Choi, K.-J. Lee, J. Jang, J. Choi, S. H. Moon and J. Cheon, Nanoparticle Assemblies as Memristors, Nano Lett., 2009, 9(6), 2229–2233, DOI:10.1021/nl900030n.
- T. H. Kim, E. Y. Jang, N. J. Lee, D. J. Choi, K.-J. Lee, J. Jang, J. Choi, S. H. Moon and J. Cheon, Nanoparticle Assemblies as Memristors, Nano Lett., 2010, 10(7), 2734, DOI:10.1021/nl101926w.
- L. L. Hench and J. K. West, The Sol-Gel Process, Chem. Rev., 1990, 90(1), 33–72, DOI:10.1021/cr00099a003.
- A. E. Danks, S. R. Hall and Z. Schnepp, The Evolution of ‘Sol–Gel’ Chemistry as a Technique for Materials Synthesis, Mater. Horiz., 2016, 3(2), 91–112, 10.1039/c5mh00260e.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O’Connor, Recent Advances in the Liquid-Phase Syntheses of Inorganic Nanoparticles, Chem. Rev., 2004, 104(9), 3893–3946, DOI:10.1021/cr030027b.
- A. Styskalik, D. Skoda, C. Barnes and J. Pinkas, The Power of Non-Hydrolytic Sol-Gel Chemistry: A Review, Catalysts, 2017, 7(6), 168, DOI:10.3390/catal7060168.
- J. Liu, Z. Wu, Q. Tian, W. Wu and X. Xiao, Shape-Controlled Iron Oxide Nanocrystals: Synthesis, Magnetic Properties and Energy Conversion Applications, CrystEngComm, 2016, 18(34), 6303–6326, 10.1039/c6ce01307d.
- C. Yang, J. Wu and Y. Hou, Fe3O4 Nanostructures: Synthesis, Growth Mechanism, Properties and Applications, Chem. Commun., 2011, 47(18), 5130, 10.1039/c0cc05862a.
- S. Majidi, F. Zeinali Sehrig, S. M. Farkhani, M. Soleymani Goloujeh and A. Akbarzadeh, Current Methods for Synthesis of Magnetic Nanoparticles, Artif. Cells, Nanomed., Biotechnol., 2014, 44(2), 722–734, DOI:10.3109/21691401.2014.982802.
- M. Usman, J. M. Byrne, A. Chaudhary, S. Orsetti, K. Hanna, C. Ruby, A. Kappler and S. B. Haderlein, Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals, Chem. Rev., 2018, 118(7), 3251–3304, DOI:10.1021/acs.chemrev.7b00224.
- J. Wallyn, N. Anton and T. F. Vandamme, Synthesis, Principles, and Properties of Magnetite Nanoparticles for In Vivo Imaging Applications — A Review, Pharmaceutics, 2019, 11(11), 601, DOI:10.3390/pharmaceutics11110601.
- R. Massart, Preparation of Aqueous Magnetic Liquids in Alkaline and Acidic Media, IEEE Trans. Magn., 1981, 17(2), 1247–1248, DOI:10.1109/tmag.1981.1061188.
- L. A. Welo and O. Baudisoh, XXXIX.Studies on Precipitated Magnetite, with Particular Reference to Hysteresis, London, Edinburgh Dublin Philos. Mag. J. Sci., 1927, 3(14), 396–410, DOI:10.1080/14786440208564209.
- W. C. Elmore, Ferromagnetic Colloid for Studying Magnetic Structures, Phys. Rev., 1938, 54(4), 309–310, DOI:10.1103/physrev.54.309.
- Y. Sahoo, H. Pizem, T. Fried, D. Golodnitsky, L. Burstein, C. N. Sukenik and G. Markovich, Alkyl Phosphonate/Phosphate Coating on Magnetite Nanoparticles: A Comparison with Fatty Acids, Langmuir, 2001, 17(25), 7907–7911, DOI:10.1021/la010703.
- G. Schikorr, Über Die Reaktionen Zwischen Eisen, Seinen Hydroxyden Und Wasser, Z. Elektrochem. Angew. Phys. Chem., 1929, 35(2), 65–70, DOI:10.1002/bbpc.19290350204.
- A. S. Drozdov, V. Ivanovski, D. Avnir and V. V. Vinogradov, A Universal Magnetic Ferrofluid: Nanomagnetite Stable Hydrosol with No Added Dispersants and at Neutral PH, J. Colloid Interface Sci., 2016, 468, 307–312, DOI:10.1016/j.jcis.2016.01.061.
- N. Pinna, S. Grancharov, P. Beato, P. Bonville, M. Antonietti and M. Niederberger, Magnetite Nanocrystals: Nonaqueous Synthesis, Characterization, and Solubility, Chem. Mater., 2005, 17(11), 3044–3049, DOI:10.1021/cm050060.
- Z. Li, H. Chen, H. Bao and M. Gao, One-Pot Reaction to Synthesize Water-Soluble Magnetite Nanocrystals, Chem. Mater., 2004, 16(8), 1391–1393, DOI:10.1021/cm035346y.
- J.-H. Lee, Y.-M. Huh, Y. Jun, J. Seo, J. Jang, H.-T. Song, S. Kim, E.-J. Cho, H.-G. Yoon, J.-S. Suh and J. Cheon, Artificially Engineered Magnetic Nanoparticles for Ultra-Sensitive Molecular Imaging, Nat. Med., 2006, 13(1), 95–99, DOI:10.1038/nm1467.
- S. Sun and H. Zeng, Size-Controlled Synthesis of Magnetite Nanoparticles, J. Am. Chem. Soc., 2002, 124(28), 8204–8205, DOI:10.1021/ja026501x.
- T. Dubois and G. Demazeau, Preparation of Fe3O4 Fine Particles through a Solvothermal Process, Mater. Lett., 1994, 19(1–2), 38–47, DOI:10.1016/0167-577x(94)90102-3.
- H. Kominami, S. Onoue, K. Matsuo and Y. Kera, Synthesis of Microcrystalline Hematite and Magnetite in Organic Solvents and Effect of a Small Amount of Water in the Solvents, J. Am. Ceram. Soc., 1999, 82(7), 1937–1940, DOI:10.1111/j.1151-2916.1999.tb02022.x.
- M.-T. Liang, S.-H. Wang, Y.-L. Chang, H.-I. Hsiang, H.-J. Huang, M.-H. Tsai, W.-C. Juan and S.-F. Lu, Iron Oxide Synthesis Using a Continuous Hydrothermal and Solvothermal System, Ceram. Int., 2010, 36(3), 1131–1135, DOI:10.1016/j.ceramint.2009.09.044.
- M. Niederberger, G. Garnweitner, N. Pinna and G. Neri, Non-Aqueous Routes to Crystalline Metal Oxide Nanoparticles: Formation Mechanisms and Applications, Prog. Solid State Chem., 2005, 33(2-4 SPEC. ISS.), 59–70, DOI:10.1016/j.progsolidstchem.2005.11.032.
- M. Niederberger and N. PinnaMetal Oxide Nanoparticles in Organic Solvents, Engineering Materials and Processes, Springer, London 2009. DOI:10.1007/978-1-84882-671-7.
- J. V. Hoene, R. G. Charles and W. M. Hickam, Thermal Decomposition of Metal Acetylacetonates: Mass Spectrometer Studies, J. Phys. Chem., 1958, 62(9), 1098–1101, DOI:10.1021/j150567a019.
- R. A. Lalancette, D. Syzdek, J. Grebowicz, E. Arslan and I. Bernal, The Thermal Decomposition and Analyses of Metal Tris-Acetylacetonates, J. Therm. Anal. Calorim., 2018, 135(6), 3463–3470, DOI:10.1007/s10973-018-7598-8.
- T. Mosmann, Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays, J. Immunol. Methods, 1983, 65(1–2), 55–63, DOI:10.1016/0022-1759(83)90303-4.
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, Unraveling Reaction Networks behind the Catalytic Oxidation of Methane with H2O2 over a Mixed-Metal MIL-53(Al,Fe) MOF Catalyst, Chem. Sci., 2018, 9(33), 6765–6773, 10.1039/c8sc02376j.
- Á. Szécsényi, E. Khramenkova, I. Y. Chernyshov, G. Li, J. Gascon and E. A. Pidko, Breaking Linear Scaling Relationships with Secondary Interactions in Confined Space: A Case Study of Methane Oxidation by Fe/ZSM-5 Zeolite, ACS Catal., 2019, 9(10), 9276–9284, DOI:10.1021/acscatal.9b01914.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. Klamt, Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena, J. Phys. Chem., 1995, 99(7), 2224–2235, DOI:10.1021/j100007a062.
- COSMOtherm. COSMOlogic Gmbh & Co KG.
- G. W. Van Oosterhout and C. J. M. Rooijmans, A New Superstructure in Gamma-Ferric Oxide, Nature, 1958, 181(4601), 44, DOI:10.1038/181044a0.
- I. David and A. J. E. Welch, The Oxidation of Magnetite and Related Spinels. Constitution of Gamma Ferric Oxide, Trans. Faraday Soc., 1956, 52, 1642, 10.1039/tf9565201642.
- T. Elder, Particle-Size Effect in Oxidation of Natural Magnetite, J. Appl. Phys., 1965, 36(3), 1012–1013, DOI:10.1063/1.1714076.
- G. D. Renshaw and C. Roscoe, Thermal Stability of γ-Ferric Oxide, Nature, 1969, 224(5216), 263–264, DOI:10.1038/224263a0.
- S. Ali, S. Khan, A. Khan and A. Bermak, Memristor Fabrication Through Printing Technologies: A Review, IEEE Access, 2021, 9, 95970–95985, DOI:10.1109/access.2021.3094027.
- T. Zaki, Catalytic Dehydration of Ethanol Using Transition Metal Oxide Catalysts, J. Colloid Interface Sci., 2005, 284(2), 606–613, DOI:10.1016/j.jcis.2004.10.048.
- L. Zhang, G. Garnweitner, I. Djerdj, M. Antonietti and M. Niederberger, Generalized Nonaqueous Sol–Gel Synthesis of Different Transition-Metal Niobate Nanocrystals and Analysis of the Growth Mechanism, Chem. – Asian J., 2008, 3(4), 746–752, DOI:10.1002/asia.200700318.
- H. M. Ismail, A Thermoanalytic Study of Metal Acetylacetonates, J. Anal. Appl. Pyrolysis, 1991, 21(3), 315–326, DOI:10.1016/0165-2370(91)80006-t.
- I. V. Zagaynov and A. K. Buryak, Mesoporous Nanoscale Ceria: Synthesis from Cerium (III) Acetylacetonate and Mechanism, J. Sol-Gel Sci. Technol., 2014, 74(1), 103–108, DOI:10.1007/s10971-014-3582-3.
- C. Toyos-Rodríguez, J. Calleja-García, L. Torres-Sánchez, A. López, A. M. Abu-Dief, A. Costa, L. Elbaile, R. D. Crespo, J. S. Garitaonandia, E. Lastra, J. A. García and F. J. García-Alonso, A Simple and Reliable Synthesis of Superparamagnetic Magnetite Nanoparticles by Thermal Decomposition of Fe(Acac)3, J. Nanomater., 2019, 2019, 1–10, DOI:10.1155/2019/2464010.
- P. Guardia, N. Pérez, A. Labarta and X. Batlle, Controlled Synthesis of Iron Oxide Nanoparticles over a Wide Size Range, Langmuir, 2009, 26(8), 5843–5847, DOI:10.1021/la903767e.
- E. M. Arnett and M. A. Mendelsohn, Destructive Autoxidation of Metal Chelates. IV. Kinetics and Mechanism, J. Am. Chem. Soc., 1962, 84(20), 3824–3829, DOI:10.1021/ja00879a008.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, Raman Microspectroscopy of Some Iron Oxides and Oxyhydroxides, J. Raman Spectrosc., 1997, 28(11), 873–878, DOI:10.1002/(sici)1097-4555(199711)28:11<873::aid-jrs177>3.0.co;2-b.
- M. Hanesch, Raman Spectroscopy of Iron Oxides and (Oxy)Hydroxides at Low Laser Power and Possible Applications in Environmental Magnetic Studies, Geophys. J. Int., 2009, 177(3), 941–948, DOI:10.1111/j.1365-246x.2009.04122.x.
- B. D. Cullity and C. D. Graham, Introduction to Magnetic Materials, John Wiley & Sons, Inc., 2008. DOI:10.1002/9780470386323.
- Y.-F. Chang, Y.-T. Tsai, G.-W. Chang, Y.-E. Syu, Y.-H. Tai and T.-C. Chang, Study of Resistive Switching Characteristics on a Temperature-Sensitive FeOx-Transition Layer in a TiN/SiO2/FeOx/Fe Structure, ECS J. Solid State Sci. Technol., 2012, 1(5), Q91–Q95, DOI:10.1149/2.003205jss.
- R. Waser and M. Aono, Nanoionics-Based Resistive Switching Memories, Nat. Mater., 2007, 6, 833–840, DOI:10.1038/nmat2023.
- D.-H. Kwon, S. Lee, C. S. Kang, Y. S. Choi, S. J. Kang, H. L. Cho, W. Sohn, J. Jo, S.-Y. Lee, K. H. Oh, T. W. Noh, R. A. De Souza, M. Martin and M. Kim, Unraveling the Origin and Mechanism of Nanofilament Formation in Polycrystalline SrTiO3 Resistive Switching Memories, Adv. Mater., 2019, 31(28), 1901322, DOI:10.1002/adma.201901322.
- A. Sawa, Resistive Switching in Transition Metal Oxides, Mater. Today, 2008, 11(6), 28–36, DOI:10.1016/S1369-7021(08)70119-6.
- Y. Aoki, C. Wiemann, V. Feyer, H.-S. Kim, C. M. Schneider, H. Ill-Yoo and M. Martin, Bulk Mixed Ion Electron Conduction in Amorphous Gallium Oxide Causes Memristive Behaviour, Nat. Commun., 2014, 5(1), 3473, DOI:10.1038/ncomms4473.
- D. Y. Guo, Y. P. Qian, Y. L. Su, H. Z. Shi, P. G. Li, J. T. Wu, S. L. Wang, C. Cui and W. H. Tang, Evidence for the Bias-Driven Migration of Oxygen Vacancies in Amorphous Non-Stoichiometric Gallium Oxide, AIP Adv., 2017, 7(6), 65312, DOI:10.1063/1.4990566.
- C. Kura, Y. Aoki, E. Tsuji, H. Habazaki and M. Martin, Fabrication of a Resistive Switching Gallium Oxide Thin Film with a Tailored Gallium Valence State and Oxygen Deficiency by Rf Cosputtering Process, RSC Adv., 2016, 6(11), 8964–8970, 10.1039/C5RA21160C.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03825g |
| This journal is © The Royal Society of Chemistry 2022 |