
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biocompatible non-leachable antimicrobial polymers with a nonionic hyperbranched backbone and phenolic terminal units†
Carlos R.
Arza‡
a,
Xiaoya
Li‡
 a,
Sedef
İlk
b,
Yang
Liu
c,
Deniz
Demircan
a and
Baozhong
Zhang
*a
a,
Sedef
İlk
b,
Yang
Liu
c,
Deniz
Demircan
a and
Baozhong
Zhang
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: baozhong.zhang@chem.lu.se
bNiğde Ömer Halisdemir University, Faculty of Medicine, Department of Immunology, TR-51240 Niğde, Turkey
cFaculty of Medicine, Department of Clinical Sciences, Orthopedics, Lund University, Lund, Sweden
First published on 9th September 2022
Abstract
This work aimed to develop biocompatible non-leachable antimicrobial polymers without ionic structures. A series of nonionic hyperbranched polymers (HBPs) with an isatin-based backbone and phenolic terminal units were synthesized and characterized. The molecular structures and thermal properties of the obtained HBPs were characterized by SEC, NMR, FTIR, TGA and DSC analyses. Disk diffusion assay revealed significant antibacterial activity of the obtained phenolic HBPs against nine different pathogenic bacteria. The presence of a methoxy or long alkyl group close to the phenolic unit enhanced the antibacterial effect against certain Gram positive and negative bacteria. The obtained nonionic HBPs were blended in polyester poly(hexamethylene terephthalate) films, which showed no noticeable leakage after being immersed in water for 5 days. Finally, these HBPs showed no cytotoxicity effect to MG-63 osteoblast-like human cells according to MTT analysis, and negligible hemolytic effect.
Introduction
Antimicrobial polymers (APs) have attracted growing attention recently, due to their high efficacy and selectivity, as well as low risk for leaching or lower skin permeation compared to small molecular antimicrobials.1–4 In the past decade, a significantly growing number of APs has been approved by the Food and Drug Administration, which highlights the importance and translational potential of this class of materials.5–8 Most APs are ionic, which interact with bacterial membranes by ionic interactions.9–18 However, the relatively high water solubility of ionic polymers could be a disadvantage for certain applications such as coatings or additives.19–21 Therefore, nonionic APs have attracted growing interest particularly in the development of new coating or additives for various materials applications.22,23Nonionic APs usually interfere with bacteria by certain non-ionic interactions with bacterial membranes (e.g. by hydrogen bonding, hydrophobic interactions, or aromatic interactions etc.).24–28 To enable such interactions, a particularly attractive strategy is to utilize naturally occurring antimicrobial molecules (e.g. aspirin, indole, istain, etc.), which can interact with bacteria. In addition, these molecules have been adopted by the natural ecosystems and thus may have lower environmental impact. This strategy has recently gained increasing attention, and a number of APs with various naturally occurring antimicrobial functionality have been reported, such as those with tropolone, aspirin, curcumin, limonene, astaxanthin, indole, or isatin structures.22,29–35
Phenolic molecules are abundant in nature and many of them are natural antimicrobials, such as phenol,36 catechol,37 guaiacol (a lignin fragment),38 pyrogallol,39 and cardanol derivatives (e.g. hydro-cardanol from cashew nut shell liquid).40,41 These phenolic molecules are potentially attractive building blocks for the design of new nonionic APs.42–44 Various phenolic thermoplastic and thermosetting polymers with antibacterial function have been reported, of which the antimicrobial effect could be measured by various methods (e.g. halo zone test, spread plate method, minimum inhibitory concentration, and minimum bactericidal concentration, etc.).42,45–49 Bio-composites with grafted phenols have also been reported with significant antibacterial activity against various Gram-positive and negative bacteria.43
Highly branched polymers such as dendrimers or hyperbranched polymers (HBPs) form a particularly important class of APs, which have a large number of densely grafted functional groups that can interact with bacteria synergistically.22,25,50–53 This can thus enhance their antimicrobial effect.50,54–63 In addition, their near spherical structures could enhance the solubility and facilitate their processing in solutions (e.g. by spin coating).64 To our knowledge, the potential of dendritic polymers with phenol termini as new antimicrobial materials have not been investigated.
Herein, we report on the synthesis of four potentially bio-based nonionic HBPs with isatin-based backbone and four different phenolic groups (i.e. phenol, catechol, guaiacol and hydro-cardanol) by a one-step polymerization of an isatin-based AB2 monomer followed by grafting various phenol units. The molecular and thermal properties of the obtained phenolic HBPs were characterized, and their antibacterial properties were evaluated and compared to that of small molecular agents. The leaching risk and cytotoxicity of the obtained polymers were also investigated.
Experimental
Materials
4-Hydroxybenzaldehyde (98%), vanillin (99%), syringaldehyde (98%), 2-oxindole (97%), methyl 2-bromoacetate (>97%), mesitylene (98%), dibutyltin oxide (DBTO) (>98%), 1,6-hexanediol (97%), 1,5-pentanediol (>97%), 1,4-butanediol (>99%), 1,3-propaneodiol (>98%) and sodium sulfate (Na2SO4) were purchased from Sigma-Aldrich. Glacial acetic acid (99.8%), hydrochloric acid (37%), N,N-dimethylformamide (DMF, ACS, Reag. Ph. Eur.) and ethyl acetate (EtOAc, ACS, Reag. Ph. Eur.) and n-heptane were purchased from VWR Chemicals. Methanol was purchased from Honeywell. Chloroform (Analytical grade, stabilized with ethanol) and xylene (Analytical grade, ACS) were purchased from Scharlau. All chemicals and reagents were used as received.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.), 1727 (CO str.), 1613 (Ar C–C str.), 1470 (Ar C–CH in-plane bend. + Ar C–C str.), 1325 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1095 (Ar C–C str. + Ar C–CH in-plane bend. + Ar C–C str.), 751 (Ar C–H out-of-plane bend).
O str.), 1727 (CO str.), 1613 (Ar C–C str.), 1470 (Ar C–CH in-plane bend. + Ar C–C str.), 1325 (Ar CCH in-plane bend. + C–N str. + Ar C–C str.), 1095 (Ar C–C str. + Ar C–CH in-plane bend. + Ar C–C str.), 751 (Ar C–H out-of-plane bend).
Measurements
Nuclear magnetic resonance (NMR) measurements were carried out on a Bruker DRX400 spectrometer at the proton frequency of 400.13 MHz and carbon frequency of 100.61 MHz. Fourier transform infrared (FTIR) spectra were measured with an attenuated total reflection (ATR) setup using a Bruker Alpha FT-IR spectrometer. Twenty-four scans were co-added using a resolution of 4 cm−1. Size exclusion chromatography (SEC) in chloroform was performed with a Viscotek 305 TDA at 35 °C with a flow rate of 1.0 mL min−1, which included a guard column and two Malvern Panalytical general purpose mixed bed columns with an exclusion limit of 20 × 106 Da for polystyrene. Detection consisted of a conventional dual cell refractive index detector, a four-capillary bridge viscometer, and a light scattering detector operating at 3 mW, at a wavelength of 670 nm, and measurements angles of 90° and 7°. The three detectors were calibrated with a polystyrene standard (96 kDa) from Polymers Laboratories. Molecular weights were determined by the triple detection method using the OmiSEC 5.12. software (Malvern). Size exclusion chromatography (SEC) in DMAc was performed using Agilent 1100/1200 Infinity HPLC System equipped with three columns (GPC column PSS GRAM 3000 Å,10 μm; GPC column PSS GRAM 1000 Å, 10 μm; GPC column PSS GRAM 30 Å, 10 μm) connected in sequence at 40 °C in DMAc with LiBr (5 g L−1) at a flow rate of 1 mL min−1. Calibration was carried out with ReadyCal-Kit poly(methyl methacrylate) standards Mp = 202–2200 kDa. Differential scanning calorimetry (DSC) measurements were performed using TA Instruments DSC Q2000. The samples were studied with a heating rate of 10 °C min−1 under nitrogen with a purge rate of 50 mL min−1. The sequence consisted of a heating ramp from 40 to 300 °C and held at that temperature for 30 s, followed by a cooling ramp to −50 °C and held at that temperature for 3 min, and finally a heating ramp to 300 °C, which was employed to determine the glass transition temperature (Tg). Thermogravimetic analysis (TGA) was performed with a thermogravimetric analyzer TA Instruments Q500 at a heating rate of 10 °C min−1 under nitrogen with a purge rate of 50 mL min−1. Water contact angle (θ) was measured in a picture (taken with a Nikon Bellows PB-6 camera) of a single water droplet placed onto the dry polymer film.Antimicrobial bioassay
MTT assay
The MG-63 human osteoblast-like cells were cultured in Dulbecco's Modified Eagle Media (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 1% streptomycin in a humidified incubator at 37 °C. The medium was replaced every 2 days. Cells were trypsinized and centrifuged at 400g for 4 min to get a concentrated cell pellet when the confluence reached 80%. 1 × 104 cells per well were seeded on a 96-well plate and cultured for 24 h before adding the materials. Test compounds (monomer 3, and HBPs) dissolved in DMSO (10 mg mL−1, 5 mg mL−1, 1 mg mL−1) were then added to the cells at a final DMSO concentration of 1% (v/v). Fresh culture medium only with 1% DMSO (v/v) was used as negative control, and each sample was replicated in four wells. After being cultured for 24 h, the cell culture medium was discarded and the cells were washed with phosphate buffer. MTT working solution (0.5 mg mL−1) was added to the cells and incubated for 2 h at 37 °C, after which DMSO (200 μL well−1) was added to the reaction products for 10 min. The solubilized contents were pipetted and transferred into a clear bottom 96-well plate. Absorbance was determined by spectrophotometry at 600 nm wavelength. To evaluate the possible interaction between polymers and MTT working solution, a control experiment was conducted by using only polymers under same conditions without adding cells.Hemolysis tests
The HaemoScan Biomaterial Haemolytic Assay (HaemoScan, Netherlands) was used to investigate the cytotoxicity of the monomer 3 and all polymers on human erythrocytes according to the manufacturer's protocol.65 Briefly, the erythrocyte was prepared by repeatedly rinsing with different wash buffer (Dilution buffer I, II and III, provided by the manufacturer) and centrifuged at 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × g for 10 min. Afterward, 5mL of the Dilution buffer III was added to re-suspend the erythrocytes. 0.5 mL erythrocyte suspension was used to test each sample. The samples were first dissolved in DMSO to form 10 mg mL−1 stock solutions. 5 μL of the stock solution was added in 0.5 mL erythrocyte suspension with a final (suspension) concentration of 100 μg mL−1 for polymers. After 24 h of incubation, the samples were centrifuged at 4500 × g for 1 min and 20μL of the supernatant pipetted into a 96-well plate along with 180μL of assay buffer. The absorbance was read at a wavelength of 450nm. Each polymer has been tested triplicate. DMSO (5 μL,1% v/v) was used as the negative control (0% hemolysis). Lysis buffer was used as the positive control (100% hemolysis). The hemolysis percentage was calculated by following equation.66
× g for 10 min. Afterward, 5mL of the Dilution buffer III was added to re-suspend the erythrocytes. 0.5 mL erythrocyte suspension was used to test each sample. The samples were first dissolved in DMSO to form 10 mg mL−1 stock solutions. 5 μL of the stock solution was added in 0.5 mL erythrocyte suspension with a final (suspension) concentration of 100 μg mL−1 for polymers. After 24 h of incubation, the samples were centrifuged at 4500 × g for 1 min and 20μL of the supernatant pipetted into a 96-well plate along with 180μL of assay buffer. The absorbance was read at a wavelength of 450nm. Each polymer has been tested triplicate. DMSO (5 μL,1% v/v) was used as the negative control (0% hemolysis). Lysis buffer was used as the positive control (100% hemolysis). The hemolysis percentage was calculated by following equation.66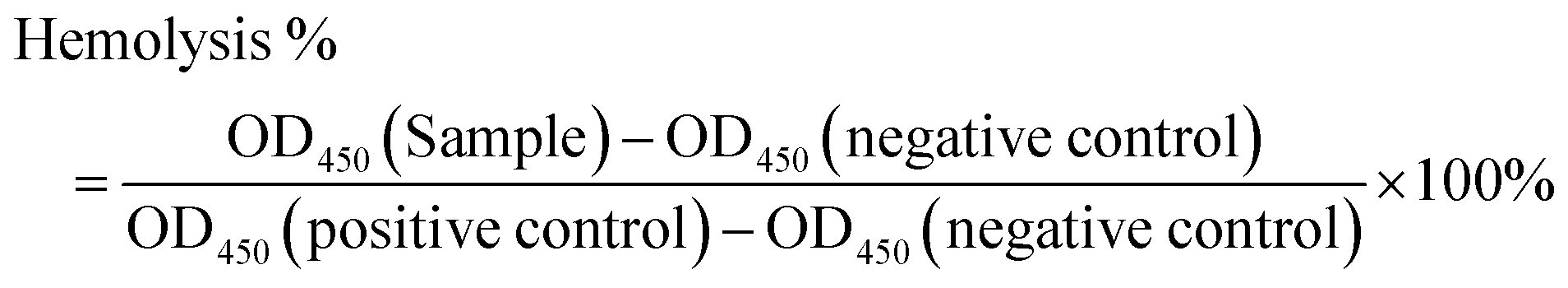
Results and discussion
Synthesis and characterization of HBPs
AB2-monomer 3 was synthesized by a straightforward SN2 reaction of isatin 1 and β-bromophenetole 2 in 89% yield (Scheme 1). Both starting molecules are potentially bio-based. Isatin (1) is widely produced in nature, and its derivatives frequently display biological activities including antimicrobial properties.67 The other starting molecule, β-bromophenetole (2), can be conveniently obtained by bromination of phenoxyethanol, which is also a bio-based molecule derived from phenol and ethylene carbonate.68 Phenoxyethanol is also widely used in industry as solvent, synthetic intermediate, and fixing agent for perfumes.69 Monomer 3 is an AB2 monomer for acid-catalysed Friedel–Crafts type polymerizations (the phenyl group can react twice with the carbonyl group in each monomer),70–72 which can yield a non-crosslinked hyperbranched polymer structure with diaryloxindoles backbone.73,74 AB2 monomer 3 was polymerized according to a previously reported polymerization protocol for a similar monomer,63 which was performed at room temperature without solvent, yielding the desired isatin-based precursor HBP1 after a simple precipitation from methanol. Afterward, HBP1 was reacted with the phenolic compounds as grafting agents (i.e. phenol, catechol, guaiacol and hydro-cardanol), yielding the four corresponding phenolic HBPs (HBP-P, HBP-C, HBP-G and HBP-H, respectively).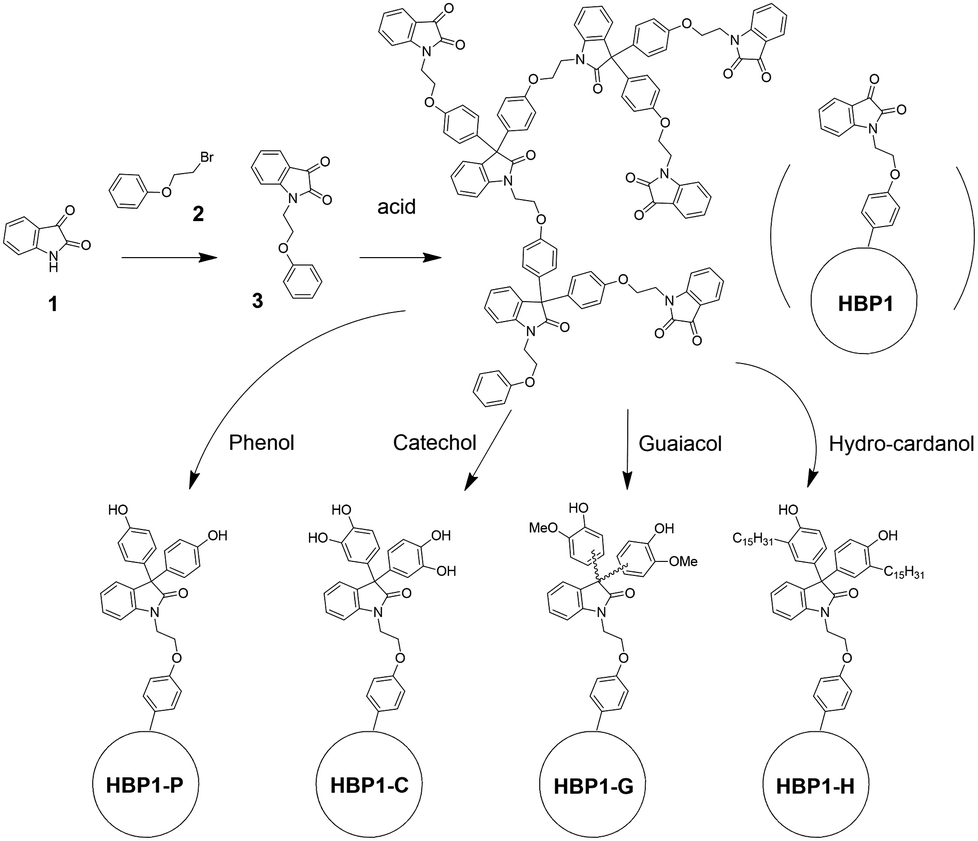 | ||
| Scheme 1 Synthesis of bio-based AB2 monomer 3, isatin-based precursor polymer (HBP1) and four phenolic HBPs, including HBP-P (with phenol), HBP-C (with catechol), HBP-G (with guaiacol), and HBP-H (with hydro-cardanol). | ||
The chemical structures of the obtained HBPs were characterized by 1H NMR spectroscopy in either CDCl3 or DMSO-d6, depending on their solubility (Fig. 1 and 2). 1H NMR spectra of monomer 3, HBP1, HBP-P, HBP-C and HBP-G were measured in DMSO-d6 (Fig. 1). HBP-H was insoluble in DMSO-d6, and its 1H NMR spectrum was measured in CDCl3 instead (Fig. 2). First, all the signals for monomer 3 in DMSO-d6 were unambiguously assigned (Fig. 1(A)). The triplets at 4.08 and 4.22 ppm (a and b, respectively) were assigned to the methylene protons (–CH2–). All the aromatic signals were also clearly assigned (c-i). When the 1H NMR spectrum of monomer 3 was measured in CDCl3, similar peaks were observed with minor changes in the chemical shifts of the peaks (Fig. 2(A)). After the polymerization of monomer 3, the 1H NMR spectrum of the resulting HBP1 in DMSO-d6 displayed broader signals, which indicated the occurrence of polymerization (Fig. 1(B)). The protons on the ethylene bridges (a and b) displayed overlapping resonances at 4.13 and 4.00 ppm, respectively. In addition, a new signal (x) appeared at 3.36 ppm (overlapping with the water peak), which was more discernible in the 1H NMR spectrum in CDCl3 at 3.54 ppm (x, Fig. 2(B)). This signal was attributed to the OCH3 groups in the polymer structures, which were formed by quenching the intermediate tertiary OH groups (partial reactions during polymerization) with methanol.63,75 The aromatic proton signals of HBP1 were broadened and overlapped at 6.74–7.69 ppm in Fig. 1(B) and 6.45–7.65 ppm in Fig. 2(B).
 | ||
| Fig. 1 1H NMR spectra of (A) monomer 3, (B) HBP1, (C) HBP-P, (D) HBP-C, and (E) HBP-G in DMSO-d6. HBP-H was insoluble in DMSO-d6, so its 1H NMR spectrum was measured in CDCl3 and compared to that of monomer 3 and HBP1 in CDCl3 (Fig. 2). | ||
 | ||
| Fig. 2 1H NMR spectra of (A) monomer 3, (B) HBP1, and (C) HBP-H in CDCl3. The inset in (C) showed the signal for phenolic group (g). | ||
The conversion of HBP1 into HBP-P, HBP-C, HBP-G and HBP-H gave rise to characteristic phenolic proton signals at 9.39, 8.87, and 8.96 (y, Fig. 1(C)–(E)), and 9.61 ppm (y, Fig. 2(C), inset), respectively. Furthermore, the most downfield aromatic signals for HBP1 (7.42–7.69 ppm in Fig. 1(B) and 7.43–7.65 ppm in Fig. 2(B)) was not observed in the 1H NMR spectra of the phenolic HBPs (Fig. 1(C)–(E) and 2(C)), which indicated the complete consumption of precursor polymer HBP1 after the phenolic grafting reactions. Moreover, the signal corresponded to the OCH3 groups due to methanol quenching in HBP1 (x in Fig. 1(B) and 2(B)) was not observed after the phenolic functionalization. This was consistent with the fact that these phenolic HBPs were synthesized in situ from monomer 3 without methanol quenching for HBP1. The 1H NMR spectrum of HBP-G in Fig. 1(E) displayed extra broad signals at 3.49–3.64 ppm (z), which were associated with the methoxy protons in the terminal guaiacol groups. Finally, the up-field signals at 0.89, 1.27, 1.46, and 2.40 ppm in the 1H NMR spectrum of HBP-H (Fig. 2(C)) corresponded to the aliphatic protons in the terminal hydro-cardanol units (m, n, p and q, respectively).
The degree of branching (DB) of HBP1 was evaluated according to the 1H NMR signal intensities that corresponded to the linear, dendritic, and terminal units (Fig. S1, ESI†). The DB values of HBP1 was calculated as 0.51 (calculations shown in ESI†), which suggested that the carbonyl group of AB2 monomer 3 showed almost the same reactivity as its intermediate with OH group.76 This is different from some other reported isatin-based AB2 monomers, which has much higher reactivity for the second reaction step.77,78 The DB values of the phenolic HBPs were expected to remain the same as that of HBP1, because they were prepared by post-polymerization functionalization of HBP1 without changing the backbone. Unfortunately, quantification of the DB values for the phenolic HBPs was unsuccessful due to the difficulty in finding discernible signals corresponding to linear units in the 1H NMR spectra.
The chemical structures of the new HBPs were further confirmed by FTIR spectroscopy. In the high wave number region (Fig. 3(A)), a broad O–H stretching band was observed at 3570–3130 cm−1 in the FTIR spectra of all the four phenolic HBPs, which was absent in the spectra of monomer 3 and HBP1. This confirmed the presence of phenolic OH groups in the polymers, which indicated the success of grafting reactions. In the carbonyl region (Fig. 3(B)), the two carbonyl groups of monomer 3 (1 and 2, Fig. 3(B)) showed an overlapped broad signal centred at ∼1728 cm−1, which was consistent with other reported isatin derivatives.79 After the polymerization of 3, the resulting HBP1 showed an additional CO stretching band at 1715 cm−1, which corresponded to the reacted isatin moieties in the backbone (3). The unreacted terminal isatin carbonyl units (1 and 2) displayed as a shoulder in the FTIR spectrum. After the phenolic grafting reactions of HBP1, the shoulder corresponded to the terminal carbonyl groups (1 and 2) in the FTIR spectrum of HBP1 disappeared, which confirmed the complete consumption of terminal isatin moieties after phenolic grafting. In addition, a new CO stretching band appeared at 1697–1671 cm−1 in the FTIR spectra of all the phenolic HBPs, which corresponded to the carbonyl group in the resulting diaryloxindole units (4). This also independently confirmed the success of phenolic grafting.
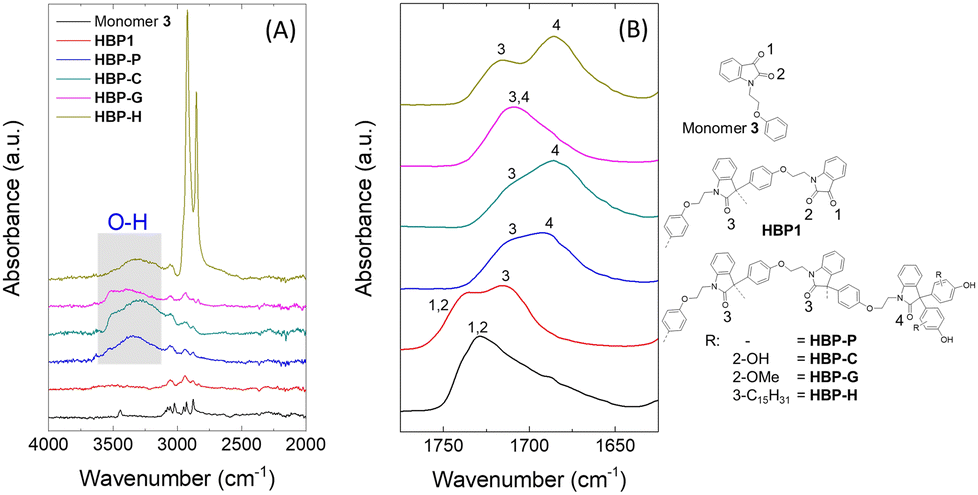 | ||
| Fig. 3 FTIR spectra of monomer 3 and HBPs in the regions of (A) 4000–2000 cm−1 and (B) 1775–1625 cm−1. Numerals 1–4 represent the different carbonyl groups. | ||
The molecular weight of HBP1 was conveniently measured by SEC in chloroform (Mn ∼ 16 kDa and Mw ∼34 kDa, Table 1). After the phenolic grafting, HBP-H with long alkyl groups remain soluble in chloroform. SEC results indicated significantly increased molecular weight of HBP-H (Mn ∼ 46.5 kDa and Mw ∼126.0 kDa) compared to its precursor HBP1, which is due to the grafted mass. The other three phenolic HBPs were insoluble or only partially soluble in chloroform or DMAc (the two commonly used SEC solvents in our group), but their soluble fraction was measured anyway to provide some insight. HBP-G was partially soluble in chloroform, and the SEC measurement of its soluble fraction showed apparently lower molecular weight than that of precursor HBP1 (Table 1), which indicated that only the low molecular weight fraction was soluble and high molecular weight fraction was insoluble and filtered off prior to SEC measurements. HBP-P was completely insoluble in chloroform, but was partially soluble DMAc. According to the SEC results in DMAc, a relatively low molecular weight was obtained (Table 1), which could also be due to the loss of insoluble high molecular weight fraction in DMAc by filtration prior to the SEC measurements. HBP-C was completely insoluble in chloroform or DMAc, which were the two solvents allowed for our SEC instruments, so its molecular weight was not measured. The SEC traces of these tested polymers are shown in Fig. S2 (ESI†).
| HBP | M n (kDa) | M w (kDa) | Đ | T g (°C) | T 5 (°C) | T d (°C) | CY (%) |
|---|---|---|---|---|---|---|---|
| HBP1 | 16.2 | 33.7 | 2.08 | 188 | 366 | 419 | 42 |
| HBP-P | 5.70 | 9.90 | 1.73 | 242 | 335 | 409 | 39 |
| HBP-C | — | — | — | 234 | 353 | 405 | 36 |
| HBP-G | 10.6 | 15.9 | 1.51 | 200 | 236 | 409 | 33 |
| HBP-H | 46.5 | 126.0 | 2.71 | 127 | 263 | 430 | 10 |
Thermal properties of the obtained HBPs were characterized by DSC and TGA measurements. According to the DSC results, the Tg value of HBP1 was 188 °C (Fig. 4 and Table 1). After phenolic grafting, the resulting HBPs with solely hydroxyl end-groups (i.e.HBP-P and HBP-C) showed higher Tg values (Tg ∼ 242 and 234 °C, respectively) due to the increased rigidity and hydrogen bonding. The Tg value of HBP-G (Tg ∼200 °C) was higher than that of HBP1, but lower than that of HBP-P and HBP-C. This could be related to the additional flexibility imparted by the methoxy groups of HBP-G, which somewhat counter-balanced the enhanced Tg by the increased rigidity and hydrogen bonding endowed by the phenolic units. When even longer aliphatic alkyl chain was incorporated (i.e.HPB-H), a rather low Tg (∼127 °C) was observed, which was caused by the lubricating effect of the flexible aliphatic alkyl chain. All the HBPs were amorphous under the conditions studied without melting endotherm in the DSC heating curves (Fig. 4).
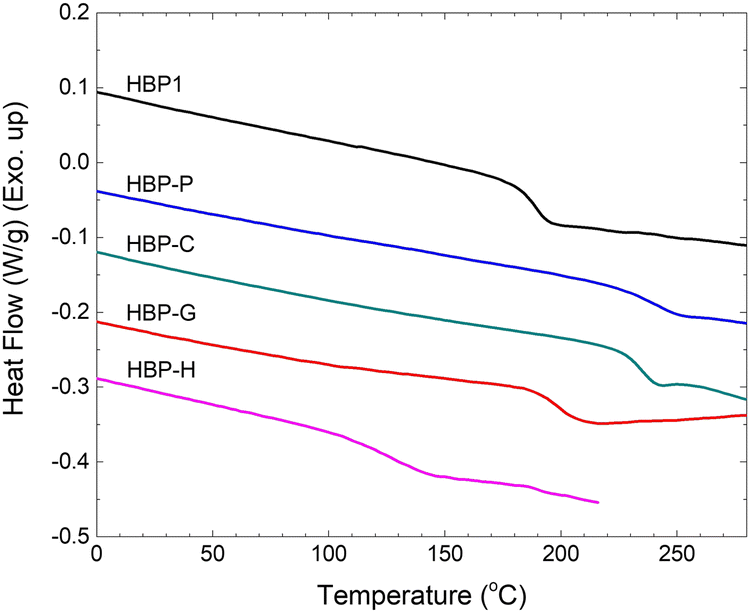 | ||
| Fig. 4 DSC curves of HBPs (second heating). | ||
All the obtained HBPs showed fairly high thermal stability according to TGA measurements (Fig. 5 and Table 1). The initial thermal decomposition temperature (T5, Table 1) for all the polymers was above 230 °C, and the temperature for the maximal decomposition rate (Td) was higher than 400 °C. In addition, the char yield (CY) at 800 °C for all the HBPs was in the range of 33–42%, except for HBP-H (CY ∼ 10%). These high CY values could be related to their high aromatic content, which suggested possible inherent flame retardance.80,81 The exceptionally low CY of HBP-H could be attributed to its relatively low aromatic content due to the presence of long alkyl units.82,83
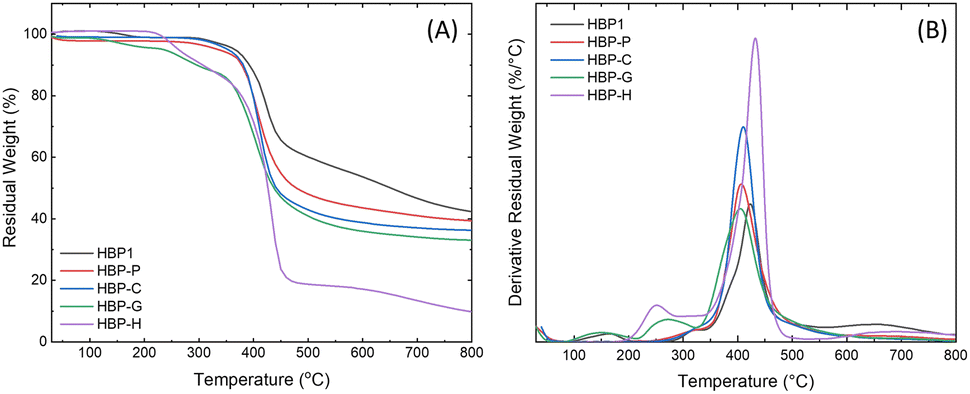 | ||
| Fig. 5 TGA (A) residual weight and (B) derivative weight loss curves of HBPs. | ||
Film casting and leaching evaluation
The short-time leaching risk of the obtained HBPs from a polymer matrix was preliminarily evaluated by measuring the UV-vis absorption of the aqueous phase after the polymer films blended with HBPs were immersed in water for 5 days. Poly(hexamethylene terephthalate) (PHT, Mn ∼ 13 kDa, Mw ∼ 21 kDa) was selected as the matrix for leaching investigations, which is softer than PET and suitable for various applications,84–86 particularly bicomponent fibres in nonwoven textiles.87 Another potential advantage of PHT is that it is synthesized using 1,6-hexanediol, which could be readily produced from various bio-resources.88,89 In this work, PHT powder was synthesized according our previously reported procedure.90,91 The thin films of pure PHT or PHT blended with HBPs were prepared according to a solution-casting protocol.92,93 The films of pure PHT or PHT with HBP1 or HBP-H were cast from their chloroform solutions (50 mg mL−1). The PHT films with HBP-P and HBP-G were cast from their HFIP solutions (50 mg mL−1). PHT film with HBP-C could not be prepared due to solubility limitations. HBP-C was only soluble in DMSO or DMF, but insoluble in many other commonly used organic solvents for film casting (e.g. chloroform, HFIP, etc.). However, PHT was insoluble in DMSO or DMF. Therefore, there was no common solvent that could dissolve both PHT and HBP-C. All the prepared films were immersed in water for 5 days, and the aqueous phase was measured by UV-vis spectroscopy. As shown in Fig. 6, none of the PHT films with HBPs (HBP1, HBP-P, HBP-G, and HBP-H) showed noticeable UV-vis absorbance, indicating their non-leachable nature from the polymer films within 5 days. On the contrary, the aqueous phase with the immersed PHT film containing monomer 3 showed significant UV-vis absorption after 5 days, indicating its leakage. This provided a preliminary insight into the non-leaching risk of the obtained polymers in pure water. In the future, it would be interesting to investigate the leaching risks under more specific chemical environment (e.g. pH, salt) for a particular application. Finally, the water contact angles of these PHT films were measured (Fig. S3, ESI†), which showed a general increase of hydrophobicity of the PHT films upon blending with HBPs. Particularly, the PHT film blended with HBP-H was the most hydrophobic, which was consistent with the presence of hydrophobic long alkyl chains in HBP-H. | ||
| Fig. 6 UV-vis absorbance spectra of the aqueous phase after the pure PHT film or PHT films containing HBPs or monomer 3 were immerged in deionized water for 5 days (photos of the films shown in Fig. S2, ESI†). UV-vis absorbance spectrum of monomer 3 in DMSO was presented as a reference. | ||
Antibacterial activity
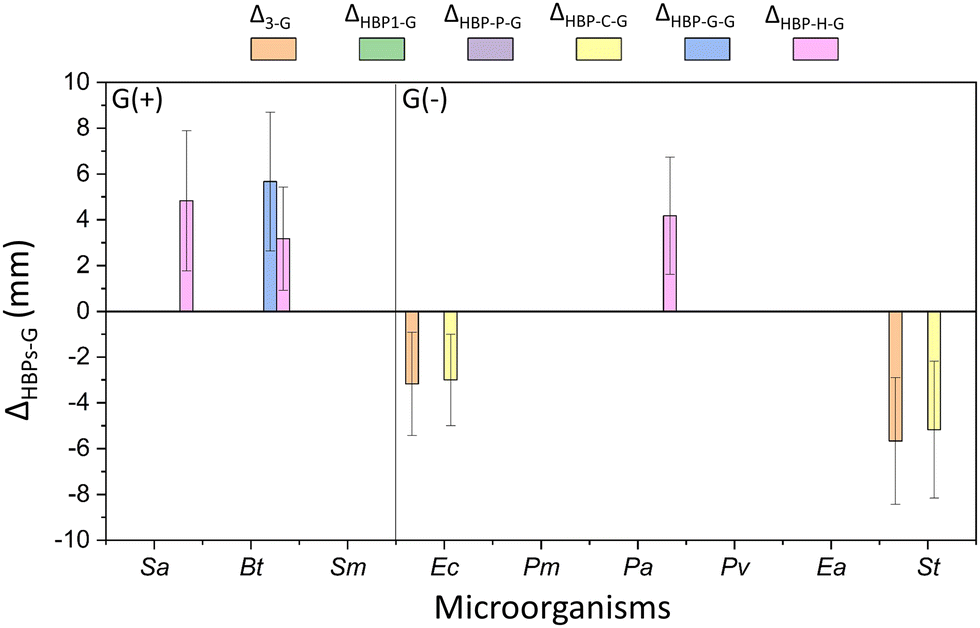
The antibacterial activity of the obtained HBPs was evaluated by a straightforward disk diffusion assay (example images in Fig. S4, ESI†). The target bacteria include six Gram-negative G(−) bacteria (Proteus mirabilis, Pseudomonas aeruginosa, Proteus vulgaris, Enterobacter aerogenes, and Salmonella typhimurium, and Escherichia coli) and three Gram-positive G(+) bacteria (Staphylococcus aureus, Bacillus thuringiensis and Streptococcus mutans). First, the isatin-functionalized polymer HBP1 and its corresponding monomer 3 were compared. As shown in Fig. S5 (ESI†), HBP1 showed significantly larger zones of inhibition than monomer 3 for two G(−) bacteria Ec and St (p < 0.05, Table S1, ESI†). For the other four tested G(−) bacteria and all the three G(+) bacteria, HBP1 showed comparable size of inhibition zones as monomer 3 (p > 0.05). One should notice that HBP1 as a polymer has considerably lower solubility and molecular diffusion rate in disk diffusion measurements, yet it showed comparable or even larger zone of inhibition compared to monomer 3. This indicated the strong antimicrobial effect of HBP1, which could be attributed to the synergistic interaction with bacteria by the densely grafted isatin groups in HBP1.63,94 It should be noted that the zones of inhibition in this work should not be simply compared to those ones we presented in our previous work about another series of isatin-functionalized polymers, due to the different solvents used for sample loading.63 The only exception is HBP-H, for which the same solvent (chloroform) was used for sample loading as in our previous work (into the paper disc prior to antimicrobial tests). In general, the inhibition zones of HBP-H were smaller than those of the isatin-functionalized polymers in our previous work. This might be related to the higher molecular weight of HBP-H, which could reduce its diffusion rate during antimicrobial experiments.26,63Next, the antibacterial effects of the obtained HBPs (including monomer 3) were compared to that of two commercial small molecular antibiotics, streptomycin and gentamicin. For simplicity, only those with significant differences (p < 0.05, Table S1, ESI†) are shown in Fig. 7 and 8. In general, monomer 3 and HBPs exhibited stronger activity (Δ > 0, p < 0.05, Table S1, ESI†) against most bacteria compared to streptomycin. There are a few exceptions where comparable effect was observed (p ≥ 0.05, Table S1, ESI†), such as for all the HBPs against G(−) bacterium Ea (shown as empty column at the corresponding places in the figure).
 | ||
| Fig. 7 Difference between the inhibition zones of the obtained polymers (or monomer 3) and streptomycin (ΔX−S, x = 3 or HBPs). Note, only those ΔX−S values with significant differences (p < 0.05, Table S1, ESI†) are shown in the figure. Positive ΔX−S values indicate more effective antibacterial effect for the investigated compounds compared with streptomycin. Absence of data column displayed in the corresponding place indicates that the inhibition zones of the monomer 3 or polymer and streptomycin were comparable without significant difference (p ≥ 0.05, Table S1, ESI†). For instance, there is no data plotted for bacterium Ea, which indicates similar antimicrobial effect for this bacterium by the obtained polymers and streptomycin. | ||
 | ||
| Fig. 8 Comparison of the inhibition zones of HBPs (or monomer 3) and gentamicin (ΔX−G, x = 3 or HBPs). Only those ΔX−G values with significant differences (p < 0.05, Table S2, ESI†) are displayed. Positive or negative ΔX−G values indicate stronger or weaker antibacterial effect for the investigated compounds compared with gentamicin, respectively. The absence of data column in the corresponding place indicates that the inhibition zones of the HBPs (or monomer 3) and gentamicin were comparable (p ≥ 0.05, Table S2, ESI†). For instance, for bacteria Pm, Pv and Ea, no data column was displayed, which indicated that for these bacteria the antimicrobial effect of the obtained polymers was comparable to that of gentamicin. | ||
The comparison of the obtained HBPs with gentamicin was more complex (Fig. 8). In most cases, no significant difference (p ≥ 0.05, Table S2, ESI†) was observed between their zones of inhibition (indicated as the absence of data column, such as for Pv). In a few cases, significantly higher (Δ > 0, p < 0.05) or lower (Δ < 0, p < 0.05) antimicrobial effects of HBPs (or monomer 3) were observed compared to that of gentamicin. For example, HBP-H was more effective (Δ > 0, p < 0.05, Table S2, ESI†) than gentamicin against two G(+) bacteria (Sa, Bt) and one G(−) bacterium (Pa). HBP-G was more effective (Δ > 0, p < 0.05, Table S2, ESI†) than gentamicin against one G(+) bacterium Bt. Monomer 3 and HBP-C were less effective than gentamicin against two G(−) bacteria Ec and St.
Furthermore, the four obtained phenolic HBPs (i.e.HBP-P, HBP-C, HBP-G and HBP-H) were compared with the isatin-functionalized HBP1 in terms of their zones of inhibition (Fig. 9). As a result, most phenolic HBPs showed comparable zones of inhibition as HBP1 (indicated by the absence of data columns in Fig. 9). Interestingly, there were three cases for G(+) bacteria, where significantly larger (Δ > 0, p < 0.05) inhibition zones were observed for phenolic HBPs. For instance, HBP-G with guaiacol moiety containing methoxy group was more effective (Δ > 0, p < 0.05, Table S3, ESI†) than HBP1 against Ec. Similarly, HBP-H with hydro-cardanol moiety containing a long alkyl group was more effective than HBP1 against Sa and Bt. These observations indicated that the presence of a methoxy or a long alkyl group could enhance the interactions with certain G(+) bacterial membranes. For certain G(−) bacteria, significantly smaller (Δ < 0, p < 0.05) inhibition zones were observed for those phenolic HBPs without ether or alkyl groups (i.e.HBP-P and HBP-C). For instance, HBP-P showed lower activity against Ec than HBP1, while HBP-C showed lower activity against both Ea and St than HBP1. This could suggest that the ether or alkyl groups of phenolic HBPs could enhance their molecular interaction with the membrane of certain G(−) bacteria, which presumably could be related to their enhanced structural hydrophobicity.64,95
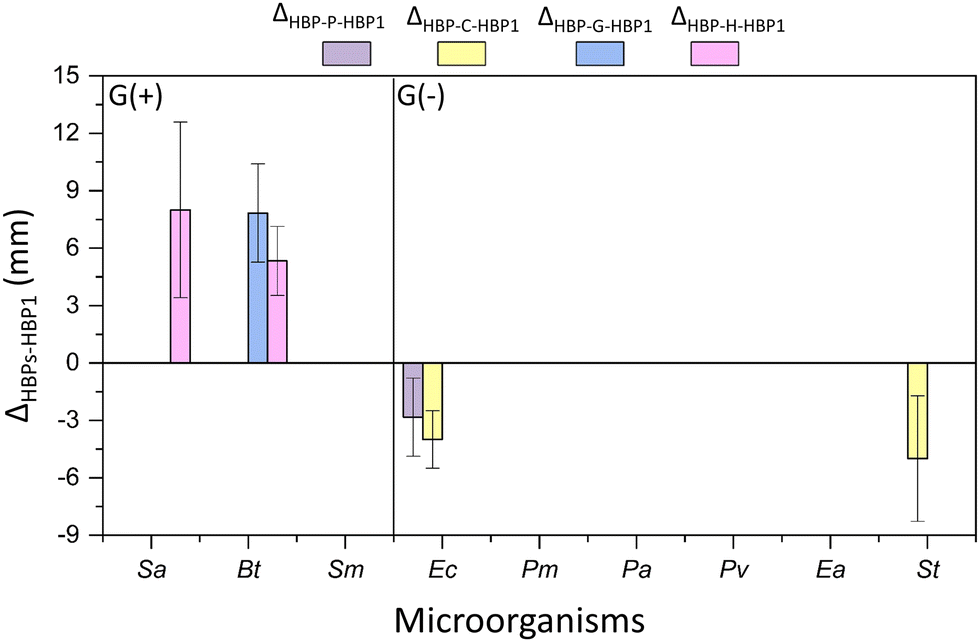 | ||
| Fig. 9 Difference between the inhibition zones of HBPs and HBP1 (ΔX-HBP-P, x = HBP-P, HBP-C, HBP-G, or HBP-H). Note, only those ΔX-HBP-1 values with significant differences (p < 0.05, Table S3, ESI†) are shown in the figure. Positive ΔX-HBP1 values indicate more effective antibacterial effect for the investigated HBPs compared with HBP1. Negative ΔX-HBP1 values indicate less effective antibacterial effect for the investigated compounds HBPs compared with HBP1. Absence of data column displayed in the corresponding place indicates that the inhibition zones of HBPs compared with HBP1 were comparable without significant difference (p ≥ 0.05, Table S3, ESI†). For instance, for bacteria Sm, Pm, Pa, Pv, and Ea, there is no data plotted, which indicates that for these bacteria the effect of the obtained polymers was very similar (no significant difference) to that of HBP1. | ||
Cytotoxicity
The cytotoxicity to MG-63 osteoblast-like human cells of the obtained monomer 3 and HBPs were evaluated using a standard MTT assay.23,96 As illustrated in Fig. 10, the cell viability of monomer 3 at 100, 500 and 1000 μg mL−1 were less than 40% (21, 24, and 31% respectively), indicating its significant toxicity to the tested cells. Compared to monomer 3, all HBPs exhibited significantly higher cell viability. For HBP-P and HBP-H, the observed cell viability values were generally in the range of 90–100%, which indicated non-toxicity. For HBP1, HBP-C and HBP-G, the observed cell viability values were higher than 100% with an increasing trend upon increased concentration. This indicated that these polymers not only were non-toxic to MG-63 osteoblast-like human cells, but also could promote their growth. Such a result was consistent with some other previously reported polymeric materials (e.g. chitosan derivatives), which could be desirable for tissue engineering and wound healing applications.97–100 To address whether the higher cell viability was related to reaction between polymers and MTT solution, control experiments were performed under the same condition without adding cells. As a result, (Fig. S6, ESI†), no significant absorbance was observed at 600 nm with all the three concentrations (as compared to the absorbance for the MTT measurements with cells), indicating no significant interaction with the MTT working solution. The exact reason for the increased cell viability remained to be unravelled. It was reported in the literature that natural polymers like chitosan could mimic the glycosaminoglycan structures in extracellular matrix, and thus could facilitate cell adhesion, migration and proliferation.99 More investigations are needed to evaluate whether synthetic HBPs may have similar effect. | ||
| Fig. 10 Cytotoxicity of monomer 3 and HBPs against osteoblast-like human cells at three concentrations (100, 500 and 1000 μg mL−1). The results were presented as relative percent viability of the treated cells compared to that of untreated control (100% of cell viability, not shown in the graph). | ||
Hemolytic activity
The in vitro toxicity on human erythrocytes of monomer 3 and all HBPs was evaluated using the Haemoscan Hemolytic Assay (HaemoScanbv, Groningen, Netherlands) according to the manufacturer's protocol.65 As shown in Fig. 11, all polymers showed low hemolytic effect (<5%) after 24 h of cultivation with the human erythrocytes, while monomer 3 showed more significant hemolytic effect (∼10%). This indicates hemocompatibility of these polymers, which may facilitate potential biomedical applications.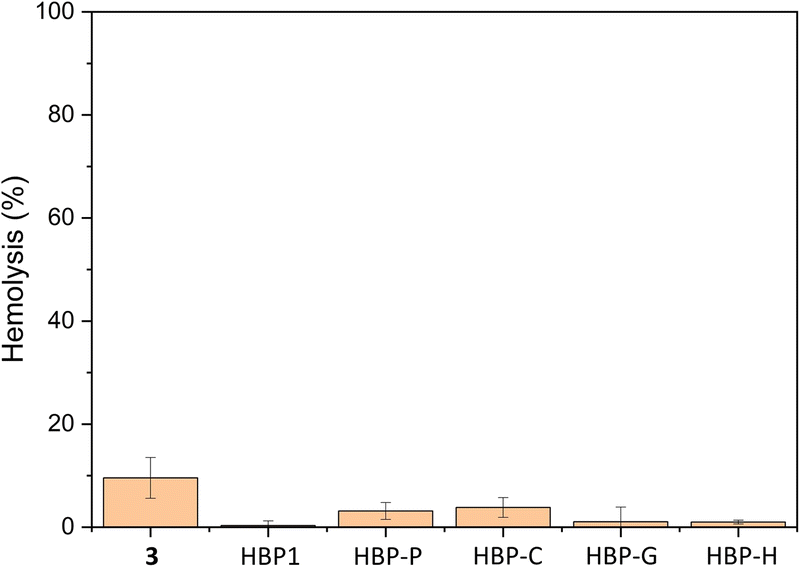 | ||
| Fig. 11 Hemolytic activity of monomer 3 and HBPs (100 μg mL−1). DMSO (1% v/v) was used as the negative control (0% hemolysis). Lysis liquid provided by the manufacture was used as the positive control (100% hemolysis). | ||
Conclusions
Four aromatic hyperbranched polymers (HBPs) with phenolic terminal groups and isatin-based backbone were synthesized via a facile solvent-free polymerization followed by functionalization with various bio-based phenolic structures. The high aromatic content of the resulting HBPs resulted in high char yields after TGA measurements, which may indicate inherent flame retardancy. As expected, the phenolic HBPs showed significant antibacterial activity against 9 different pathogenic bacteria, as well as negligible leakage from polyester films into water. Interestingly, we discovered that the presence of a methoxy or long alkyl group close to the phenolic unit could enhance the antibacterial effect for certain Gram positive or negative bacteria. Finally, these bio-based HBPs were non-cytotoxic to the MG-63 osteoblast-like human cells, neither did they show any significant hemolytic activity. These results indicated that these new polymers could be potentially suitable antimicrobial materials for various biomedical applications. In the future, synthetic work toward more structural variations is planned to better understand the structure-property relationship of this new class of sustainable antimicrobial materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financed by the ÅForsk Foundation (No. 16-479), Mistra Foundation (the “STEPS” project, No. 2016/1489), Swedish Research Council for Sustainable Development (Formas, No. 2021-01107), the Crafoord Foundation (No. 20160774 and 20180939), the Royal Physiographic Society in Lund, and Guangzhou Elite Project (GEP). We thank Sofia Essén and Katja Bernfur for mass spectrometry measurements, Niklas Warlin and Bona AB for SEC measurements, and Jingyi Rao, Sujeesh Sebastian, and Deepak Raina for valuable discussions.References
- K. Peng, T. Zou, W. Ding, R. Wang, J. Guo, J. J. Round, W. Tu, C. Liub and J. Hu, RSC Adv., 2017, 7, 24903–24913 RSC.
- W. Chin, G. Zhong, Q. Pu, C. Yang, W. Lou, P. F. De Sessions, B. Periaswamy, A. Lee, Z. C. Liang, X. Ding, S. Gao, C. W. Chu, S. Bianco, C. Bao, Y. W. Tong, W. Fan, M. Wu, J. L. Hedrick and Y. Y. Yang, Nat. Commun., 2018, 9, 1–14 CrossRef CAS.
- T. Zhu, Y. Sha, J. Yan, P. Pageni, M. A. Rahman, Y. Yan and C. Tang, Nat. Commun., 2018, 9, 917 CrossRef PubMed.
- A. L. Hook, C. Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies and M. R. Alexander, Nat. Biotechnol., 2012, 30, 868–875 CrossRef CAS PubMed.
- G. N. Tew, R. W. Scott, M. L. Klein and W. F. DeGrado, Acc. Chem. Res., 2010, 43, 30–39 CrossRef CAS PubMed.
- T. Chemistry and S. Review, Biomacromolecules, 2007, 8, 1359–1384 CrossRef.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46–71 CrossRef CAS.
- D. Raafat and H. Sahl, Microb. Biotechnol., 2009, 2, 186–201 CrossRef CAS PubMed.
- G. Li and J. Shen, J. Appl. Polym. Sci., 2000, 676–684 CrossRef CAS.
- F. Ferrero, M. Periolatto and S. Ferrario, J. Cleaner Prod., 2015, 96, 244–252 CrossRef CAS.
- V. W. L. Ng, J. P. K. Tan, J. Leong, Z. X. Voo, J. L. Hedrick and Y. Y. Yang, Macromolecules, 2014, 47, 1285–1291 CrossRef CAS.
- N. D. Koromilas, G. C. Lainioti, G. Vasilopoulos, A. Vantarakis and J. K. Kallitsis, Polym. Chem., 2016, 7, 3562–3575 RSC.
- Y. Jiao, L. Niu, S. Ma, J. Li, F. Tay and J. Chen, Prog. Polym. Sci., 2017, 7, 53–90 CrossRef PubMed.
- E.-R. Kenawy and Y. R. Abdel-Fattah, Macromol. Biosci., 2002, 2, 261–266 CrossRef CAS.
- E. F. Palermo, K. Lienkamp, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2019, 58, 3690–3693 CrossRef CAS PubMed.
- A. Kuroki, A. Kengmo Tchoupa, M. Hartlieb, R. Peltier, K. E. S. Locock, M. Unnikrishnan and S. Perrier, Biomaterials, 2019, 217, 119249 CrossRef CAS PubMed.
- B. Wang, G. Feng, M. Seifrid, M. Wang, B. Liu and G. C. Bazan, Angew. Chem., Int. Ed., 2017, 56, 16063–16066 CrossRef CAS PubMed.
- R. Costa, J. L. Pereira, J. Gomes, F. Gonçalves, D. Hunkeler and M. G. Rasteiro, Chemosphere, 2014, 112, 177–184 CrossRef CAS PubMed.
- K. Lewandowska, J. Solution Chem., 2013, 42, 1654–1662 CrossRef CAS PubMed.
- K. Izutsu and K. Shigeo, Phys. Chem. Chem. Phys., 2000, 2, 123–127 RSC.
- S. Karpagam and S. Guhanathan, Prog. Org. Coat., 2014, 77, 1901–1910 CrossRef CAS.
- X. Li, I. Sedef, J. A. Linares-pasten, Y. Liu, D. B. Raina, D. Demircan and B. Zhang, Biomacromolecules, 2021, 22, 2256–2271 CrossRef CAS PubMed.
- M. B. Patel, S. A. Patel, A. Ray and R. M. Patel, J. Appl. Polym. Sci., 2002, 89, 895–900 CrossRef.
- D. Demircan and B. Zhang, Carbohydr. Polym., 2017, 157, 1913–1921 CrossRef CAS PubMed.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS.
- W. S. Moon, K. H. Chung, D. J. Seol, E. S. Park, J. H. Shim, M. N. Kim and J. S. Yoon, J. Appl. Polym. Sci., 2003, 90, 2933–2937 CrossRef CAS.
- L. Martin, P. Gurnani, J. Zhang, M. Hartlieb, N. R. Cameron, A. M. Eissa and S. Perrier, Biomacromolecules, 2019, 20, 1297–1307 CrossRef CAS PubMed.
- R. J. Cornell and L. G. Donaruma, J. Med. Chem., 1965, 8, 388–390 CrossRef CAS PubMed.
- L. Erdmann and K. E. Uhrich, Biomaterials, 2000, 21, 1941–1946 CrossRef CAS PubMed.
- R. Jabara, N. Chronds and K. Robinson, Catheter. Cardiovasc. Interv., 2008, 72, 186–194 CrossRef PubMed.
- N. Shpaisman, L. Sheihet, J. Bushman, J. Winters and J. Kohn, Biomacromolecules, 2012, 13, 2279–2286 CrossRef CAS PubMed.
- H. Tang, C. J. Murphy, B. Zhang, Y. Shen, E. A. Van Kirk, W. J. Murdoch and M. Radosz, Biomaterials, 2010, 31, 7139–7149 CrossRef CAS.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- S. Weintraub, T. Shpigel, L. G. Harris, R. Schuster, E. C. Lewis and D. Y. Lewitus, Polym. Chem., 2017, 8, 4182–4189 RSC.
- J. J. Lucchini, J. Corre and A. Cremieux, Res. Microbiol., 1990, 141, 499–510 CrossRef CAS PubMed.
- E. Y. Jeong, J. H. Jeon, C. H. Lee and H. S. Lee, Food Chem., 2009, 115, 1006–1010 CrossRef CAS.
- H. Liu, B. Lepoittevin, C. Roddier, V. Guerineau, L. Bech, J. M. Herry, M. N. Bellon-Fontaine and P. Roger, Polymer, 2011, 52, 1908–1916 CrossRef CAS.
- K. K. Gaikwad, S. Singh and Y. S. Lee, J. Coat. Technol. Res., 2019, 16, 147–157 CrossRef CAS.
- M. C. Lubi and E. T. Thachil, Des. Monomers Polym., 2012, 3, 123–153 CrossRef.
- P. Boonsai, P. Phuwapraisirisan and C. Chanchao, Int. J. Med. Sci., 2014, 11, 327–336 CrossRef PubMed.
- T. Nonaka, Y. Uemura, K. Ohse, K. Jyono and S. Kurihara, J. Appl. Polym. Sci., 1997, 66, 1621–1630 CrossRef CAS.
- H. M. N. Iqbal, G. Kyazze, I. C. Locke, T. Tron and T. Keshavarz, Green Chem., 2015, 17, 3858–3869 RSC.
- L. Bouarab-Chibane, V. Forquet, P. Lantéri, Y. Clément, L. Léonard-Akkari, N. Oulahal, P. Degraeve and C. Bordes, Front. Microbiol., 2019, 10, 829 CrossRef PubMed.
- K. Huang, X. Fan, R. Ashby and H. Ngo, Prog. Org. Coat., 2021, 155, 106228 CrossRef CAS.
- K. Huang, R. Ashby, X. Fan, R. A. Moreau, G. D. Strahan, A. Nuñez and H. Ngo, Prog. Org. Coat., 2020, 141, 105536 CrossRef CAS.
- G. Elegir, A. Kindl, P. Sadocco and M. Orlandi, Enzyme Microb. Technol., 2008, 43, 84–92 CrossRef CAS.
- A. Nagaraja, Y. M. Puttaiahgowda, A. Kulal, A. M. Parambil and T. Varadavenkatesan, Macromol. Res., 2019, 27, 301–309 CrossRef CAS.
- E. S. Park, W. S. Moon, M. J. Song, M. N. Kim, K. H. Chung and J. S. Yoon, Int. Biodeterior. Biodegrad., 2001, 47, 209–214 CrossRef CAS.
- H. Bakhshi, S. Agarwal, T. Hayashi, N. Zhu, X. Ni, Z. Shen, F. Padella, H. Wang, J. Soliveri, R. Gómez and F. J. de la Mata, J. Mater. Chem. B, 2017, 5, 6827–6834 RSC.
- D. Konkolewicz, M. J. Monteiro and S. Perrier, Macromolecules, 2011, 44, 7067–7087 CrossRef CAS.
- D. Wang, T. Zhao, X. Zhu, D. Yan and W. Wang, Chem. Soc. Rev., 2015, 44, 4023–4071 RSC.
- P. Zhao, F. Mecozzi, S. Wessel, B. Fieten, M. Driesse, W. Woudstra, H. J. Busscher, H. C. Van Der Mei and T. J. A. Loontjens, Langmuir, 2019, 35, 5779–5786 CrossRef CAS PubMed.
- C. Z. Chen and S. L. Cooper, Biomaterials, 2002, 23, 3359–3368 CrossRef CAS PubMed.
- S. Sathiyaraj, A. Shanavas, K. A. Kumar, A. Sathiyaseelan, J. Senthilselvan, P. T. Kalaichelvan and A. S. Nasar, Eur. Polym. J., 2017, 95, 216–231 CrossRef CAS.
- P. Ortega, J. L. Copa-Patiño, M. A. Muñoz-Fernandez, J. Soliveri, R. Gomez and F. J. de la Mata, Org. Biomol. Chem., 2008, 6, 3264 RSC.
- C. Abid and S. Chattopadhyay, J. Appl. Polym. Sci., 2010, 116, 1640–1649 CAS.
- S. Charles, N. Vasanthan, D. Kwon and G. Sekosan, Tetrahedron Lett., 2012, 53, 6670–6675 CrossRef CAS.
- D. Gangadharan, N. Dhandhala and D. Dixit, J. Appl. Polym. Sci., 2012, 124, 1384–1391 CrossRef CAS.
- E. Fuentes-Paniagua, J. M. Hernández-Ros, M. Sánchez-Milla, M. A. Camero, M. Maly, J. Pérez-Serrano, J. L. Copa-Patiño, J. Sánchez-Nieves, J. Soliveri, R. Gómez and F. Javier de la Mata, RSC Adv., 2014, 4, 1256–1265 RSC.
- H. Bakhshi and S. Agarwal, Polym. Chem., 2016, 7, 5322–5330 RSC.
- J. Lin, S. Qiu, K. Lewis and A. M. Klibanov, Biotechnol. Bioeng., 2003, 83, 168–172 CrossRef CAS PubMed.
- C. R. Arza, S. Ilk, D. Demircan and B. Zhang, Green Chem., 2018, 20, 1238–1249 RSC.
- X. Li, X. Wang, S. Subramaniyan, Y. Liu, J. Rao and B. Zhang, Biomacromolecules, 2022, 23, 150–162 CrossRef CAS PubMed.
- K. V. Nemani, K. L. Moodie, J. B. Brennick, A. Su and B. Gimi, Mater. Sci. Eng., C, 2013, 33, 4453–4459 CrossRef CAS PubMed.
- H. Yu, L. Liu, H. Yang, R. Zhou, C. Che, X. Li, C. Li, S. Luan, J. Yin and H. Shi, ACS Appl. Mater. Interfaces, 2018, 10, 39257–39267 CrossRef CAS.
- H. Guo, Eur. J. Med. Chem., 2019, 164, 678–688 CrossRef CAS PubMed.
- P. Ziosi, T. Tabanelli, G. Fornasari, S. Cocchi, F. Cavani and P. Righi, Catal. Sci. Technol., 2014, 4, 4386–4395 RSC.
- Y. Teruo, I. Shigeru and I. Yoshiharu, Bull. Chem. Soc. Jpn., 1973, 46, 553–556 CrossRef.
- D. A. Klumpp, K. Y. Yeung, G. K. S. Prakash and G. A. Olah, J. Org. Chem., 1998, 63, 4481–4484 CrossRef CAS.
- J. Khan, A. Tyagi, N. Yadav, R. Mahato and C. K. Hazra, J. Org. Chem., 2021, 86, 17833–17847 CrossRef CAS PubMed.
- H. M. Colquhoun, M. G. Zolotukhin, L. M. Khalilov and U. M. Dzhemilev, Macromolecules, 2001, 34, 1122–1124 CrossRef CAS.
- M. L. Yang, Y. X. Wu, Y. Liu, J. J. Qiu and C. M. Liu, Polym. Chem., 2019, 10, 6217–6226 RSC.
- Y. Zheng, S. Li and C. Gao, Chem. Soc. Rev., 2015, 44, 4091–4130 RSC.
- J.-Y. Chen, Z.-L. Xiang, F. Yu, B. F. Sels, Y. Fu, T. Sun, M. Smet and W. Dehaen, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2596–2603 CrossRef CAS.
- C. J. Hawker, R. Lee and J. M. J. Frechet, J. Am. Chem. Soc., 1991, 113, 4583–4588 CrossRef CAS.
- M. Smet and E. Schacht, Angew. Chem., Int. Ed., 2002, 41, 4729–4732 CrossRef.
- Y. Fu, J. Chen, H. Xu, C. Van Oosterwijck, X. Zhang, W. Dehaen and M. Smet, Macromol. Rapid Commun., 2012, 33, 798–804 CrossRef CAS PubMed.
- A. Bigotto and V. Galasso, Spectrochim. Acta, 1979, 35, 725–732 CrossRef.
- X. L. Wang, T. Fu, D. M. Guo, J. N. Wu, X. L. Wang, L. Chen and Y. Z. Wang, Polym. Chem., 2016, 7, 1584–1592 RSC.
- N. Warlin, E. Nilsson, Z. Guo, S. V. Mankar, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, Polym. Chem., 2021, 12, 4942–4953 RSC.
- N. M. Barkoula, B. Alcock, N. O. Cabrera and T. Peijs, Polym. Polym. Compos., 2008, 16, 101–113 CAS.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- N. González-Vidal, A. M. de Ilarduya, S. Muñoz-Guerra, P. Castell and M. T. Martínez, Compos. Sci. Technol., 2010, 70, 789–796 CrossRef.
- N. González-Vidal, A. M. De Ilarduya, V. Herrera and S. Muñoz-Guerra, Macromolecules, 2008, 41, 4136–4146 CrossRef.
- X. Lefèbvre, M. H. J. Koch, H. Reynaers and C. David, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1–18 CrossRef.
- Z. Guo, N. Warlin, S. V. Mankar, M. Sidqi, M. Andersson, B. Zhang and E. Nilsson, ACS Sustainable Chem. Eng., 2021, 9, 16778–16785 CrossRef CAS.
- E. Kovács, G. Turczel, L. Szabó, R. Varga, I. Tóth, P. T. Anastas and R. Tuba, ACS Sustainable Chem. Eng., 2017, 5, 11215–11220 CrossRef.
- J. He, S. P. Burt, M. Ball, D. Zhao, I. Hermans, J. A. Dumesic and G. W. Huber, ACS Catal., 2018, 8, 1427–1439 CrossRef CAS.
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S. H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- P. Wang, C. R. Arza and B. Zhang, Polym. Chem., 2018, 9, 4706–4710 RSC.
- C. R. Arza and B. Zhang, ACS Omega, 2019, 4, 15012–15021 CrossRef CAS PubMed.
- R. C. Goy, S. T. B. Morais and O. B. G. Assis, Rev. Bras. Farmacogn., 2016, 26, 122–127 CrossRef CAS.
- T. Nonaka, L. Hua, T. Ogata and S. Kurihara, J. Appl. Polym. Sci., 2003, 87, 386–393 CrossRef CAS.
- X. Li, S. İlk, Y. Liu, D. B. Raina, D. Demircan and B. Zhang, Polym. Chem., 2022, 13, 2307–2319 RSC.
- B. Duan, X. Yuan, Y. Zhu, Y. Zhang, X. Li, Y. Zhang and K. Yao, Eur. Polym. J., 2006, 42, 2013–2022 CrossRef CAS.
- W. Y. Chuang, T. H. Young, C. H. Yao and W. Y. Chiu, Biomaterials, 1999, 20, 1479–1487 CrossRef CAS PubMed.
- D. O. Zamora, S. Natesan and R. J. Christy, J. Visualized Exp., 2012, 1–7 Search PubMed.
- J. Sun, Y. Fan, W. Ye, L. Tian, S. Niu, W. Ming, J. Zhao and L. Ren, Chem. Eng. J., 2021, 417, 128049 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01233b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |