
A perylene diimide-based nanoring architecture for exogenous and endogenous ATP detection: biochemical assay for monitoring phosphorylation of glucose†
Prabhpreet
Singh
 *a,
Poonam
Sharma
a,
Neha
Sharma
b and
Satwinderjeet
Kaur
b
*a,
Poonam
Sharma
a,
Neha
Sharma
b and
Satwinderjeet
Kaur
b
aDepartment of Chemistry, UGC Centre for Advanced Studies II, Guru Nanak Dev University, Amritsar 143 005, India. E-mail: prabhpreet.chem@gndu.ac.in; Tel: +91-84271-01534
bDepartment of Botanical and Environmental Sciences, Guru Nanak Dev University, Amritsar 143 005, India
First published on 24th November 2021
Abstract
Positively charged amphiphiles hold great significance in supramolecular chemistry due to their good solubility, and physiochemical and molecular recognition properties. Herein, we report the synthesis, characterization and molecular recognition properties of the dicationic amphiphile based on perylene diimide–tyrosine alkyl amide amine (PDI 3). PDI 3 showed the formation of a nanoring architecture in the self-assembled aggregated state (90% H2O–DMSO mixture) as observed by SEM and TEM studies. The diameter of the nanoring is around 30–50 nm with a height varying from 1 to 2 nm. The self-assembled aggregates of PDI 3 are very sensitive towards nucleoside triphosphates. Upon addition of ATP, PDI 3 showed a decrease in the absorbance and emission intensity at 535 and 580 nm (due to the monomer state), respectively. The lowest detection limit for ATP is 10.8 nM (UV) and 3.06 nM (FI). Upon interaction of ATP with PDI 3, the nanoring morphology transformed into a spherical structure. These changes could be attributed to the formation of ionic self-assembled aggregates between dicationic PDI 3 and negatively charged ATP via electrostatic and H-bonding interactions. The complexation mechanism of PDI 3 and ATP was confirmed by optical, NMR, Job's plot, DLS, SEM and AFM studies. PDI 3 displays low cytotoxicity toward MG-63 cells and can be successfully used for the detection of exogenous and endogenous ATP. The resulting PDI 3 + ATP complex is successfully used as a ‘turn-on’ biochemical assay for monitoring phosphorylation of glucose.
Introduction
Adenosine triphosphate (ATP), which consists of adenine as a purine ring, ribose as sugar and three negatively charged phosphate groups, is the main source of energy used by organisms and cells for performing primary and secondary metabolic reactions and for the biosynthesis of complex macromolecules.1,2 The high energy phosphate groups in ATP upon hydrolysis release a large amount of energy. The important functions of ATP include active transport of macromolecules in and out of the cells, cell signalling and signal transduction, structural maintenance, muscle contraction, synthesis of DNA and RNA and substrate phosphorylation. As ATP regulates cellular metabolism, alteration of the ATP level can be considered as an indicator for cell injury, hypoglycaemia, tumours, Alzheimer's disease, angio cardiopathy and many other diseases.3–5 Therefore, the detection of ATP is important for clinical diagnosis and biochemical purpose. In the literature, besides the bioluminescence method involving the firefly luciferase enzyme and high-performance liquid chromatography (HPLC),6 other technical methods such as surface plasmon resonance (SPR),7 colorimetry,8 fluorescence assays,9–12 surface-enhanced Raman scattering (SERS),13 chemiluminescence,14 and electrochemical15 and nuclear magnetic resonance16 are used for the determination of ATP. In contrast, the fluorescence technique acts as an easy, cost effective, time-saving method with a rapid response and high selectivity to detect analytes up to the nano- and picomolar range and is beneficial over other traditional methods.17Recent reports demonstrate that strong electrostatic interactions along with π–π, hydrophobic and H-bonding interactions can play a significant role in the recognition of ATP in aqueous medium.2 The ATP detection strategy involves (i) chemosensors complexed with metal ions such as Zn2+, Cd2+, Cu2+ and Tb3+ (known as ‘ensemble’) and (ii) chemosensors possessing complementary electronic interactions to form (ionic)self-assembly. Chemosensors with incorporated metal ion create positively charged centres for electrostatic interactions with phosphate anions of ATP, but this method requires the use of heavy metal ions.18,19 On the other hand, for the detection of ATP using the (ionic)self-assembly approach we need a positively charged building block (host) which can interact with the phosphate groups of ATP and result in changes in the photophysical and microscopic properties.20,21 We further need to add π–π, and Watson–Crick base pairing to augment selectivity amongst different nucleoside polyphosphates (NPPs).22
Perylene diimide, a polyaromatic compound which consists of large π-conjugation, is a versatile scaffold because of many unique properties such as long wavelength absorption with molar absorptivity and red to near-IR emission with high quantum yield, biocompatibility, low cytotoxicity, ease of chemical functionalization, thermal, chemical and photostability, electron accepting and having the tendency to self-assemble, are available on a single platform.23,24 In the last decade only a few examples of PDI based chemosensors for the fluorescence detection of ATP in aqueous medium (fit-for-purpose) have appeared in the literature due to the insolubility of PDIs in aqueous medium.25–27 The solubility of PDI derivatives can be increased by introducing water soluble functional moieties such as positively charged groups (polyamines, pyridinium, and sulfonates), surfactants, dendrimers and (poly)heteroatoms.28–32 In the literature, perylene diimide-based (bola)amphiphiles have been scarcely explored for their (ionic)self-assembly with surfactants or biomolecules. In 2010, Kramer's group developed N-imide functionalized probes for the detection of heparin in blood serum and plasma.33 Also, the same group in 2017 reported bay-functionalized PDI derivatives for the detection of dermatan sulfate.34 Duan and Wang's groups synthesized N-imide functionalized cationic propylamine derivative for the detection of cholesterol.35 Recently, our group also reported positively charged PDI based architects of the N-aryl benzimidazolium (BIm) group for GTP, sodium dodecyl sulfate and serum albumin protein detection in aqueous medium using the ionic self-assembled approach.36,37
To the best of our knowledge, no example is known, where tyrosine alkyl amide amine has been used for the bay-functionalization of PDIs. In continuation of our interest in developing PDI-based probes for molecular recognition,38–42 herein, we report the synthesis and characterization of bay-functionalized dicationic perylene diimide–tyrosine alkyl amide amine hybrid (PDI 3) for the detection of ATP. The lowest limits of detection were 10.8 nM and 3.06 nM using absorbance and fluorescence mode, respectively. The microscopic studies revealed the formation of the nanorings of PDI 3 alone whereas upon addition of ATP, nanoring of PDI 3 transformed to spherical aggregates. DLS studies also indicated an increase in size of the aggregates. The up-field NMR chemical shifts for PDI aromatic signals were observed upon addition of ATP to the solution of PDI 3. All these results point to the formation of ionic self-assembly between PDI 3 and ATP driven by electrostatic and H-bonding interactions. We have successfully utilized PDI 3 for (a) exogenous and (b) endogenous detection of ATP in MG-63 live cells, and (c) the PDI 3 + ATP complex in a biochemical assay for monitoring of the phosphorylation of glucose.
Experimental section
Chemicals and solvents were of reagent grade and used without further purification. All solvents used, such as dimethylformamide (DMF) and CH3OH were of HPLC grade. Coupling reagents and N,N-diisopropylethylamine (DIEA) were purchased from Spectrochem (India). TLC was performed on aluminium sheets coated with silica gel 60 F254 (Merck, Darmstadt). Chromatographic purification was done using a silica gel 60–120 mesh. Deionized water was obtained from ULTRA UV/UF Rions Lab Water System Ultra 370 series devices. Nuclear magnetic resonance (NMR) spectra were recorded at 500 MHz for 1H; 125 MHz for 13C (Bruker Avance III 500 MHz spectrometer) and 400 MHz for 1H; 100 MHz for 13C (Jeol) using CDCl3 and DMSO-d6 as solvent. The peak values were obtained as ppm (δ) and referenced to the TMS in 1H NMR and deuterated solvent in the 13C NMR spectra. Chemical shift values are reported in ppm, coupling constant (J) in Hz and abbreviations used for splitting patterns are s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublet, t = triplet, q = quartet and m = multiplet. Fourier transform infra-red (FTIR) spectra were recorded in the solid state (ATR) on a Cary 630 FTIR spectrometer (Agilent Technologies). High-resolution mass spectrometry (HRMS) data were determined on a BRUKER DALTONIK micrOTOFQ11 mass spectrometer. All theoretical calculations were carried out using the density functional theory (DFT) method at the hybrid B3LYP/6-31G* level using the Gaussian 09 package. TGA data were recorded on HITACHI STA7200 under N2. UV-Visible and emission spectra were recorded on a Shimadzu-1900 spectrophotometer (UV) and Shimadzu-RF 6000 spectrofluorometer (Shimadzu, Japan), respectively, using quartz cuvettes of 1 cm in length at room temperature. For collecting the absorbance data quartz cells of 1 cm in length were used with a fixed spectral bandwidth (2 nm) and scan rate (140 nm min−1). The following parameters were kept constant for all spectroscopic investigations such as excitation wavelength = 490 nm; excitation and emission slit widths = 10 and 10 nm, respectively. The dynamic light scattering experiment was recorded using a light scattering apparatus, Zetasizer, Nano-ZS, Nano Series, Malvern Instruments (Malvern, U.K.) at 25.0 ± 0.1 °C. The specific optical rotation values were studied at 25 °C using an Anton Paar Opto tech polarimeter (model MCP 150) using a 10 mm quartz cell in the concentration range of 0.1/100 ml–0.4/100 ml. A Metrohm Autolab PGSTAT302N electrochemical workstation was employed for recording cyclic voltammetry (CV) data of PDI 2, PDI 3 at room temperature. Three-electrodes cell system containing a Pt working electrode, Pt wire as a counter electrode and Ag/Ag+ (0.1 M AgNO3 in CH3CN) as reference electrode have been used. Atomic force microscopy (AFM) imaging was performed using a Tosca 400 instrument in tapping mode to image the 2-D and 3-D textures of surfaces. FE-SEM measurements were performed on a ZEISS SUPRA™ 55, operated at an acceleration voltage of 10 kV with a tungsten filament (electron source). Sputtering of dried samples was performed with silver prior to FE-SEM imaging. PDI–Br was prepared according to our previous reports.Procedure for NMR Titration
1H NMR titration of PDI 3 with ATP and UTP was performed in DMSO-d6 solvents on a JEOL-NMR 400 spectrophotometer. Data were processed using delta/MestreNova software to stack the spectra of PDI 3, PDI 3 + ATP at different concentrations.Procedure for the model reaction
For the model reaction, PDI 3 (1 mM) was dissolved in a 50% H2O–DMSO mixture (1 mL) and to this solution, ATP (1 equivalent) was added. The mixture was stirred for 30 min, and the precipitates formed were filtered and dried. The NMR spectra of these precipitates were recorded in the DMSO(d6) solution.Detection limit
The detection limit was calculated based on the absorbance or fluorescence titrations. To determine the signal-to-noise ratio, the absorbance/emission intensity of PDI 3 (10 μM) without ATP was measured by 3 times and the standard deviation of blank solution was determined. The detection limit (DL) was then calculated using the equation DL = 3σ/m where σ is the standard deviation of blank solution measurements; and m is the slope of the linear curve between absorbance/emission intensities of PDI 3versus ATP concentration.Cell culture and treatment
The human osteosarcoma (MG-63) cell line was obtained from the National Centre for Cell Science (NCCS, Pune, India). These cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS) and antibiotic–antimycotic solution. The cells were cultured in cell culture flasks and incubated in a humidified atmosphere at 37 °C under 5% CO2 and 95% air mixture. These cells were subcultured for further experimental purposes. The cytotoxic potential of PDI 3 towards the human osteosarcoma MG-63 cell line was determined using MTT assay. The cells were cultured at the standard density of 8 × 103 cells per well in 96 well microplates in 100 μL of DMEM medium. These cultured cells were then incubated for 24 hours to allow adherence of cells. After incubation for 24 hours, cells were treated with various concentrations of the PDI 3 samples. On completion of another 12 hours, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye solution was added in each well and incubation was continued for an additional 2 h for measuring the ability of viable cells to reduce it into purple coloured formazan. Subsequently, the supernatant MTT solution was removed and then 100 μl of dimethyl sulfoxide (DMSO) was added to dissolve the intracellular insoluble purple-coloured formazan. Then absorbance was taken at 570 nm using a multi-well plate reader (BioTek Synergy HT). The cell viability (%) was calculated using the formula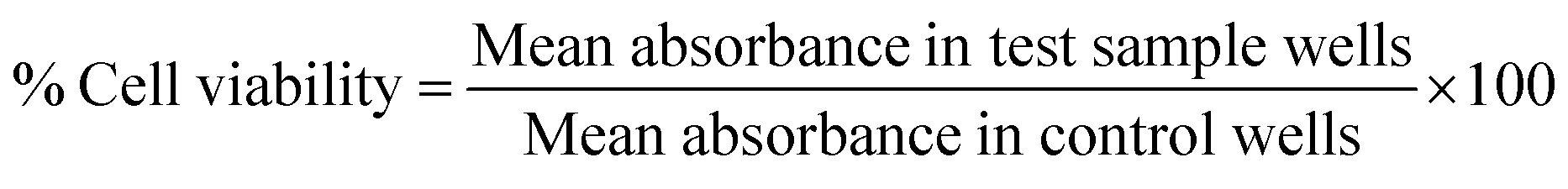
Cells of two wells were treated with PDI 3 (5 μM prepared in DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :media; 1:99); cells of four wells were treated with PDI 3 (5 μM) for 30 minutes and then these cells were treated with exogenous AMP, ADP (0.5 equivalents), ATP and UTP (0.5 and 1.0 equivalents, respectively) in duplicate and incubated at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies and two wells serving as controls. For endogenous detection of ATP, MG-63 cells were seeded in 24 well plates to make three sets (in duplicate) of cells: cells treated with PDI 3 (5 μM) as control, cells treated with Ca2+ (5 mM) and then incubated with PDI 3 and cells treated with sodium azide (NaN3; 5 mM) and then incubated with PDI 3 (5 μM) at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies. After treatment, the cells were washed two times with 1× PBS and fixed with 4% paraformaldehyde for 10–15 minutes. After this, the cells were washed with 1× PBS thrice and then the coverslips having cells were mounted on the glass slides containing a drop of anti-fading reagent (Fluoromount; Sigma). The picture observed under the fluorescence microscope (Nikon Eclipse TS2, Japan) was further analysed using software version 4.11.00 of NIS Elements AR analysis (Nikon Corporation, Japan). The synthesis of tyrosine-based alkyl amide (1) is given in the supporting information.
:media; 1:99); cells of four wells were treated with PDI 3 (5 μM) for 30 minutes and then these cells were treated with exogenous AMP, ADP (0.5 equivalents), ATP and UTP (0.5 and 1.0 equivalents, respectively) in duplicate and incubated at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies and two wells serving as controls. For endogenous detection of ATP, MG-63 cells were seeded in 24 well plates to make three sets (in duplicate) of cells: cells treated with PDI 3 (5 μM) as control, cells treated with Ca2+ (5 mM) and then incubated with PDI 3 and cells treated with sodium azide (NaN3; 5 mM) and then incubated with PDI 3 (5 μM) at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies. After treatment, the cells were washed two times with 1× PBS and fixed with 4% paraformaldehyde for 10–15 minutes. After this, the cells were washed with 1× PBS thrice and then the coverslips having cells were mounted on the glass slides containing a drop of anti-fading reagent (Fluoromount; Sigma). The picture observed under the fluorescence microscope (Nikon Eclipse TS2, Japan) was further analysed using software version 4.11.00 of NIS Elements AR analysis (Nikon Corporation, Japan). The synthesis of tyrosine-based alkyl amide (1) is given in the supporting information.
Synthesis of PDI 2
A tyrosine-based amide derivative 1 (407 mg, 0.9 mmol) and K2CO3 (125 mg, 0.9 mmol) were dissolved in the minimum quantity of DMF (3 ml). To the above reaction mixture, PDI–Br (500 mg, 0.82 mmol) was added under N2. The reaction mixture was heated at 80 °C for 3 h to complete the reaction and the reaction progress was monitored by TLC. After completion of the reaction, DMF was removed on a rotary evaporator and CHCl3 was added. The CHCl3 layer was extracted with water, brine and dried over Na2SO4 and concentrated under vacuum. The crude product obtained was further purified by column chromatography (SiO2, 3:97 EtOAc:CHCl3) to isolate the pure product PDI 2, as a red solid, Rf = 0.59 in (3:97 EtOAc:CHCl3); Yield 77%; 1H NMR (500 MHz, CDCl3, 25 °C) δ 9.49 (d, J = 8.5 Hz, 1H), 8.70–8.58 (m, 5H), 8.21 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.11 (d, J = 8.0 Hz, 2H), 6.27 (bs, 1H, –NH), 5.21 (bs, 1H, –NH), 5.09–4.98 (m, 2H), 4.74 (bs, 1H, –NH), 4.35 (bt, 1H, tyrosine –CH), 3.33–3.21 (m, 2H), 3.15–3.06 (m, tyrosine –CH2 and –CH2 of the butyl chain, 4H), 2.31–2.16 (m, 4H), 1.99–1.86 (m, 4H), 1.52–1.47 (m, 4H), 1.44 (s, 9H), 1.40 (s, 9H), 0.94–0.88 (m, 12H); 13C NMR (125 MHz, CDCl3, 25 °C) δ 171.08, 156.15, 156.04, 153.65, 134.33, 133.60, 131.63, 129.29, 128.60, 128.46, 126.97, 126.89, 125.86, 123.59, 122.39, 119.84, 80.31, 79.13, 57.84, 57.66, 51.50, 40.00, 39.17, 38.07, 33.80, 29.70, 29.08, 27.51, 26.38, 26.25, 25.03, 25.00, 11.36; IR spectrum (ATR): νmax [cm−1] = 3317.3, 2967, 2877.5, 2363.1, 2117, 1692.2, 1654.9, 1595.3, 1505.8, 1453.6, 1401.4, 1334.3, 1326.9, 1252.3, 1162.9, 939.3, 805, 745.47.
Synthesis of PDI 3
PDI 2 (100 mg, 0.102 mmol) was treated with 4 M HCl in dioxane (1 ml) and reaction was stirred for 1 h at room temperature. Reaction progress and completion was checked using TLC. After solvent evaporation, dark red precipitates separated out upon addition of diethyl ether. The precipitates were filtered to get pure PDI 3. Rf = 0.22 in 15% MeOH:CHCl3; yield = 89%; 1H NMR (500 MHz, DMSO-d6, 25 °C) δ 9.06 (d, J = 8.3 Hz, 1H), 8.82 (bt, 1H), 8.59 (dd, J = 17.5, 8.0 Hz, 2H), 8.45–8.30 (m, 5H), 8.23 (d, J = 7.5 Hz, 1H), 8.01 (s, 3H), 7.84 (s, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 4.96–4.79 (m, 2H), 4.08 (bt, 1H), 3.22–3.01 (m, 4H), 2.83–2.74 (bt, 2H), 2.22–2.04 (m, 5H), 1.96–1.80 (m, 4H), 1.64–1.56 (m, 2H), 1.55–1.44 (m, 2H), 0.91–0.80 (m, 12H); 13C NMR (125 MHz, DMSO-d6, 25 °C) δ 168.32, 155.40, 153.54, 133.51, 133.17, 132.93, 132.69, 132.40, 128.32, 127.47, 125.72, 124.84, 124.40, 123.45, 121.97, 120.25, 57.20, 54.02, 38.75, 38.32, 36.77, 26.08, 24.88, 24.87, 24.69, 11.87, 11.81; IR spectrum (ATR): νmax [cm−1] = 3056, 2959, 2922, 2870, 1692, 1654, 1587, 1505, 1401, 1326, 1252, 805, 745; HRMS-ESI calculated for C49H51N4O8+m/z 803.3659 (observed m/z 803.5146).Results and discussion
Design, structural characterization and properties of PDI 3
The bay-functionalized dicationic amphiphile PDI 3 consists of perylene diimide as the hydrophobic unit and tyrosine alkyl amide amine as the hydrophilic unit (Scheme 1). For the synthesis of PDI 3, the Boc-L-tyrosine was first condensed with tert-butyl (4-aminobutyl)carbamate using 1-ethyl-3-(3′-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl) and hydroxybenzotriazole (HOBt) coupling reagent to obtain compound 1 which was subsequently reacted with PDI–Br using K2CO3–DMF mixture to afford PDI 2 (Scheme 1). The tert-butoxycarbonyl (Boc) groups of PDI 2 were cleaved using 4 M HCl in dioxane to get PDI 3 as chloride salt. The synthesized compounds were characterized by 1H, 13C NMR (1D, 2D), mass and IR spectroscopies (Fig. S1–S3, ESI†). The 1H NMR spectrum of PDI 3 shows aromatic proton signals at δ 9.06 (d, 1H), 8.45–8.30 (m, 5H) and 8.23 (d, 1H) ppm (PDI); δ 7.33 (d, 2H) and 7.12 (d, 2H) ppm (tyrosine) along with C–H proton at δ 4.08 ppm. The mass spectrum of PDI 3 showed a peak at m/z 803.5146. | ||
| Scheme 1 Synthesis of PDI 3: reagents and conditions: (a) EDC.HCl, HOBt, DIEA, DMF, RT, N2, 4 h (b) K2CO3, DMF, 70–80 °C, 3 h; (c) 4 M HCl in 1,4 dioxane, RT, 1 h. | ||
The theoretical calculation of PDI 3 has been performed using the Gaussian 09 package using density functional theory (DFT) and time dependent density functional theory (TD-DFT) at the B3LYP/6-31G* level. The energy optimized geometry of the PDI 3 revealed twisting of the naphthalene core with a dihedral angle of ∼1.7°, whereas PDI 2 showed a dihedral angle of ∼3.1° between two-naphthalene cores. Both the highest occupied molecular orbital (HOMO, −8.94 eV) and lowest unoccupied molecular orbital (LUMO, −6.66 eV) in PDI 3 are situated on the perylene core and energy gap was −2.28 eV. Similarly, for PDI 2, HOMO (−5.85 eV) and LUMO (−3.44 eV) are situated on the perylene core and the energy gap is −2.41 eV [Fig. 1(i–ii,a–c)]. The time dependent density functional theory (TD-DFT) calculations predict λex = 553 nm for PDI 3 and λex = 529 for PDI 2 which is in consonance with experimental results (Fig. S4, ESI†). This result indicates that HOMO and LUMO levels in PDI 3 drop in energy and gain more stability in comparison to PDI 2.
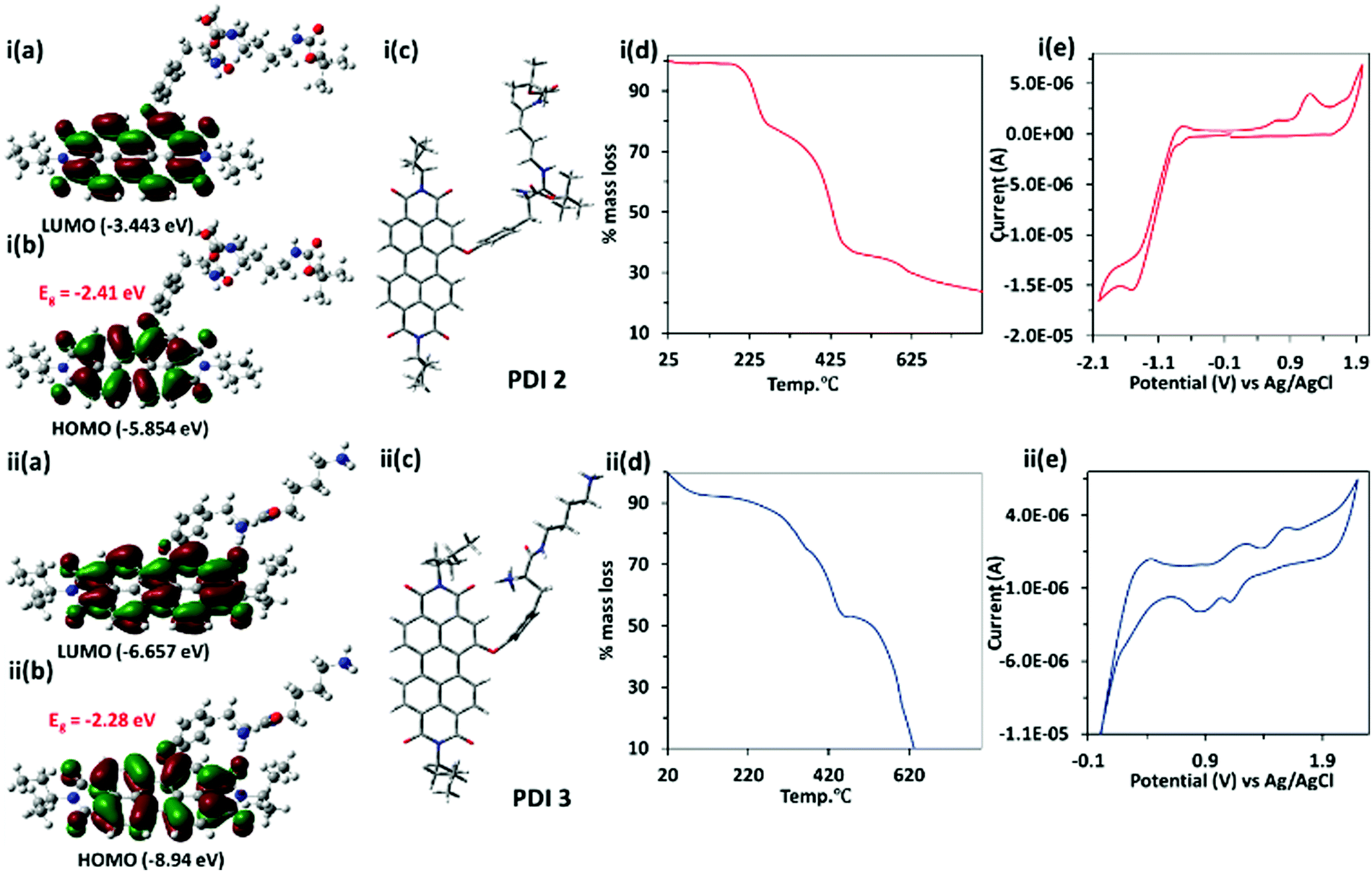 | ||
| Fig. 1 Contour plot of HOMO and LUMO and energy optimized structure (front view) of i(a–c) PDI 2 and ii(a–c) PDI 3 using DFT calculation at the B3LYP/6-31G* basis set; TGA curves of i(d) PDI 2 and ii(d) PDI 3; cyclic voltammogram (CV) of i(e) PDI 2 and ii(e) PDI 3 (0.5 mM) in CH2Cl2; supporting electrolyte 0.1 mol L−1 tetrabutylammonium perchlorate (TBAP); scan rate 0.05 V s−1. | ||
The thermal stability of perylene diimide based dicationic amphiphile PDI 3, its Boc protected derivative PDI 2 and compound 1 (as control) (Fig. S5, ESI†) were assessed using thermo-gravimetric analysis. The TGA of PDI 2 give rise to two step decompositions corresponding to loss of tert-butoxycarbonyl (Boc) groups at <240 °C (∼18% mass loss) and the tyrosine alkyl amide chain at 330–430 °C (∼40% mass loss). In the case of PDI 3, only one step mass loss (∼50%) at <450 °C was observed [Fig. 1(i–ii,d)]. This could be attributed to the fact that PDI 3 is devoid of any tert-butoxycarbonyl (Boc) groups. The mass loss at >450 °C in the TGA graph of both PDI 2 and PDI 3 corresponds to the decomposition of the PDI core.
The cyclic voltammetric (CV) data for PDI 2 and 3 have been recorded using three-electrode cell systems containing a Pt working electrode, Pt wire as the counter electrode and Ag/Ag+ (0.1 M AgNO3 in CH3CN) as the reference electrode. The CV studies of PDI 2 and PDI 3 showed reversible oxidation and reduction peaks. The reduction potential of PDI 3 is more positive with value ca. 1.25 V and 0.7 V in comparison to PDI 2 (−1.4 V). The more positive value of reduction potential of PDI 3 correlates with the dicationic nature of PDI 3. The onset reduction and oxidation potential peaks were utilized to calculate HOMO and LUMO energy levels using the equation EHOMO = −[Eox(onset) + 4.4] eV and ELUMO = −[Ered(onset) + 4.4] eV. The electrochemically (CV) calculated energies corresponding to HOMO and LUMO levels are −8.47 eV and −6.35 eV, respectively, which are in close agreement with DFT data [Fig. 1(i–ii,e)]. The specific optical rotation ([α]D25) of PDI 2 is +27.5° (25 °C, path length: +10 mm; concentration: 4 mg/100 mL; solvent: CHCl3), whereas specific optical rotation of PDI 3 is slightly changed to +25.0° (Fig. S6, ESI†).
Spectroscopic and pH studies of PDI 3
The absorbance spectrum of PDI 3 recorded in DMSO showed absorption bands at 535 and 503 nm corresponding to 0–0 and 0–1 vibronic level transition for the So–S1 electronic level. In DMSO, PDI 3 exhibits normal Franck–Condon ratio (A0–0/A0–1) ∼1.42 which indicates that it exists in the molecularly dissolved state (monomer state). As soon as the fraction of water added in the DMSO solution of PDI 3, we observed a continuous decrease in the A0–0/A0–1 ratio and the value of the A0–0/A0–1 ratio ca. 0.83 was reached at 90% H2O–DMSO mixture (Fig. 2a). So, in >80% H2O–DMSO mixture, the lowest value of A0–0/A0–1 ratio indicates that PDI 3 exists in an aggregated state. In DMSO, no significant aggregation was observed (degree of aggregation, αagg = 0), whereas the αagg value increased to 0.97 in the >80% H2O–DMSO mixture. The value of ΔG* steadily increased from −0.28 (DMSO) to −0.11 (90% H2O–DMSO). In consonance with this aggregation process, we observed a broad tail of the absorbance band in the range 560–615 nm in 90% H2O–DMSO. In contrast, we observed that for PDI 2, which is a Boc protected (neutral) analogue of PDI 3, the Franck–Condon ratio (A0–0/A0–1) ∼ 1.44 in 0–10% H2O–DMSO abruptly decreased to 0.80 in 30% H2O–DMSO and then a plateau was achieved. The αagg follows the same trend with the value increased from 0 (0–10% H2O–DMSO) to 0.94 (30% H2O–DMSO). It means that PDI 3 due to its cationic nature is more soluble in water and it tends to aggregate slowly in comparison to the more hydrophobic PDI 2 (Fig. S7–S8, ESI†). | ||
| Fig. 2 (a) Absorption and (b) emission changes of PDI 3 (10 μM) after the incremental addition of 10 vol% H2O in DMSO; [Inset of (a)] (top) Plot of Franck–Condon (A0–0/A0–1) values; (bottom) Plot of the degree of aggregation (αagg) versus water fraction in DMSO; [Inset of (b)] Magnified view of the fluorescence spectra shown in (b). | ||
The fluorescence spectra of PDI 3 recorded in DMSO and in the 0–90% H2O–DMSO mixture, matched the absorption data, where λem at 580 nm (corresponding to the monomer state) in DMSO slowly decreased as the fraction of water in DMSO increased to 0–90%. In the 90% H2O–DMSO mixture, a new broad band in the range 550–700 nm (λem = 630 nm) was observed which corresponds to the aggregated state of PDI 3. This decrease in fluorescence intensity in 90% H2O–DMSO is referred to as aggregation caused quenching (ACQ) (Fig. 2b).
The negative charge on the phosphate groups of ATP (guest) and positive charge on the ammonium groups of PDI 3 (host) are pH dependent and it is important to consider a suitable range of pH to maximize the ion-pairing (electrostatic) interactions between the charged functional groups of ATP and PDI 3. ATP is stable in aqueous solutions between pH 6.8 and 7.4.43 So, we carried out only pH titration of PDI 3 to determine the pH range for studying the complexation of PDI 3 with ATP. The pH-dependent fluorescence spectra of PDI 3 measured in 50% H2O–DMSO mixture reveal that as the pH of the solution decreases (pH < 7), the emission intensity at 580 nm increases (Fig. S9, ESI†). On the other hand, as the pH of the solution increases (pH > 7), the emission intensity at 580 nm decreases and at 635 nm increases. This could be attributed to the fact that under basic conditions (pH > 7), ammonium groups are deprotonated (charge neutralization) which reduces electrostatic repulsion between ammonium groups and consequently the π–π stacking interactions of perylene chromophore enhanced due to the close approach of PDI 3 molecules. This resulted in the decrease of the monomer intensity at 580 nm and increase in the emission intensity of aggregates at 635 nm. However, under acidic conditions, opposite relations exist because the accumulation of more positive charge on ammonium groups increases the electronic repulsions between perylene chromophore and PDI 3 mostly exist in the monomer state. So, the complexation studies of PDI 3 with ATP have been carried out in a 50% HEPES buffered–DMSO mixture at pH 7.
ATP recognition with PDI 3
In preliminary studies, we have added different nucleoside polyphosphates (NPPs) such as AMP, CMP, GMP, UMP (monophosphates); ADP, CDP, GDP, UDP (diphosphates), ATP, CTP, GTP, UTP (triphosphates), H2PO4−, HPO42−, PO43−, P2O74−, CH3COO−, NO3−, SO42−, CO32−, S2− ions (0.5 equiv.) in PDI 3 (10 μM for UV-Vis and 5 μM for FI, pH 7.0) and optical spectra were recorded. It was found that PDI 3 showed marked decrease of absorbance and emission intensity with addition of ATP and UTP only. The magnitude of fluorescence quenching of PDI 3 by triphosphates is higher (65–70%) in comparison to fluorescence quenching by diphosphates (12–18%) and monophosphates (6–11%) (Fig. S10 and S11, ESI†).Next, we studied the interaction of PDI 3 with ATP using absorbance and fluorescence spectroscopy. The successive addition of ATP (0–40 μM) to the 50% HEPES buffered solution of PDI 3 (10 μM, pH 7.0), resulted in a gradual decrease in the absorption intensity at 535 and 515 nm, corresponding to vibronic levels of 0–0 and 0–1 for S0–S1 transition (Fig. 3a and b). A plateau was achieved upon addition of 1 μM of ATP (0.1 equivalent). The Franck–Condon ratio (A0–0/A0–1) drastically changed from ca. 1.02 (PDI 3) to 0.8 (PDI 3 + ATP). A visible colour change in PDI 3 from pink to light pink upon addition of ATP was observed (Fig. 3b, Inset). The absorbance ratio (A/Ao) plot drawn against [ATP] shows two linear response curves between 0–5 × 10−8 M (R2 = 0.9968) and 5–60 × 10−8 M (R2 = 0.9826). The limit of detection of PDI 3 towards ATP was found to be 10.8 nM (DL = 3σ/m). This aggregation could be attributed to the formation of (ionic) self-assembled aggregates via electrostatic and H-bonding interactions between positively charged PDI 3 and negatively charged ATP.
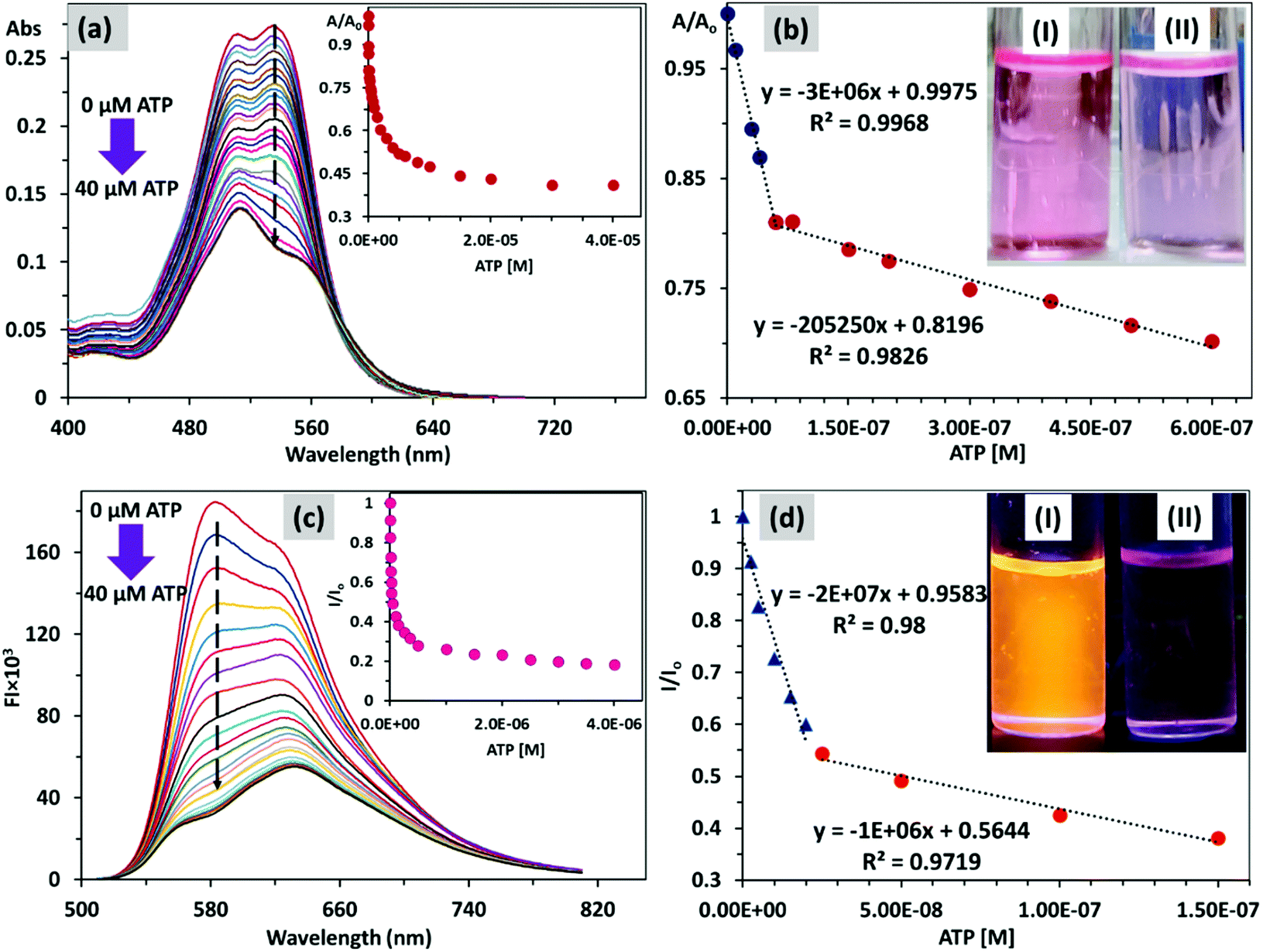 | ||
| Fig. 3 Changes in (a) absorbance (10 μM) and (c) emission (5 μM) spectra of PDI 3 with gradual increase of ATP concentrations in 1:1 HEPES buffer:DMSO medium; (λex = 490 nm); slit width (Ex/Em) = 10/10 nm; [Inset of (a,c)] plot of absorbance (A/Ao) and emission (I/Io) changes with change in ATP concentration, respectively; Linear correlation plot of (b) A/Ao and (d) I/Ioversus the concentration of ATP to determine the lowest limit of detection; [Inset of (b,d)] visible and fluorescence (under UV lamp, λ = 365 nm) colour change of PDI 3 (I) before and (II) after addition of ATP. | ||
We also observed that addition of ATP resulted in a dramatic change in the fluorescence spectrum of PDI 3 (5 μM). Upon addition of ATP, the emission intensity of the PDI 3 at 580 nm gradually decreases (emission band due to the monomer state), whereas small shoulder peaks at 635 nm in PDI 3 gradually become prominent (emission band due to the aggregated state). The saturation point was reached upon addition of 0.5 μM (0.1 equivalent) of ATP and fluorescence quenching reached 82.5% (Fig. 3c and d). A plot of emission intensity ratio (I/Io) against [ATP] provides a linear correlation at very low concentrations 0–2.5 × 10−8 M and 2.5–15 × 10−8 M. The limit of detection using (DL = 3σ/m) was calculated as 3.06 nM. The Stern–Volmer (Ksv) quenching constant for interaction of the PDI 3 with ATP was found to be 1.96 × 107 M−1. The fluorescence quantum yield for complexation of PDI 3 with ATP was found to decrease from 45% (in PDI 3) to 13% (PDI 3 + ATP). The Benesi–Hildebrand (B–H) plot of the emission data reveals the formation of a 1:1 (PDI 3 + ATP) stoichiometric complex with binding constants of 6.98 × 107 M−1 and 1.05 × 107 M−1. Job's plot analysis revealed formation of 1:1 (PDI 3:ATP) stoichiometry with a binding constant value of logβ 7.3 ± 0.4 (1:1, PDI 3:ATP) (Fig. S12, ESI†).
The UV-Vis and fluorescence titrations of PDI 3 with ATP were also performed in 90% HEPES buffered solution where PDI 3 exists in the aggregated state. The results showed that PDI 3 interacts strongly with ATP with decrease in absorbance and emission intensities at 511 and 635 nm (aggregated state). The limits of detection were 1.89 nM (UV-Vis) and 5.21 nM (FI). The plot of A/Ao (absorbance data) and I/Io (fluorescence data) against [ATP] shows linear correlation at low concentrations such as 0–20 nM and 0–5 nM (Fig. S13, ESI†). As a control, response of tyrosine towards ATP was also studied which showed no change upon addition of 1 equivalent of ATP (Fig. S13a, ESI†).
Subsequently, we have also performed fluorescence titration of PDI 3 in the presence of other triphosphate anions, including CTP, GTP and UTP. A response towards these phosphates with the magnitude of fluorescence quenching follows the order of ATP (85.3%) > UTP (73.6%) > GTP (63.5%) ∼ CTP (59%) (Fig. 4). The comparison of ATP with ADP and AMP follows the order of ATP (85.3%) ≫ ADP (24.1%) > AMP (13.5%). This selectivity of PDI 3 for ATP in comparison to ADP/AMP is an important implication because the modulation in ATP/ADP ratio in cells is required for maintaining cellular events. To check the photobleaching, 50% HEPES buffered–DMSO solution of PDI 3 was exposed to a 300 W xenon lamp for 1 h and then fluorescence intensity was recorded. The loss of fluorescence intensity was <10%.
 | ||
| Fig. 4 Fluorescence point graph of PDI 3 showing the effect of addition of different nucleoside polyphosphates recorded in HEPES buffer–DMSO (1:1, pH 7.0) solution. | ||
NMR titration of PDI 3 with ATP
The (ionic)self-assembly based interaction between PDI 3 and ATP was further evidenced by 1H NMR titration in DMSO-d6 (Fig. 5). The perylene diimide aromatic protons at δ 9.46, 8.99–9.05, 8.64–8.55, 8.05 ppm and tyrosine phenyl protons at δ 7.43, 7.33 ppm of PDI 3 showed up-field shifts to new positions at δ 9.26, 8.75, 8.49–8.35, 7.88 ppm and δ 7.39, 7.18 ppm, respectively, upon addition of 0.1 equivalents of ATP. Moreover, broad singlet protons at δ 8.05, 7.82 ppm due to NH3+ groups completely disappeared upon addition of 0.5 equivalents of ATP with concomitant broadening of all the signals in the NMR spectrum. | ||
| Fig. 5 Partial 1H NMR spectra (stacked) of PDI 3 (1 mM) after incremental addition of ATP in DMSO-d6 solution. | ||
Similar up-field shifts of perylene diimide aromatic protons and tyrosine phenyl protons were observed in the NMR spectrum of PDI 3 (1 mM) upon addition of UTP in DMSO-d6 solution (Fig. S14, ESI†). This signal broadening and up-field chemical shift of aromatic protons is consistent with strong electrostatic and H-bonding interactions of PDI 3 with ATP leading to the formation of ionic self-assembled aggregates. To further validate these chemical shift changes, we have isolated the PDI 3 + ATP complex and recorded its 1H NMR spectrum in DMSO-d6 solution. The up-field shift of the aromatic signals along with the disappearance of NH3+ signals was clearly observed (Fig. 5, top spectra).
Dynamic light scattering and microscopic studies
The formation of (ionic) self-assembled supramolecular aggregates between PDI 3 and ATP was further confirmed by dynamic light scattering (DLS), scanning electron microscopy (SEM) and atomic force microscopy (AFM) techniques (Fig. 6). The DLS data of PDI 3 alone and those after addition of ATP were recorded in a 90% H2O:DMSO mixture and are presented in Fig. 6a. We observed that the hydrodynamic size of the PDI 3 is in the range of 10–20 nm with a polydispersity index of 0.3. Upon addition of ATP, the size of the PDI 3 aggregates increased drastically, the diameter of these aggregates are in the range of 200–350 nm and polydispersity index remains 0.18 (Fig. 6d) which indicates that binding of ATP with the tyrosine alkyl amide amine chain of PDI 3 triggered ion-pairing interactions via ionic self-assembly. The dicationic (positively charged) nature of PDI 3 was further confirmed by recording the zeta (ζ) potential measurements. The ζ-potential of PDI 3 was calculated to be 24 ± 0.7 mV. Upon addition of ATP to the solution of PDI 3, the ζ-potential value decreases to 6.5 ± 0.2 mV. This result is in consonance with DLS data and could be attributed to neutralization of positive charge of PDI 3 with negatively charged phosphate groups through ion-pairing.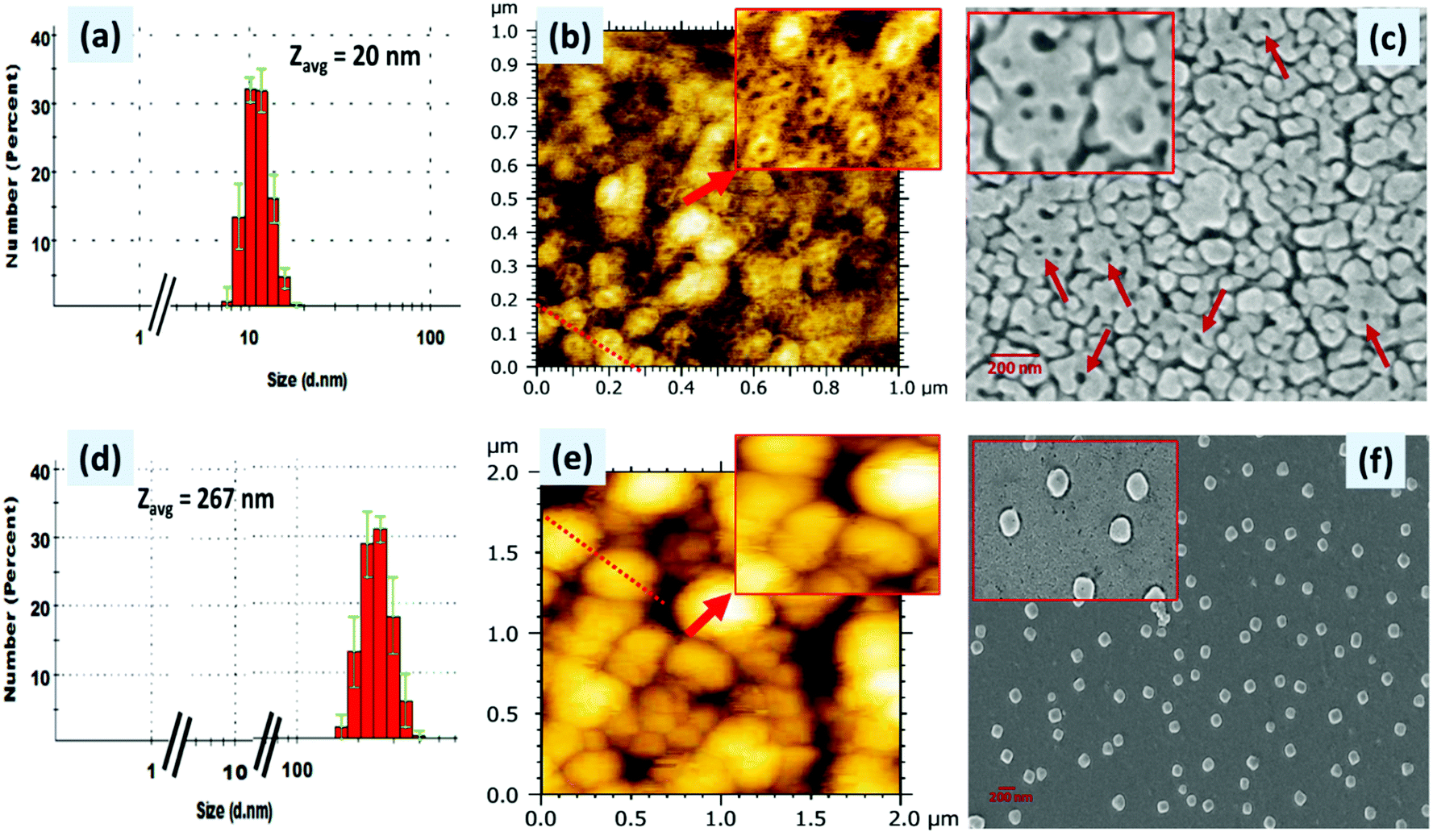 | ||
| Fig. 6 (a) DLS data (b) AFM and (c) SEM images of PDI 3 alone and (d) DLS data (e) AFM and (f) SEM images of PDI 3 after addition of ATP in 90% H2O–DMSO solution; [Inset of (b,c and e,f)] magnified view of nanoring of PDI 3 and spherical morphology of PDI 3 + ATP. | ||
These DLS data were further supported by AFM and SEM studies (Fig. 6). The samples for AFM and SEM studies were prepared in a 90% H2O: DMSO mixture and deposited onto a clean glass slide using the drop-casting method. In AFM images, it was observed that PDI 3 self-assembled into a supramolecular nanoring like architecture all over the surface with the thickness of the nanoring ranging from 2.2 to 3.8 nm. The diameter of the regular nanoring is in the range of 30–50 nm and its height lies between 1 and 2 nm. The analysis of the cross-sectional height and the diameter profile of these AFM images revealed that the diameter width of the internal empty cavity of a single nanoring is around 45 nm. Multiple nanorings were seen placed on the surface without any definite pattern. We also deposited 50% H2O–DMSO solution of PDI 3 on the glass surface and observed the partial formation of a nanoring on the surface (Fig. S15, ESI†). In comparison to AFM, the SEM micrographs showed instead globular and unresolved nanoring-like features with large thickness although the diameter is in the range of 32–43 nm (Fig. 6b and c). The diameter of the internal cavity is drastically decreased to 15–20 nm. Probably, the nanoring structure was formed in the aggregated state of PDI 3 and π–π interactions between the PDI aromatic ring and H-bonding play important role.
Interestingly, upon addition of ATP into 90% H2O–DMSO solution of PDI 3, the AFM images showed the transformation of the nanoring morphology into a spherical morphology all over the surface. The diameter of these spherical aggregates is in the range of 100–280 nm in consonance with DLS data (Fig. 6d–f). The cross-sectional height and diameter analysis revealed spherical aggregates with a height varying from 20 to 40 nm. These results are further confirmed by the SEM data. The SEM micrographs revealed segregated spherical structure all over the surface. The diameter of these spherical structures is in the range of 110–130 nm. This is probably due to the impact of formation of self-assembled aggregates wherein the repulsive interaction between PDI 3 and ATP helps them to arrange in a nanoring structure. Upon addition of ATP, charge neutralization favours the close approach of PDI 3 with ATP and compact (ionic) self-assembled aggregates in the form of spheres are observed.
Exogenous and endogenous cellular imaging of ATP
We next applied PDI 3 for cellular imaging of ATP (exogenous and endogenous) in the live cells (Fig. 7,8). The MTT assay was performed on human osteosarcoma (MG-63) live cells to check the cytotoxic tolerance of PDI 3. We observed that within the concentration range of 0–12 μM of PDI 3, >85% MG-63 cells remain viable after incubation for 12 h. The IC50 value is 50 μM. It means that 12 μM concentration of the PDI 3 is slightly toxic towards the cell; however, at the working concentration of 5 μM of PDI 3, we observed >92% cell viability (Fig. S16, ESI†). | ||
| Fig. 7 Fluorescence microscopic images of NPPs in MG-63 live cells (a–b) fluorescence and brightfield image of MG-63 cells incubated with PDI 3 alone; fluorescence images of MG-63 cells first treated with PDI 3 and then incubated with (c) AMP, (d) ADP, (e–f) ATP and (g–h) UTP for 30 minutes; [Inset of (a,c–e,g)] showing a magnified view of MG-63 cells. (Scale bar: 10× and 20×). | ||
 | ||
| Fig. 8 (a) Brightfield image of MG-63 live cells; Fluorescence images of (b) MG-63 live cells incubated with PDI 3 (5 μM) for 30 minutes; MG-63 live cells first incubated with (c) NaN3 (5 mM) and (d) Ca(NO3)2 (5 mM) and then incubated with PDI 3 for 30 minutes. | ||
Fig. 7a and b shows the fluorescence images of MG-63 cells incubated with PDI 3 (working concentration = 5 μM) and brightfield image of MG-63 cells. Bright red fluorescence was observed emitting from the cytoplasm of the cells, indicating that PDI 3 successfully penetrated the cell membrane. The cell penetration might come from intrinsically aromatic nature of the perylene diimide (hydrophobic) and the presence of dicationic ammonium groups (hydrophilic). The morphology and shape of the cells remain intact as seen in the brightfield image. We further performed selective exogenous detection of ATP amongst different NPPs (AMP and ADP) in MG-63 cells. For this purpose, MG-63 cells were seeded in 24 well plates to make four sets of cells: cells treated with exogenous AMP and ADP (0.5 equivalent), cells treated with exogenous ATP and UTP (as control) (0.5 and 1.0 equivalents) and incubated at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies.
We observed that the fluorescence image of MG-63 cells treated with PDI 3 gives a bright red fluorescence which hardly changed upon addition of 0.5 equivalent of AMP whereas the addition of ADP resulted in a slight change in the fluorescence intensity (Fig. 7c and d). On the other hand, fluorescence image of MG-63 cells treated with PDI 3 gives a bright red fluorescence which gets completely quenched upon addition of ATP (0.5 equivalents) (Fig. 7e and f). In comparison to ATP, UTP at the same concentration resulted in less fluorescence quenching in the cells (Fig. 7g and h).
Encouraged by the higher affinity and good selectivity of PDI 3 towards ATP amongst different NPPs, next we applied PDI 3 to monitor the intracellular (endogenous) ATP level in MG-63 live cells (Fig. 8). For this purpose, MG-63 cells were seeded in 24 well plates to make three sets (in duplicate) of cells: cells treated with PDI 3 (5 μM) as control, cells treated with Ca2+ (5 mM) and then incubated with PDI 3 and cells treated with sodium azide (NaN3; 5 mM) and then incubated with PDI 3 (5 μM) at 37 °C for 30 min in a CO2 incubator prior to cell imaging studies. As reported in the literature, NaN3 inhibit cytochrome oxidase that prevent mitochondrial oxidative phosphorylation, resulting in an abrupt depletion of the intracellular ATP level.44 In contrast, Ca2+ ions stimulate dehydrogenase activity that enhances O2 flux in the mitochondria, thereby enhancing respiration and, consequently, ATP and NADH levels.45
As shown in Fig. 8c and d, we observed that the fluorescence image of MG-63 cells treated with NaN3 followed by incubation with PDI 3 gives a bright red fluorescence which hardly changed while MG-63 cells which were treated with Ca2+ ions followed by incubation with PDI 3 showed 80–85% decrease in the fluorescence intensity of PDI 3, as compared to control. This means that PDI 3 can be selectively used for the sensitive detection of the endogenous ATP level.
Biochemical assay for monitoring phosphorylation of glucose
In biochemistry, phosphorylation of proteins is an important step for many cellular processes such as regulation of protein function and signals transmittance throughout the cells. Moreover, in the literature, the hexokinase method, which involves phosphorylation of glucose, is a tedious lab protocol for the detection of blood glucose. Several papers on electrochemical glucose sensors and on the development of chemical tools to monitor the activity of protein kinases are known in the literature. Moreover, commercially available kits such as BioProfile Analysers (Nova Biomedical), Biochemistry Analyzers (YSI Life Sciences) and Seahorse extracellular flux analyser (Seahorse Bioscience) can measure glucose and lactate in cell culture media but these involve tedious procedures.46 So, the development of an easy to use real time biochemical assay for monitoring phosphorylation of glucose as an indirect method for glucose detection is important.We have successfully used PDI 3 + ATP complex in a fluorescence ‘turn-on’ biochemical assay for monitoring the phosphorylation of glucose in 50% H2O–DMSO solution. The assay mixture contained PDI 3 (5 μM), glucose (5 μM), ATP (0.5 equivalent) and hexokinase enzyme (20 U mL−1). The pH of the solution assay is 7. The PDI 3 + ATP complex showed weak fluorescence at 580 nm due to the formation of (ionic) self-assembled aggregates (vide supra). The enzyme hexokinase catalyses the phosphoryl group transfer reaction between glucose and ATP. The phosphorylation of glucose results in the formation of glucose-6-phosphate and ADP or AMP. The conversion of ATP to AMP would result in the dissociation of PDI 3 + ATP ionic self-assembled aggregates and PDI 3 would be released in the solution in the monomer state that ultimately leads to fluorescence enhancement at 580 nm.
The real time change in fluorescence intensity at 580 nm as a function of time is depicted in Fig. 9. The PDI 3 (5 μM) showed high fluorescence at 580 nm in 50% H2O–DMSO solution. Upon addition of glucose (5 μM), a minor decrease (<8%) in the fluorescence intensity was observed. Subsequent addition of ATP (2.5 μM) caused a drastic decrease (> 77%) in the fluorescence intensity of PDI 3.
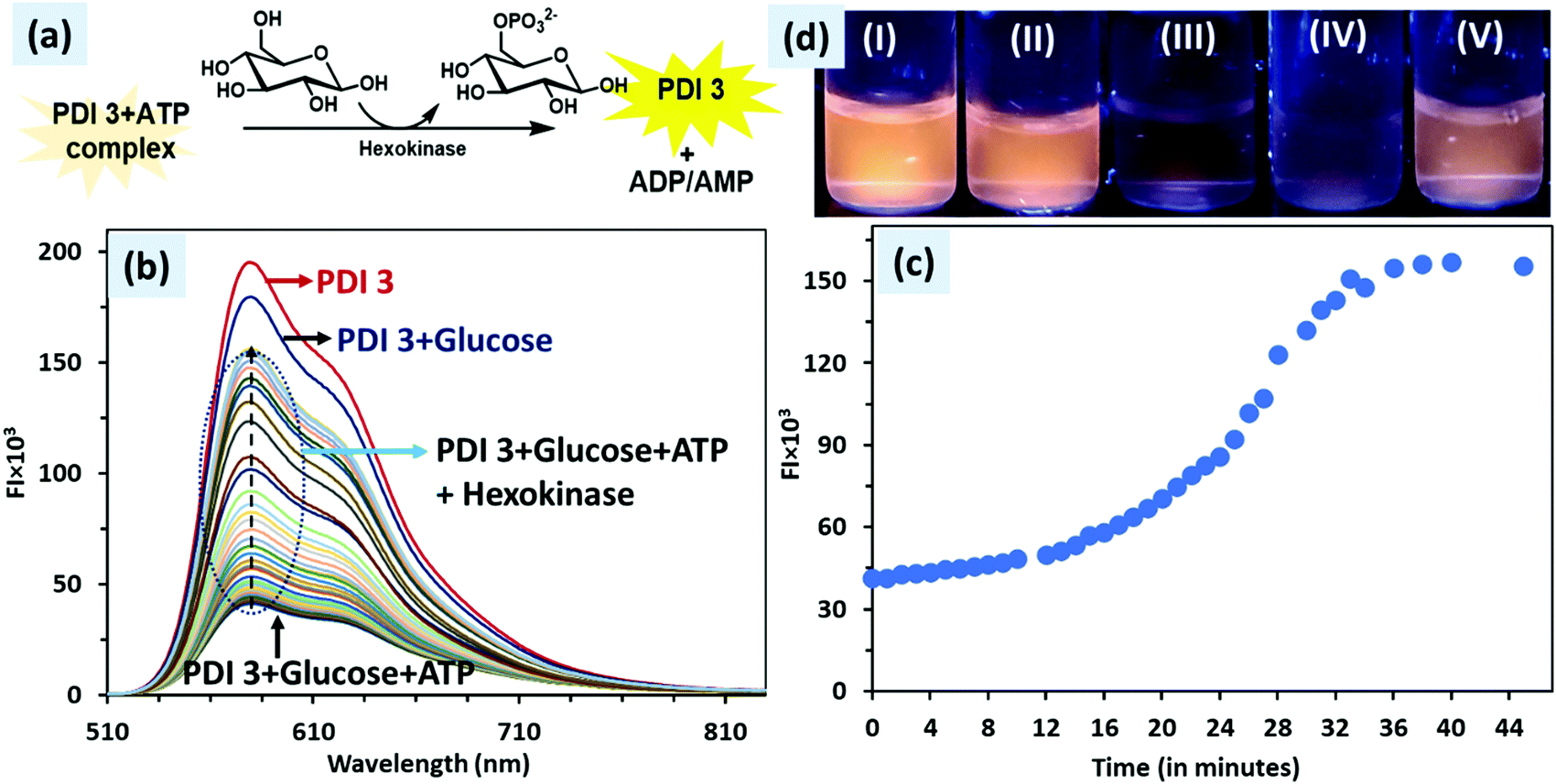 | ||
| Fig. 9 (a) Schematic illustration of glycosylation reaction performed with PDI 3 + ATP complex, glucose and hexokinase; (b) emission spectrum of PDI 3 + ATP + glucose upon addition of hexokinase at different time durations (0–45 min); (c) plot of fluorescence intensity (at 580 nm) of PDI 3 + ATP + glucose upon addition of hexokinase and time (in minutes) and (d) photographs of (I) PDI 3, (II) PDI 3 + glucose, (III) PDI 3 + glucose + ATP, (IV) PDI 3 + glucose + ATP + hexokinase (0 min) and PDI 3 + ATP + glucose + hexokinase (45 min) under 365 nm UV lamp illumination. | ||
In this assay, when the hexokinase enzyme was added, a small increase in the fluorescence intensity (∼25%) was observed in the 0–15 min time interval. However, after 15 min, the fluorescence intensity shot up and a plateau was achieved in 35 minutes; the overall recovery of the fluorescence enhancement was 73%. To ascertain that these changes are due to the glycosylation reaction by ATP rather than hydrolysis of ATP alone under the experimental conditions, we recorded the fluorescence spectrum of the PDI 3 + ATP complex alone after a regular interval of time (Fig. S17, ESI†). No change in the fluorescence spectrum of the PDI 3 + ATP complex was observed in 1 h. This means that the change in the fluorescence intensity is due to the glycosylation reaction and not due to the hydrolysis of ATP. The results clearly illustrate that this PDI 3 + ATP complex based biochemical assay could be utilized for monitoring of phosphorylation of glucose in an attempt to develop a tool for determining the glucose level.
Conclusions
In conclusion, a novel dicationic amphiphile based on perylene diimide–tyrosine alkyl amide amine (PDI 3) was successfully applied for the detection of (i) nanomolar ATP concentrations in 50% and 90% HEPES buffered–DMSO solution; (ii) exogenous and endogenous detection of ATP and (iii) fabrication of ‘turn-on’ biochemical assay for monitoring phosphorylation of glucose using the hexokinase method. The transformation of the self-assembled nanoring architecture of PDI 3 into spherical aggregates upon addition of ATP was also observed. The mechanism of interaction involves electrostatic and H-bonding interactions between positively charged PDI 3 and negatively charged ATP via (ionic) self-assembly. DLS, NMR and microscopic studies truly supported this mechanism. We believe that this biochemical assay can be used as a tool to determine the glucose level in biofluids in the near future.Author contributions
Prabhpreet Singh: conceptualization, supervision, methodology, writing – original draft, writing – review & editing, funding acquisition. Poonam Sharma: investigation. Neha Sharma and Satwinderjeet Kaur: investigation (cell imaging).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work is supported by a grant from University Grant Commission (UGC) to Guru Nanak Dev University, Amritsar, under the RUSA 2.0 Component 4.0 scheme. We also thank DST for FIST and PURSE programmes to the university. P. Sharma is thankful to the Council of Scientific and Industrial Research (CSIR), India, for Senior Research Fellowship (grant no. 09/254(0303)/2020-EMR-I).Notes and references
- (a) J. R. Knowles, Annu. Rev. Biochem., 1980, 49, 877–919 CrossRef CAS; (b) F. M. Ashcroft and F. M. Gribble, Diabetologia, 1999, 42, 903–919 CrossRef CAS PubMed; (c) X. Shen, G. Mizuguchi, A. Hamiche and C. Wu, Nature, 2000, 406, 541–544 CrossRef CAS.
- (a) Y. Zhou, Z. Xu and J. Yoon, Chem. Soc. Rev., 2011, 40, 2222–2235 RSC; (b) Y. Hu, S. Long, H. Fu, Y. She, Z. Xu and J. Yoon, Chem. Soc. Rev., 2021, 50, 589–618 RSC; (c) A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS.
- (a) Z. Gao, Z. Qiu, M. Lu, J. Shu and D. Tang, Biosens. Bioelectron., 2017, 89, 1006–1012 CrossRef CAS; (b) W. D. Bostick and B. S. Ausmus, Anal. Biochem., 1978, 88, 78–92 CrossRef CAS.
- (a) S. L. Cai, Y. B. Zheng, S. H. Cao, X. H. Cai and Y. Q. Li, Chem. Commun., 2016, 52, 12450–12453 RSC; (b) J. K. Ahn, H. Y. Kim, K. S. Park and H. G. Park, Anal. Chem., 2018, 90, 11340–11343 CrossRef CAS PubMed.
- (a) G. R. Dubyak and C. El-Moatassim, Am. J. Physiol., 1993, 265, C577–C606 CrossRef CAS; (b) E. H. Abraham, P. Okunieff, S. Scala, P. Vos, M. J. S. Oosterveld, A. Y. Chen, B. Shrivastav and G. Guidotti, Science, 1997, 275, 1324–1325 CrossRef CAS PubMed; (c) H. Yokoshiki, M. Sunagawa, T. Seki and N. Sperelakis, Am. J. Physiol., 1998, 274, C25–C37 CrossRef CAS.
- S. V. Khlyntseva, Y. R. Bazel, A. B. Vishnikin and V. Andruch, J. Am. Chem. Soc., 2009, 64, 657–673 CAS.
- L. Xie, X. Yan and Y. Du, Biosens. Bioelectron., 2014, 53, 58–64 CrossRef CAS.
- D. Cheng, Y. Li, J. Wang, Y. Sun, L. Jin, C. Li and Y. Lu, Chem. Commun., 2015, 51, 8544–8546 RSC.
- (a) Y. Zhong and T. Yi, J. Mater. Chem. B, 2019, 7, 2549–2556 RSC; (b) Y. Y. Fan, Z. L. Mou, M. Wang, J. Li, J. Zhang, F. Q. Dang and Z. Q. Zhang, Anal. Chem., 2018, 90, 13708–13713 CrossRef CAS; (c) S. Farshbaf and P. Anzenbacher, Chem. Commun., 2019, 55, 1770–1773 RSC.
- (a) X. Zheng, R. Peng, X. Jiang, Y. Wang, S. Xu, G. Ke, T. Fu, Q. Liu, S. Huan and X. Zhang, Anal. Chem., 2017, 89, 10941–10947 CrossRef CAS PubMed; (b) S. Shrivastava, N. M. Triet, Y. M. Son, W. I. Lee and N. E. Lee, Biosens. Bioelectron., 2017, 90, 450–458 CrossRef CAS.
- (a) X. Ji, J. Wang, S. Niu and C. Ding, Chem. Commun., 2019, 55, 5243–5246 RSC; (b) R. Villamil-Ramos, P. Gomez-Tagle, J. C. Aguilar-Cordero and A. K. Yatsimirsky, Anal. Chim. Acta, 2019, 1057, 51–59 CAS; (c) H. Ma, M. Yang, C. Zhang, Y. Ma, Y. Qin, Z. Lei, L. Chang, L. Lei, T. Wang and Y. Y. Yuan, J. Mater. Chem. B, 2017, 5, 8525–8531 RSC.
- (a) N. H. Kim, B. W. Kim, H. Moon, H. Yoo, R. H. Kang, J. K. Hur, Y. Oh, B. M. Kim and D. Kim, Anal. Chim. Acta, 2021, 1152, 338269 CrossRef CAS; (b) Z. Wang, Y. Zhang, J. Yin, M. Li, H. Luo, Y. Yang, X. Xu, Q. Yong and S. Wang, Sens. Actuators, B, 2020, 320, 128249 CrossRef CAS; (c) X. Zhou, J. Li, L.-L. Tan, Q. Li and L. Shang, J. Mater. Chem. B, 2020, 8, 3661–3666 RSC.
- Y. Wu, F. Xiao, Z. Wu and R. Yu, Anal. Chem., 2017, 89, 2852–2858 CrossRef CAS.
- Y. P. Dong, Y. Zhou, J. Wang and J. J. Zhu, Anal. Chem., 2016, 88, 5469–5475 CrossRef CAS.
- H. Xie, Y. Chai, Y. Yuan and R. Yuan, Chem. Commun., 2017, 53, 8368–8371 RSC.
- G. A. Nagana Gowda, L. Abell, C. F. Lee, R. Tian and D. Raftery, Anal. Chem., 2016, 88, 4817–4824 CrossRef CAS.
- (a) S. Dhiman, M. Ahmad, N. Singla, G. Kumar, P. Singh, V. Luxami, N. Kaur and S. Kumar, Coord. Chem. Rev., 2020, 405, 213138 CrossRef CAS; (b) K. Kaur, R. Saini, A. Kumar, V. Luxami, N. Kaur, P. Singh and S. Kumar, Coord. Chem. Rev., 2012, 256, 1992–2028 CrossRef CAS.
- (a) H. Singh, R. Sharma, G. Bhargava, S. Kumar and P. Singh, ChemistrySelect, 2018, 3, 7840–7848 CrossRef CAS; (b) C. Patra, C. Sen, A. D. Mahapatra, D. Chattopadhyay, A. Mahapatra and C. Sinha, J. Photochem. Photobiol., A, 2017, 341, 97–107 CrossRef CAS.
- (a) C. Patra, A. K. Bhanja, A. Mahapatra, S. Mishra, K. D. Saha and C. Sinha, RSC Adv., 2016, 6, 76505–76513 RSC; (b) M. Schaeferling, T. Lang and A. Schnettelker, J. Fluoresc., 2014, 24, 251–256 CrossRef CAS PubMed.
- Z. Xu, N. J. Singh, J. Lim, J. Pan, H. N. Kim, S. Park, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2009, 131, 15528–15533 CrossRef CAS.
- (a) S. Atilgan and E. U. Akkaya, Tetrahedron Lett., 2004, 45, 9269–9271 CrossRef CAS; (b) G. Zong, L. Xian and G. Lu, Tetrahedron Lett., 2007, 48, 3891–3894 CrossRef CAS.
- (a) H. Abe, Y. Mawatari, H. Teraoka, K. Fujimoto and M. Inouye, J. Org. Chem., 2004, 69, 495–504 CrossRef CAS PubMed; (b) P. P. Neelakandan, M. Hariharan and D. Ramaiah, Org. Lett., 2005, 7, 5765–5768 CrossRef CAS.
- (a) P. Singh, A. Hirsch and S. Kumar, TrAC, Trends Anal. Chem., 2021, 138, 116237 CrossRef CAS; (b) L. S. Mittal, P. Sharma, N. Kaur and P. Singh, Anal. Methods, 2019, 11, 5320–5327 RSC.
- (a) F. Wurthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2015, 115, 11967–11998 CrossRef; (b) C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- (a) L. Yan, Z. Ye, C. Peng and S. Zhang, Tetrahedron, 2012, 68, 2725–2727 CrossRef CAS; (b) L. Zhong, F. Xing, Y. Bai, Y. Zhao and S. Zhu, Spectrochim. Acta, Part A, 2013, 115, 370–375 CrossRef CAS.
- B. Muthuraj, S. R. Chowdhury, S. Mukherjee, C. Patra and P. K. Iyer, RSC Adv., 2015, 5, 28211–28218 RSC.
- L. Lu, H.-J. Sun, Y.-T. Zeng, Y. Shao, M. V. Bermeshev, Y. Zhao, B. Sun, Z.-J. Chen, X.-K. Ren and M. Zhu, J. Mater. Chem. C, 2020, 8, 10422–10430 RSC.
- (a) V. Grande, B. Soberats, S. Herbst, V. Stepanenkoa and F. Wurthner, Chem. Sci., 2018, 9, 6904–6911 RSC; (b) J. Baram, H. Weissman, Y. Tidhar, I. Pinkas and B. Rybtchinski, Angew. Chem., Int. Ed., 2014, 53, 4123–4126 CrossRef CAS; (c) E. Cohen, H. Weissman, I. Pinkas, E. Shimoni, P. Rehak, P. Král and B. Rybtchinski, ACS Nano, 2018, 12, 317–326 CrossRef CAS PubMed.
- (a) H. Cheng, Y. Zhao, H. Xu, Y. Hu, L. Zhang, G. Song and Z. Yao, Dyes Pigm., 2020, 180, 108456 CrossRef CAS; (b) M. Rappold, U. Warttinger and R. Kramer, Molecules, 2017, 22, 768–778 CrossRef.
- J. M. Englert, F. Hauke, X. Feng, K. Mullen and A. Hirsch, Chem. Commun., 2010, 46, 9194–9196 RSC.
- (a) G. Abellen, P. Ares, S. Wild, E. Nuin, C. Neiss, D. R.-S. Miguel, P. Segovia, C. Gibaja, E. G. Michel, A. Gçrling, F. Hauke, J. Glmez-Herrero, A. Hirsch and F. Zamora, Angew. Chem., Int. Ed., 2017, 56, 14389–14394 CrossRef; (b) S. Rehm, V. Stepanenko, X. Zhang, T. H. Rehm and F. Würthner, Chem. – Eur. J., 2010, 16, 3372–3382 CrossRef CAS PubMed.
- (a) C. Backes, U. Mundloch, C. D. Schmidt, J. N. Coleman, W. Wohlleben, F. Hauke and A. Hirsch, Chem. – Eur. J., 2010, 16, 13185–13192 CrossRef CAS PubMed; (b) C. D. Schmidt, N. Lang, N. Jux and A. Hirsch, Chem. – Eur. J., 2011, 17, 5289–5299 CrossRef CAS.
- (a) H. Szelke, S. Schubel, J. Harenberg and R. Kramer, Chem. Commun., 2010, 46, 1667–1669 RSC; (b) H. Szelke, S. Schübel, J. Harenberg and R. Kramer, Bioorg. Med. Chem. Lett., 2010, 20, 1445–1447 CrossRef CAS PubMed.
- M. Rappold, U. Warttinger and R. Krämer, Molecules, 2017, 22, 768–778 CrossRef PubMed.
- M. Han, G.-C. Wang and H.-Q. Duan, Chin. Chem. Lett., 2014, 25, 51–54 CrossRef CAS.
- (a) M. Marcia, P. Singh, F. Hauke, M. Maggini and A. Hirsch, Org. Biomol. Chem., 2014, 12, 7045–7058 RSC; (b) P. Singh, L. S. Mittal, S. Kaur, S. Kaur, G. Bhargava and S. Kumar, Sens. Actuators, B, 2018, 255, 478–489 CrossRef CAS; (c) P. Singh, L. S. Mittal, G. Bhargava and S. Kumar, Chem. – Asian J., 2017, 12, 890–899 CrossRef CAS.
- (a) L. S. Mittal, P. Sharma, N. Kaur and P. Singh, Anal. Methods, 2019, 11, 5320–5327 RSC; (b) P. Singh, L. S. Mittal, V. Vanita, R. Kumar, G. Bhargava, A. Walia and S. Kumar, Chem. Commun., 2014, 50, 13994–13997 RSC.
- K. Kumar, S. Kaur, S. Kaur, G. Bhargava, S. Kumar and P. Singh, J. Mater. Chem. B, 2020, 8, 125–135 RSC.
- K. Kumar, S. Kaur, S. Kaur, G. Bhargava, S. Kumar and P. Singh, J. Mater. Chem. B, 2019, 7, 7218–7227 RSC.
- P. Singh, K. Kumar, N. Kaur, S. Kaur and S. Kaur, Dyes Pigm., 2020, 180, 108448 CrossRef CAS.
- P. Singh, L. S. Mittal, V. Vanita, R. Kumar, G. Bhargava, A. Walia and S. Kumar, J. Mater. Chem. B, 2016, 4, 3750–3759 RSC.
- P. Singh and N. Kaur, New J. Chem., 2021, 45, 16784–16793 RSC.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman., New York, 5th edn, 2002, p. 376 Search PubMed.
- M. W. Bowler, M. G. Montgomery, A. G. Leslie and J. E. Walker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8646–8649 CrossRef CAS.
- (a) R. M. Denton, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 1309–1316 CrossRef CAS PubMed; (b) B. Glancy and R. S. Balaban, Biochemistry, 2012, 51, 2959–2973 CrossRef CAS.
- (a) T. TeSlaa and M. A. Teitell, Methods Enzymol., 2014, 542, 91–114 CAS; (b) A. Heller and B. Feldman, Chem. Rev., 2008, 108, 2482–2505 CrossRef CAS; (c) M. K. Tarrant and P. A. Cole, Annu. Rev. Biochem., 2009, 78, 797–825 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and additional photophysical and microscopic data of PDI 3 and the PDI 3 + ATP complex. See DOI: 10.1039/d1tb02235k |
| This journal is © The Royal Society of Chemistry 2022 |