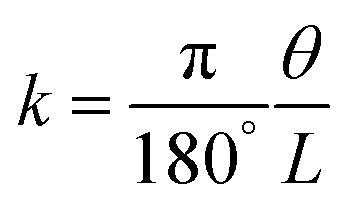A programmable bilayer hydrogel actuator based on the asymmetric distribution of crystalline regions
Received
3rd September 2021
, Accepted 17th November 2021
First published on 18th November 2021
Abstract
A novel strategy to fabricate bilayer hydrogel actuators based on the asymmetric distribution of crystalline regions across the bilayer structures was proposed. By employing PVA polymer chains into an alkali solvent-derived chitosan hydrogel matrix, chitosan/PVA hybrid bilayer hydrogels with both excellent responsive bending and mechanical properties were obtained as pH-controlled manipulators. In the design, the chitosan/PVA hydrogels upon treatment with freeze-thawing cycles were taken as the first monolayer, where excessive crystalline regions appeared. The original chitosan/PVA hydrogel as the second monolayer was then integrated into one bilayer device through the chemical-crosslinking of epichlorohydrin at the interface. The results showed that the resultant chitosan/PVA bilayer hydrogel actuator with a weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 displayed better sensitivity upon exposure to stimuli. The actuation behaviors are strongly dependent on experimental parameters such as the pH, PVA content and the chemical-crosslinking density. It is proposed that the driving force originates from the asymmetric distribution of crystalline regions, thus resulting in differential swelling ratios between the monolayers. In addition, programmable 3D shape transformations were achieved by using the bilayer hydrogel with designed 2D geometric patterns, and the tailored gripper-like hydrogel actuator can successfully capture and transport the cargo. Moreover, this actuation behavior can be erased and re-written on demand under certain conditions. Taking advantage of this universal strategy, more attractive actuators derived from synthetic or natural polymers in combination with PVA are highly expected, which can be used as smart soft robots in various fields such as manipulators, grippers, and cantilever sensors.
:1 displayed better sensitivity upon exposure to stimuli. The actuation behaviors are strongly dependent on experimental parameters such as the pH, PVA content and the chemical-crosslinking density. It is proposed that the driving force originates from the asymmetric distribution of crystalline regions, thus resulting in differential swelling ratios between the monolayers. In addition, programmable 3D shape transformations were achieved by using the bilayer hydrogel with designed 2D geometric patterns, and the tailored gripper-like hydrogel actuator can successfully capture and transport the cargo. Moreover, this actuation behavior can be erased and re-written on demand under certain conditions. Taking advantage of this universal strategy, more attractive actuators derived from synthetic or natural polymers in combination with PVA are highly expected, which can be used as smart soft robots in various fields such as manipulators, grippers, and cantilever sensors.
Introduction
In nature, numerous living creatures can be capable of hardening, rotating, winding and curving in response to environmental stimuli, for the sake of self-protection or preying.1 Inspired by these, various artificial intelligent devices have emerged as promising candidates for biomimicry. Particularly, hydrogel actuators have gained considerable attention in the fields of bionics,2,3 sensors,4–6 intelligent valves,7 soft robots,8,9 and biomedicines,10,11 because they exhibit excellent biocompatibility and moduli ranges similar to biological tissues. These soft materials were well-organized and could reversibly display predicted transformation or movement with respect to external stimuli, i.e., temperature,12,13 light,14 pH,15 electrical fields,16,17 humidity18 and ionic strength.19,20 Usually, the actuation was achieved due to the heterogeneous structure in the hydrogel matrix, where the non-uniform swelling and de-swelling process occurred when exposed to these stimuli.21,22 Hence, designing and integrating the heterogeneous features into a hydrogel are critical for manipulating their actuation behaviors in a given region.
Up to now, several typical inhomogeneous structures including bilayer structures,15,23 gradient structures,12,24 patterned structures,25,26 oriented structures27,28 and others29 have been readily introduced to fabricate actuator devices. The most common way is the bilayer strategy, where a passive hydrogel layer and an active layer are integrated into a single device. Controllable deformations such as bending and bucking would take place as a result of the asymmetrical responsive properties of these two parts. In order to endow the bilayer hydrogel networks with different responsive abilities, various methods featuring the construction of inhomogeneity in the layers have been developed. Generally, they can be divided into three categories. First, the actuation of hydrogel bilayer devices can be mediated by employing different contents of nanofillers in each layer,30–34 such as clay, graphene, SiO2, nano-fibrillated cellulose or MoO2 nanosheets. For example, Chu's group has developed a series of poly(N-isopropylacrylamide)–clay nanocomposite hydrogels as temperature-controlled manipulators.30 The introduction of clay nanosheets into the hydrogel can not only act as a physical crosslink to enhance the mechanical strength, but also modulate the thermo-induced shrinking rate. The thermo-responsive actuation of the PNIPAM–clay hydrogel was thereby achieved by differentiating the contents of clays across the hydrogel thickness. In a similar way, a novel kind of MoO2/LAPONITE®/pNIPAM ternary nanocomposite bilayer hydrogel actuator with different contents of 2D-MoO2 has also been successfully fabricated.33 The as-fabricated hydrogel actuator with a low loading of 2D-MoO2 (1.5 mg mL−1) in one layer can produce a significant temperature increase, thus enabling flexibly controllable and reversible deformation upon exposure to a NIR light. Secondly, hydrogel layers with different polymer chains or functions can be taken as the building blocks to develop bilayer actuators. Zhang et al. have proposed intelligent bilayer hydrogel actuators from chitosan (CS) and cellulose/carboxy-methylcellulose (C/CMC).15 They can perform programmed transformations such as the ‘S’ shape, helices, tube-like, bamboo-like, wave-like, and flower-like shapes under different pH conditions. The ionization of carboxyl groups in the C/CMC layer (pH > 3.8) and the protonation of amino groups in the CS layer (pH < 3.8) commonly contributed to these phenomena. Huang's group presented a salt-responsive interpenetrating network (IPN) hydrogel consisting of a cationic pTMAEMA layer and a zwitterionic pSBVI layer.20 These two polymer networks showed opposite swelling behaviors due to the polyelectrolyte and antipolyelectrolyte effects in salt solution. Therefore, this as-prepared hydrogel could demonstrate a series of switchable bulk and interfacial properties, including the structural modulation, tunable antimicrobial properties, and surface regeneration by ionic strength. Moreover, a hydrogel layer (pAA) can be assembled with an organogel layer (pMBA) into a binary copolymer film via one-step polymerization of layered immiscible monomer solutions.35 Attributed to the cooperative asymmetric swelling/shrinking of the hydrogel and organogel networks, this Janus copolymer film can display bidirectional and site-specific bending behavior in both aqueous and organic solutions. Thirdly, the actuation of hydrogel bilayer devices can be achieved by altering the crosslinking density of each layer to build a bilayered-structure.36–38 For instance, in the presence of Fe3+, a pAAc/clay hydrogel with excessive cross-linking density exhibited a lower swelling degree and higher modulus.36 Combining the pAAc/clay layer with the Fe3+–PAAc/clay layer, they could be manipulated into bilayer soft actuators. By varying the concentration of Al3+ in the (Na-alginate/pNIPAM)/(Al-alginate/pNIPAM) bilayer hydrogel matrix, it would regulate the mechanical properties of the tough hydrogel, and modulate its lower critical solution temperature as well.37 The as-prepared four-arm gripper can be demonstrated to be a prototype of all-hydrogel soft robots.
The bilayer strategy has been proved to be an effective and convenient way to construct hydrogel actuators. With the necessity for the development of artificial intelligent devices, more innovative concepts of bilayer structures are still required: for example, on the premise of the same polymer chains and same contents of polymers and crosslinking agents, how to generate inhomogeneous structures in the hydrogel matrix without addition of any nanofillers. Hence, in this work, polyvinyl alcohol (PVA) as a fascinating polymer was tentatively introduced into the hydrogel matrix. PVA shows a characteristic of crystallization via inter-/intra-molecular hydrogen bonds during the freeze–thawing cycles.39 The resulting crystalline domains for PVA were expected to render the as-prepared hydrogel layer with differential swelling behaviors. To test the feasibility, a sample of the chitosan/PVA hybrid hydrogel was prepared from the alkali/urea solvent system as the pH-responsive actuator. In the hydrogel, chitosan plays an active role as the pH sensitive component, while PVA was used to modulate the swelling ability in a controlled manner. After the freeze-thawing treatment of chitosan/PVA (denoted as FH-Gel), it was integrated with the original chitosan/PVA hydrogel layer (labeled as H-Gel). The proposed bilayer hydrogels (FH-Gel/H-Gel) can operate as responsive actuators by varying the pH conditions. The actuation behaviors strongly depend on the weight ratio of chitosan/PVA, pH, and initial cross-linking density. In addition, programmable 3D transformations such as flowers, helices, S-like shapes, etc. were obtained by designing 2D geometric patterns. Furthermore, the gripper-like hydrogel actuator can successfully capture and transport the target object, revealing the actual loading ability. To my best knowledge, crystalline domains in the hydrogel mainly provide extra physical cross-linked domains to largely enhance the mechanical strength, but hardly used in the fabrication of actuators. This report provides a universal and simple way to construct bilayer hydrogel actuators, which is suitable for both synthetic and natural polymers. It will have great significance in the design of more novel intelligent devices for practical applications.
Experimental section
Materials and reagents
All of the reagents were used as received unless otherwise noted. Chitosan with a degree of deacetylation of about 89% was purchased from Ruji Biotech (Shanghai, China). Lithium hydroxide monohydrate (LiOH·H2O, 98%), epichlorohydrin (denoted as ECH) and poly(vinyl alcohol) (PVA, molecular weight 145000) were of analytical grade and purchased from Aladdin Chemical Co. Ltd (Shanghai, China). Potassium hydroxide and urea were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Other chemical reagents including HCl and NaOH obtained from commercial sources in China were of analytical grade and used without further purifications. All the solutions were prepared with distilled water.
Preparation of chitosan/PVA hybrid hydrogels
The chitosan/PVA hydrogels containing ECH were prepared as follows. Firstly, a fixed amount of 4 g chitosan powder was dissolved in 100 g solvent system consisting of LiOH/KOH/urea/H2O in a weight ratio of 4.5:7.8:80.5 via a freezing–thawing process. Then, certain amounts of 12 wt% PVA aqueous solution and ECH loaded in separated funnels were slowly added to 4% chitosan solution under vigorous agitation, to achieve the desired weight ratio of chitosan to PVA (i.e., 3:0.5, 3:1, 3:2, and 3:3). ECH was taken as the chemical-crosslinking agent, and the weight ratio to chitosan ranges from 1.0 to 4.0. The temperature during the whole process was maintained at 0 °C. After being degassed by centrifugation for 7 min at 4 °C, the obtained transparent solution was cast into a Petri dish with a desired thickness to obtain the pre-gel sheet, which then was maintained at ambient temperature for 12 h for the chemical cross-linking reaction of the hydroxyl groups on the polymer chains with ECH. Finally, the as-prepared hydrogels were removed from the mold and thoroughly washed with deionized water to remove residuals. The obtained chitosan/PVA hydrogel was denoted as the H-Gel. By subjecting the H-Gel to freeze–thawing treatment 6–7 times below −20 °C, chitosan/PVA hydrogels with more nanocrystalline domains (labeled as FH-Gel) were collected.
Preparation of chitosan/PVA bilayer hydrogels
Double-layered chitosan/PVA hydrogels were constructed as depicted in Fig. 1. With the help of PTFE frames, the transparent chitosan/PVA pre-gel solution was directly cast on the surface of the FH-Gel. The bilayer pre-gel sheets were allowed to cure at room temperature for 12 hours, and the upper and lower sections will be connected together at the contact interfaces via chemical cross-linking. After thoroughly washing with deionized water to remove any residuals, a bilayer hydrogel was obtained.
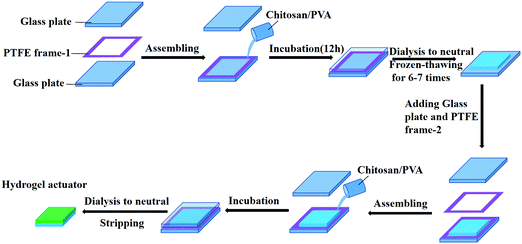 |
| | Fig. 1 Schematic illustration of the preparation of a double-layered chitosan/PVA hydrogel actuator. | |
Preparation of patterned hydrogel actuators
Smart actuators were prepared by a multistep molding process. The flower-like hydrogel actuators shown in Fig. 5d and 6 can be directly tailored from the bilayer hydrogel sheets. For the other programmed transformations, the actuators can be fabricated as follows. Firstly, the FH-Gel/H-Gel bilayer hydrogel strips (width ∼5 mm, thickness ∼1.5 mm) were paralleled or lined up into the Petri dish as shown in Fig. 5a–c. Then the chitosan/PVA pre-gel solution as the matrix was cast into the Petri dish and filled the space among the covered bilayer hydrogel strips. After completely curing at room temperature for 12 h, patterned actuators will be obtained with further dialysis and solidification in pure water.
Characterization
The morphologies of dry hydrogel samples were characterized by using scanning electron microscopy (SEM, Hitachi S-4300, Japan) at an accelerating voltage of 40 kV. The wide-angle X-ray diffraction (XRD) patterns of the dried hydrogel samples were recorded in a reflection mode on a Rigaku SmartLab diffractometer equipped with a CuKα radiation source (λ = 1.542 Å) operated at 40 kV and 30 mA. The samples were scanned at 1° min−1 and at a step size of 2.5° in 2θ. The FT-IR spectra were recorded in the wavenumber range from 4000 to 500 cm−1 using an attenuated total reflection Fourier transform infrared spectroscope (ATR-FTIR, PerkinElmer, USA) at room temperature. The hydrogels were freeze-dried in a conventional freezer–dryer and then dried at 45 °C in a vacuum chamber to eliminate water from the samples. Raman spectroscopy and spatial Raman mapping were performed using a Raman imaging microscope (Thermo Scientific DXR xi, USA). The wavelength of the excitation laser was 532 nm. Raman maps were collected using a spatial resolution of 500 nm. The MCR method developed by the OMNIC xi software was applied for calculating the proportion of physically cross-linked and chemically crosslinked domains.
Mechanical testing
The tensile mechanical measurement of the bilayer hydrogel was performed on a universal tensile-compression tester (Instron 5900, USA) at room temperature. The test was carried out on dumbbell-shaped samples with a size of 11.5 mm (L) × 2 mm (d) × 3.3 mm (W), and the stretching rate was 5.0 mm min−1. The interfacial adhesion of bilayer hydrogels was evaluated by using the 180°-peeling method. A 40.0 × 10.0 × 4.0 mm bilayer hydrogel was prepared, where one end of the monolayer was detached from the other one. During the testing, the free ends were clamped by the upper and lower gripper, respectively. The peeling rate was set as 5.0 mm min−1.
Swelling behaviors
The equilibrium swelling ratio (ESR) of each hydrogel under certain conditions was studied at room temperature. After removing the surface moisture with filter paper, the weight of the expanded hydrogel was measured. The expanded hydrogel is then dried to a constant weight to obtain the dry weight of the sample. ESR can be calculated as| | | ESR = (Ws − Wd)/Wd × 100% | (1) |
where Ws is the weight of the expanded hydrogel and Wd is the weight of the corresponding hydrogel in the dry state. All ESRs are the averages of three repeated measurements.
Deformation of pH-responsive bilayer hydrogels
The 2D-to-3D transformation and object loading performance were displayed in pH = 2 aqueous solution, and recorded with a digital camera. In the load bearing test, a small ball object (3.0 g) with a diameter of 16 mm was used.
Results and discussion
Concept of the design and the fabrication process of bilayer hydrogel actuators
This concept of novel bilayer hydrogel actuators was achieved by incorporating the FH-Gel layer (higher content of crystalline fibrils) with the H-Gel layer (lower content) into one device. It has been reported that PVA chains exhibit the powerful ability to form crystalline nanofibrils upon freeze–thawing cycles.40,41 By in situ producing more crystalline domains in one of the chitosan/PVA hybrid hydrogel layers, an asymmetrical distribution of crystalline fibrils across the bilayer structure was obtained. The nanocrystalline domains, similar to the nanoclays,30 not only can render the hydrogel device with much better mechanical properties as a result of excessive physical-crosslinking, but also may give rise to different swelling behaviors between two layers. Thus, the chitosan/PVA hydrogel actuators possess both excellent mechanical and pH-responsive actuation properties.
The fabrication process is schematically illustrated in Fig. 1. Firstly, the mixed chitosan/PVA pre-gel solution containing ECH as the chemical-crosslinking agent was prepared from the alkali/urea system at 0 °C,42,43 then it was poured into a PTFE frame and cured at room temperature for 12 h to form a hydrogel. The alkali/urea solvent system has been identified to be a better candidate to dissolve chitosan than acidic solution, since it would endow the as-formed chitosan hydrogel with excellent swelling behavior and high mechanical strength.43 After dialysis to neutral in deionized water, the as-prepared hydrogel sheet was subjected to the freeze-thawing treatment for 6–7 cycles in a refrigerator to obtain the FH-Gel layer. For the construction of the second layer (H-Gel layer), the FH-Gel layer was returned into the PTFE frame, and another PTFE frame with the same thickness was placed on top of it. Subsequently, the chitosan/PVA pre-gel solution was injected again. During the period of further 12 h incubation at room temperature, two layers were chemically bonded together via the function of ECH. After thoroughly washing with deionized water to remove the residues, the designed double-layered hydrogel actuator was obtained from the mold. The thickness of each hydrogel layer depends on the thickness of the PTFE frame, while the composition is controlled by the weight ratio of chitosan to PVA.
Structural characterization of the hydrogel layers
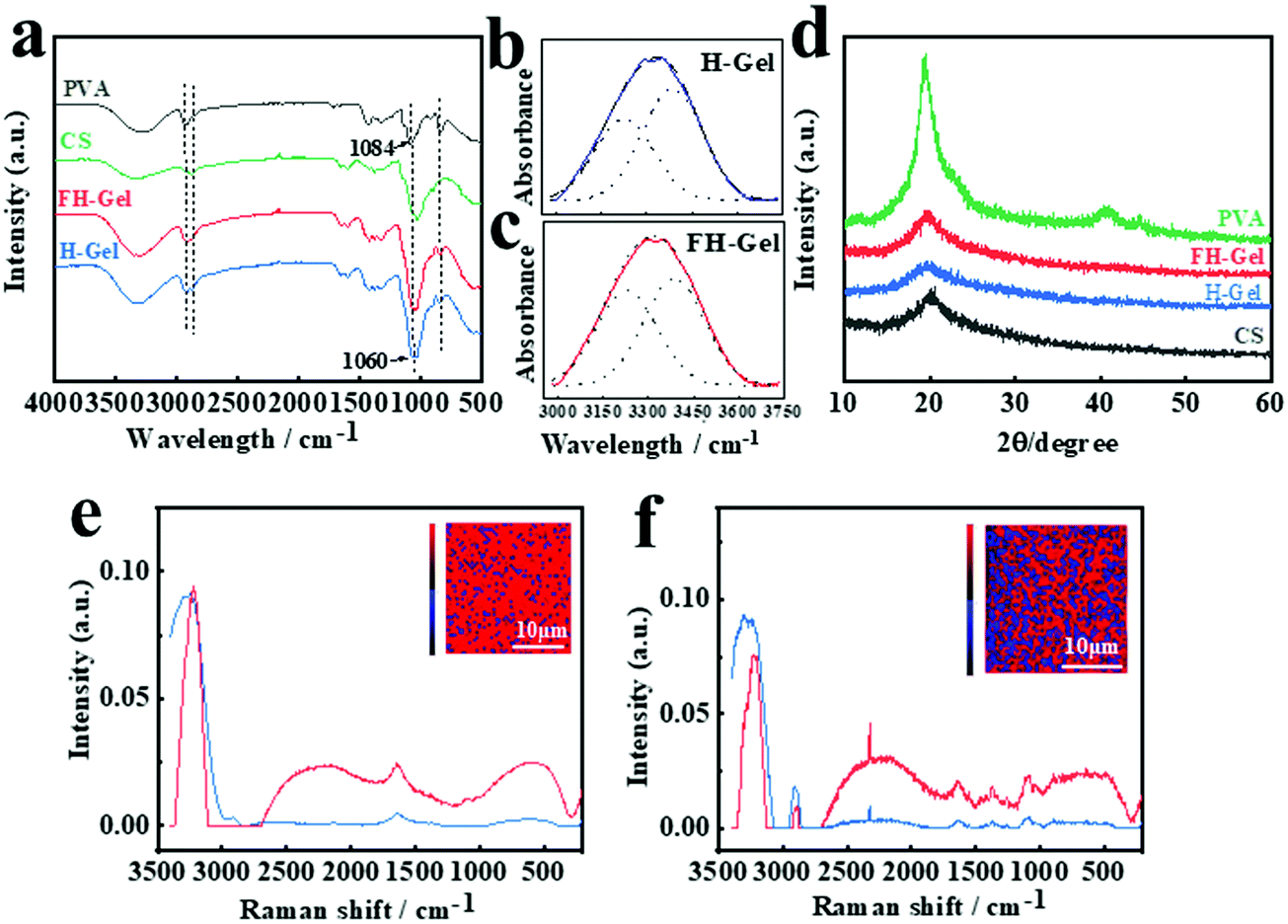
PVA possess abundant hydroxyl groups, which will facilitate the polymer chains to form intrachain/interchain hydrogen bonding under certain conditions. Upon exposure to a low temperature (<0 °C), the as-formed ice crystals in the matrix would expel PVA chains to form regions of high polymer concentrations. As the adjacent PVA chains are widely involved in hydrogen bond formation, nanocrystalline domains appear abundantly, leading to excessive physically cross-linked networks of nanofibrils.39,41 To confirm the appearance and asymmetrical distribution of nanocrystalline domains across the hydrogel layers, ATR-FTIR spectra were firstly employed (Fig. 2a–c). As shown, PVA shows prominent absorption bands at around 3237–3317 cm−1 for O–H stretching vibration, 2909–2940 cm−1 for the stretching of –CH in the backbone and 1084 cm−1 for C–O stretching vibration.44,45 For both cases of the FH-Gel and H-Gel, the characteristic peaks for the –CH and C–O stretching were red-shifted to 2868–2914 cm−1 and 1060 cm−1, respectively. Meanwhile, the intensity of the absorption bands at 1060 cm−1 was greatly enhanced in comparison with PVA or chitosan alone. These variations demonstrate strong interactions (chemical crosslinking interactions by ECH and hydrogen bonding) between the chitosan and PVA polymer chains in hybrid hydrogel networks. In order to clarify the different contents of nanocrystalline domains in the FH-Gel and H-Gel, the broad peak for the O–H stretching vibration in each case was further fitted and split into two peaks at 3227 and 3385 cm−1, corresponding to the hydrogen bonds of PVA–PVA and PVA–water, respectively (Fig. 2b and c).46 The area ratio of the peak at ∼3227 cm−1 to the sum of the peak at ∼3227 cm−1 and the peak at ∼3385 cm−1 was calculated to quantify the contribution of hydrogen bonding of PVA–PVA hydroxyl groups. As indicated, the area ratio was determined to be 47.1% for the FH-Gel, which is slightly higher than that for the H-Gel with a value of 41.2%. This small difference may be attributed to the fact that all the testing samples were once manipulated by the freeze–drying treatment. Some nanocrystalline domains would be generated in the H-Gel during that process. Nevertheless, this result can still reveal that the hydrogen bonding of PVA–PVA was more profound in the FH-Gel layer. More nanocrystalline domains exist in the FH-Gel layer.
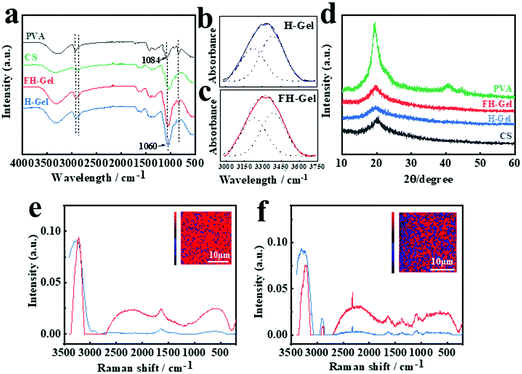 |
| | Fig. 2 (a) ATR-FTIR spectra of chitosan, PVA, H-gel and FH-gel samples. H-Gel denotes the original chitosan/PVA (weight ratio = 3:1) hydrogel. FH-Gel refers to the chitosan/PVA (weight ratio = 3:1) hydrogel upon freeze-thawing treatment for 6–7 cycles. ATR-FTIR patterns for the O–H stretching vibration and the corresponding peak-split fitting located at 3227 and 3385 cm−1 for (b) H-Gel and (c) FH-Gel. (d) Wide angle X-ray diffraction spectra of each sample. Raman spectra of individual chemically and physically cross-linked domains within H-Gel (e), and (f) FH-Gel samples. The inset shows the reconstructed Raman images of the hydrogel obtained from the OH stretching mode intensities (3000–3400 cm−1). Red corresponds to the chemically cross-linked domains, and blue indicates the physically cross-linked domains. | |
Fig. 2d presents the XRD patterns of chitosan, PVA, FH-Gel and H-Gel, respectively. The typical pattern of PVA exhibits a strong diffraction peak at 2θ = 19.5°, corresponding to the typical reflection plane of (101) in semicrystalline PVA.47 Chitosan shows the characteristic peak at 20.1°, which is assigned to (110) reflections. It is attributed to the regular crystal lattice of chitosan.43 For the hybrid hydrogels, a diffraction peak at 19.8° was observed in the patterns of both cases, suggesting the formation of C–O–C between chitosan and PVA. The values of FWHM (Full Width at Half Maxima) for the FH-Gel and H-Gel were estimated to be 5.9° and 7.6°, respectively. This observation indicates that more crystallite aggregations for PVA via strong hydrogen bonds formed in the FH-Gel.
Further evidence for the differentiated contents of nanocrystalline domains in the H-Gel and FH-Gel was obtained using 3D confocal Raman spectra as depicted in Fig. 2e and f, respectively, which enables direct visualization of the variations in the physically and chemically cross-linked domains inside various samples in a non-invasive fashion.48,49 More physically cross-linked domains demonstrate the higher content of nanocrystalline domains on the premise of the same chemical crosslinking density within each independent hydrogel layers. The proportion of the physically cross-linked domains was also evaluated by the multivariate curvilinear resolution (MCR) (Fig. 2e and f, inset). The reconstructed Raman images show the respective intensities of the O–H stretching (3000–3400 cm−1) in blue and red colors. The obvious intensity of 3242 cm−1 in the blue spectrum represents the OH-rich region (the physical crosslinking region), while the corresponding peak in red color is related to the OH-deficient region (the chemical crosslinking domain). By exposing chitosan/PVA to freeze–thawing for several cycles, the intensity of the O–H stretching (blue spectrum) for the FH-Gel was greatly enhanced as indicated in Fig. 2f. According to chemometric analysis, the ratio of physically cross-linked domains to the chemically cross-linked domains for the H-Gel was evaluated to be 12.4%, whereas a considerable increase was observed in the FH-Gel with a value of 35.9%. This confirms strong hydrogen bonding interactions between PVA chains, thus resulting in more nanocrystalline domains in the FH-Gel layer.
On the basis of the FTIR, Raman and XRD analysis, more nanocrystalline domains were generated in situ through the freeze–thawing cycles, leading to excessive physically cross-linked networks of nanofibrils within the FH-Gel layers. This occurrence will contribute to an asymmetrical distribution of crystalline fibrils across the bilayer structure, ultimately resulting in the anisotropic swelling behaviors.
pH-Responsive swelling behaviors of the hydrogel layers
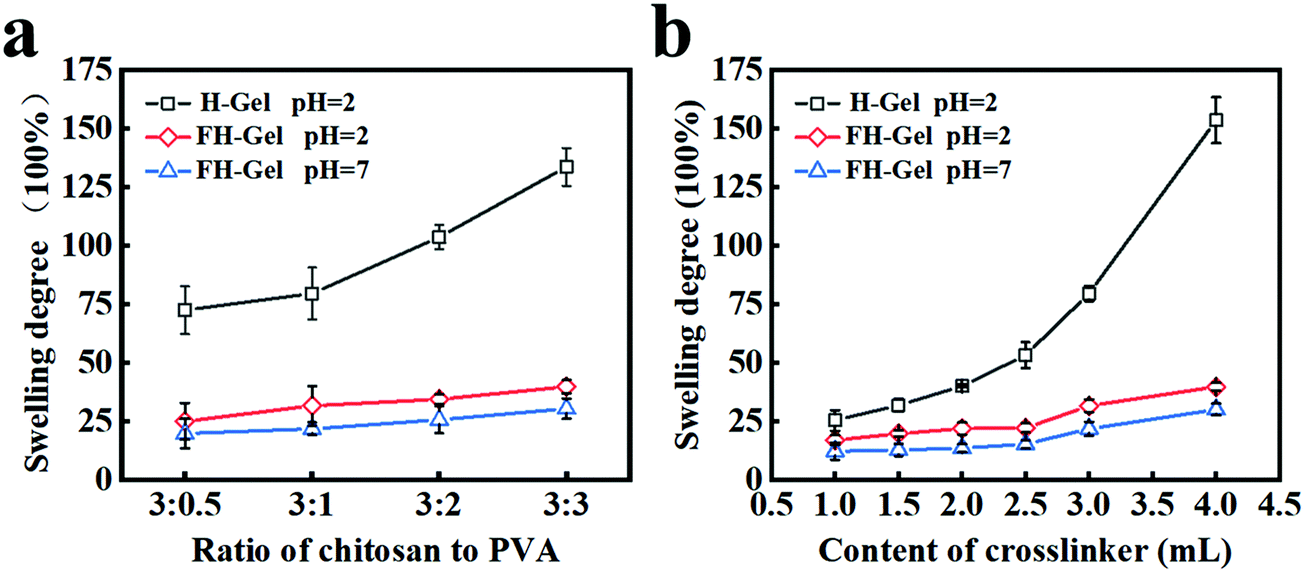
As a result of the protonation of the amino groups in chitosan under acidic conditions, the as-prepared chitosan/PVA hybrid hydrogels could display different pH-responsive swelling behaviors. As indicated in Fig. 3a, dramatic differences in the equilibrium swelling degree were observed for the H-Gel and FH-Gel layers at each given weight ratio of chitosan to PVA. As is expected, the H-Gel layers showed a higher swelling degree than the FH-Gel layers at pH = 2. This difference was further enlarged with the decrease in the chitosan/PVA weight ratio. As the weight ratios varied from 3:0.5 to 3:3, the swelling degrees for the H-Gel and FH-Gel layers increased from 73 to 133 and 25 to 38, respectively. By comparison, the swelling behavior for the FH-Gel layer hardly changed, since the existence of excessive nanocrystalline domains as the physical-crosslinking joints could inhibit the relative movement of the polymer chains against the electrostatic repulsion. Moreover, the swelling behaviors for the chitosan/PVA hydrogels also strongly depend on the chemical-crosslinking density of the matrix. As shown in Fig. 3b, by mediating the feeding ratio of ECH to chitosan from 1.0 to 4.0, both the H-Gel and FH-Gel layers exhibited enhanced swelling degrees from 25 to 153 and 15 to 38, respectively. Since the H-Gel layer would display lower mechanical strength at higher content of the ECH crosslinker (data not shown), a feeding ratio of 3.0 was applied in the following construction, which could be an ideal candidate to meet the requirements of appropriate mechanical and anisotropic swelling properties. As mentioned above, the resulting nanocrystalline domains readily play an important role in controlling the pH-responsive swelling and mechanical characteristics of monolayer chitosan/PVA hydrogels, which is fundamental to fabricate the pH-triggered asymmetric response of bilayer soft actuators.
 |
| | Fig. 3 (a) The swelling behaviors for the chitosan/PVA hydrogels with different weight ratios under each condition. (b) Dependence of the swelling degrees on the content of the crosslinking agent (ECH) for the chitosan/PVA hydrogel layers with a fixed weight ratio of 3:1. | |
pH-Responsive actuation behaviors of the hydrogel actuators
With respect to the differential swelling ratios, the H-Gel and FH-Gel monolayers were assembled into one integrated bilayer device, and the resultant pH-responsive actuation behaviors were investigated. Fig. 4a presents the variation of bending behaviors for the bilayer hydrogel actuators with different weight ratios of chitosan to PVA in pH = 2 aqueous solution. As indicated, the bending angles gradually increased along with the extension of incubation time. Specifically, the soft manipulator with a weight ratio of 3:1 demonstrated faster response with a larger bending angle in the process, implying the maximum anisotropic characteristic across the hydrogel layers. This occurrence can be ascribed to the competitive effect between the swelling and the resistant monolayers. During the bending process, the expansion of the volume for the H-Gel layer has to overcome the resistance of the FH-Gel layer originating from the mechanical stiffness of the interpenetrated network. Although the asymmetric differences in the swelling degree would be enlarged along with the increment of PVA in the chitosan/PVA hydrogel (i.e. 3:2, 3:3), the enhancement of mechanical stiffness can display the adverse effects on the bending actuation, thereby giving rise to slower responsive speed and smaller bending angle.
 |
| | Fig. 4 (a) The variation of bending behaviors for the chitosan/PVA hydrogel actuators with different weight ratios in pH = 2 aqueous solution. (b) Effects of pH on the bending angles for chitosan/PVA (weight ratio = 3:1) in aqueous solution. (c) The corresponding bending images along with the incubation time for the bilayer hydrogel actuator (weight ratio = 3:1) in pH = 2 aqueous solution. (d) SEM images of the bilevel cross-sections for the hydrogel actuator after swelling, H-Gel layer (left) and FH-Gel layer (right). | |
This competitive effect can be further depicted by Timoshenko theory,50 where a correlation between material properties can be well established. The bending curvature (k) of a bilayer actuator is determined as a function of its overall thickness (h), and the stiffness and thickness ratios of the two layers (m and n, respectively):
| | 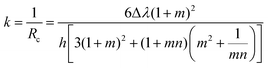 | (2) |
| | 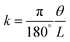 | (3) |
where
k indicates the curvature,
θ represents the bending angle,
L refers to the strip's free bending length,
m =
h1/
h2 and
n =
E1/
E2.
E1 and
E2 are the Young's moduli of layers 1 and 2, while
h1 and
h2 are the thicknesses of layers 1 and 2. The parameter Δ
λ in
eqn (2) can be expressed as Δ
λ =
λ1 −
λ2, where
λ1 and
λ2 are the linear expansion ratios of each separated layer. Assuming that the hydrogel actuators consist of two layers with equal thickness, in the given system with the fixed value of
L and
h, the combined equation can be simplified as
| |  | (4) |
where
k′ is considered to be constant. Based on
eqn (4), it can be concluded that the actuation behaviors for the bilayer hydrogel device are mainly determined by the swelling differentiation and mechanical stiffness of each layer. More importantly, this analytical model can be used to predict the final deformation shape, and will also give us some suggestions on what parameters should be available for the fabrication process to create a desired 3D structure.
In addition, the influences of pH on the actuation behavior for the bilayer hydrogel actuator were identified, as depicted in Fig. 4b. The chitosan/PVA bilayer actuator (weight ratio = 3:1) shows better sensitivity under lower pH conditions with a larger bending angle and relatively faster bending speed. In the case of the environmental pH value below the pKa of chitosan (∼6.5),51 a lower pH is beneficial to the ionization of amino groups. As a consequence, the positive charge density of chitosan increases, and the enhanced electrostatic interaction triggers a faster bending behavior. Fig. 4c shows the visual evidence for this dynamic actuation behavior at pH = 2. After soaking the bilayer device into the acidic solution, the gel strip took a big deformation and bent into a circle in 300 s. The transparent segment represents the H-Gel layer, while the opalescent one refers to the FH-Gel layer. From the pictures, a highly swollen state with an increased thickness for the H-Gel layer was reached during the actuation period. Hence, the bilayer actuator can easily bend toward to the direction of the FH-Gel layer. The corresponding SEM images at a constant state of actuation were also collected (Fig. 4d). As indicated, a clear boundary line across the section of the bilayer device was observed. Compared to the FH-Gel layer (right) with the compact nanofibril network, the H-Gel layer shows the characteristic of a large continuous pore structure, definitely implying its higher swelling ratio in the actuation process.
Multiple transformations for the programmed hydrogel actuators
This basic bending/unbending motion can be manipulated to create multiple transformations with sophisticated structures. By precisely controlling the distribution of responsive domains in one, two or even three dimensions to design programmable or predictable shape morphing of hydrogel devices, various geometric transformations can be predicted. For example, we incorporated the FH-Gel/H-Gel bilayer hydrogel strips into the H-Gel matrix with periodic alignment, where the bilayer strips displayed an oblique angle of 70° with respect to the long axis of the sheet (Fig. 5a). Due to the different sensitivity to external pH stimulus (Fig. 3), the transformation of the planar sheet could yield a 3D helical architecture. When joining one bilayer hydrogel strip with the other one at a reversed direction along the long axis (Fig. 5b), an S-like shape formed in a low pH medium. By assembling the H-Gel layer onto the both ends of the FH-Gel strip, as indicated in Fig. 5c, a special variation with desired shape was achieved. Furthermore, as presented in Fig. 5d, the bilayer hydrogel can be tailored into a flower shape. Arising from the bending movement of seven hydrogel petals in acidic solution, the blooming hydrogel flower gradually closed.
 |
| | Fig. 5 Self-deformation for the bilayer hydrogel actuators with different architectures in pH = 2 aqueous solution. (a1–d1) The corresponding models for illustrating the multiple variations. (a2–d2) The original images of hydrogel actuators before deformation. (a3–d3) The predicted transformations of hydrogel actuators. In the geometric design (a), the blue color represents the FH-Gel/H-Gel bilayer hydrogel strips, while the green sections stand for the H-Gel matrix. In the designs (b–d), the blue color denotes the FH-Gel layer, while the green sections indicate the H-Gel hybrid hydrogel. | |
From a practical application point of view, an intelligent actuator with appropriate tailored size could be applied as an underwater robot for cargo transportation. As shown in Fig. 6, a bionic manipulator that can grab a heavy object is simulated by crossing “X”. A four-arm bilayer hydrogel is fixed on the bottom end of the wire, and then placed on top of the spherical object (∼3.0 g) in an acidic water solution (pH = 2). Due to the larger swelling ratio of the H-Gel layer, this double-layered manipulator can bend down to capture the heavy object within 2 min, and further bring it from the aqueous medium. Finally, by placing the actuator in an alkaline medium, it can return to its original shape and release the cargo. It can be seen that this intelligent manipulator with a rapid response deformation can complete simple mechanical operations in harsh acidic media.
 |
| | Fig. 6 The process of loading an object with an “X” type gripper in an acidic aqueous solution at pH = 2. | |
Mechanical properties of the hydrogel actuator
The asymmetric deformation in the interface of the bilayer hydrogel can lead to delamination.52 Thus, it is of great importance to create a robust interface between the layers to ensure reliable actuation without any destruction. In our finding, the H-Gel and FH-Gel layers were crosslinked by ECH through covalent bonds as illustrated in Fig. 7a. Due to the fact that a large amount of –OH and –NH2 groups exist in the PVA and chitosan chains, the pre-gel solution of the H-Gel layer could be chemically bonded to free groups such as –OH or –NH2 in the FH-Gel layer at the interface during the preparation of bilayer actuators. In addition, the possible hydrogen bonding interactions between these hydrophilic polymer chains also occurred at the interface of layers, demonstrating the enhancement of interfacial adhesion. To further estimate the adhesion strength in the interface, the H-Gel layer was separated with the FH-Gel at one end of the bilayer strip, and then carried out the 180° peeling test. The peeling curve between the hydrogel layers is presented in Fig. 7b. The interfacial toughness (G) was defined as53where Fave is the average force at the plateau in the steady-state region of the peeling process, and w is the width of the tested hydrogel sheet. Based on eqn (5), the interfacial toughness for this bilayer device (2 mm thickness) was evaluated to be 93.1 J m−2. In spite of being not too high, it is enough to ensure the integrity of the bilayer film in the shape-shifting process.
 |
| | Fig. 7 (a) Schematic interface interactions in the double-layered hydrogel actuator. (b) The 180° peeling force curve of two as-contacted hydrogel sheets. (c) Photographs of the hydrogel actuator under tensiling, twisting and knitting. (d) Typical tensile stress–strain curve of the hydrogel actuator. In the as-prepared hydrogel actuator, the weight ratio of chitosan to PVA was 3:1, and the content of the ECH crosslinking agent was fixed to be 3 mL per 4 g chitosan. | |
For the demand of bearing load, the mechanical properties of the bilayer actuator were explored. As shown in pictures, the as-prepared bilayer hydrogels were transparent/translucent and colorless. Under stretching, twisting, and knitting, the hydrogels could withstand large deformations without any rupture or delamination (Fig. 7c). When the external stress was removed, they could recover their original shape, demonstrating the excellent flexibility and elasticity. The typical tensile stress–strain curve (Fig. 7d) reveals that the tensile fracture stress for the chitosan/PVA double-layered hydrogel reaches 62.3 kPa with a tensile fracture strain of 216%, when the weight ratio of chitosan to PVA is 3:1. According to the linear fitting in the initial region, the Young's modulus was estimated to be 0.26 kPa. The introduction of the crystalline regions in a hydrogel has a profound effect on its mechanical strength. On one hand, these in situ as-formed crystalline regions could provide extra physical-crosslinking junctions to maintain the network during stretching. On the other hand, the crystalline structures in the hydrogel could act as “load carriers” that effectively absorb energy and sustain large deformations. Similar to those microgel-reinforced systems,54,55 the resultant crystalline nanofibrils can be employed as biobased reinforcement for various potential applications. For example, muscle-like fatigue-resistant hydrogels have been achieved by aligning the PVA nanofibrillar architectures in synthetic hydrogels through the mechanical training.56 The crystalline nanofibrils endowed the materials with the combinational properties of high fatigue resistance, high strength, low Young's modulus, and high water content.
Reversible actuation behaviors for the bilayer hydrogel actuator
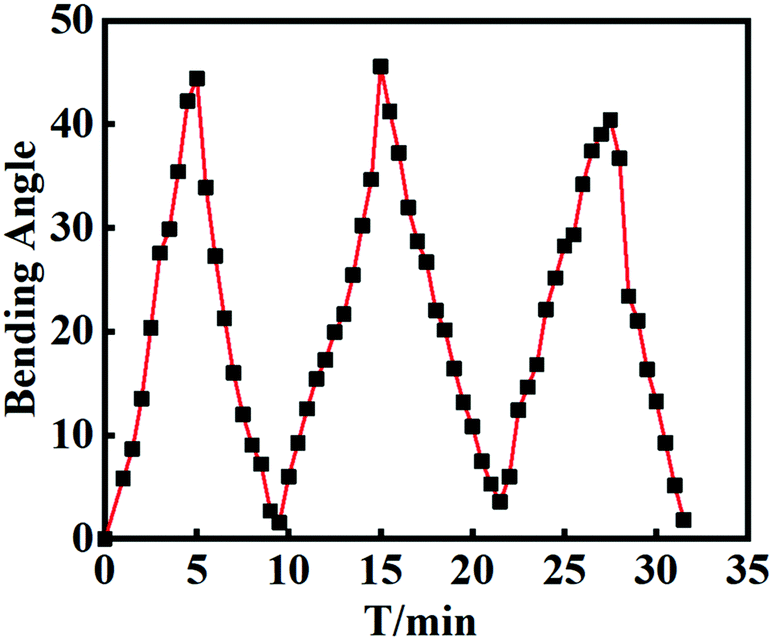
The ability to perform reversible action is another important parameter for the hydrogel manipulators. To evaluate the stability, repeatability and reversibility during the deformation cycles, the bilayer hydrogel actuator was alternately and repeatedly impregnated in aqueous solutions with pH = 2 and pH = 12. As shown in Fig. 8, the bilayer actuator could bend back and forth quickly as the gel strip was exposed to alternating pH conditions. During the repeated cycles, the bilayer hydrogel displayed almost the same bending angles at the same incubation time. After each cycle, the bending angle can return to its original state. The pattern of each cycle is fairly similar, indicating the good reversibility. The results further supported the strong interface adhesion between H-Gel and FH-Gel layers, and the excellent mechanical stability of the bilayer hydrogels during the repeated deformation.
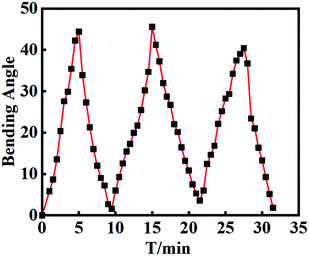 |
| | Fig. 8 The variation of bending angles for the bilayer hydrogel actuator after soaking equilibrium alternately in pH = 2 and pH = 12 aqueous solutions for 3 cycles. | |
Erasable and rewritable actuation ability for the bilayer hydrogel actuator
The crystallized PVA domains formed in the freezing/thawing treatment can melt at 60 °C in water,57 hence, the actuation behavior for the chitosan/PVA bilayer hydrogel can be easily erased on demand by incubating the device in a hot water bath (>60 °C) for a certain period (2–9 h). As shown in Fig. 9a, the original bilayer actuator can quickly bend into a circle in pH = 2 aqueous solution during the short period. However, the erased bilayer hydrogel strip could hardly undergo any bending deformation, only displaying a slightly swollen state in 120 s. This phenomenon indicates that the actuation ability of the bilayer sample has been readily removed. Interestingly, the actuation ability can be re-written under certain conditions. To testify the feasibility, we placed the erased hydrogel sheet on top of the refrigerated planar steel board for a few minutes. As expected, the crystallized PVA domains could be regenerated in the matrix of the down layer in a controllable manner. After several cycles of treatment, it would give rise to an asymmetric distribution of crystalline domains across the top and down segments. As a result, the hydrogel strip regained the actuation ability, and could gradually bend towards one direction with a slower response speed in an acidic environment (as shown in Fig. 9b). All the results indicated that by asymmetrically distributing the PVA crystalline domains across the bilayer hydrogel matrix is an effective and convenient way to concept the hydrogel actuators.
 |
| | Fig. 9 (a) The process of bending deformation in pH = 2 aqueous solution for the erased and original chitosan/PVA bilayer hydrogel samples (weight ratio = 3:1). (b) The bending process for the re-written chitosan/PVA hydrogel strip in pH = 2 aqueous solution. | |
Conclusions
In this work, we presented a convenient method for fabricating bilayer hydrogel actuators based on the asymmetric distribution of crystalline domains across the bilayer structure. Benefitting from the characteristic of easy crystallization for PVA polymer chains during the freeze–thawing cycles, the as-prepared chitosan/PVA bilayer hydrogel actuator with the inhomogeneous structure displayed an asymmetric deformation when exposed to low pH conditions. This pH-sensitive bilayer hydrogel can be further manipulated into various devices to perform multiple desired, programmable 3D shape transformations, such as flower, helix, S-like shape and other particular variations. Moreover, the proposed gripper-like hydrogel actuator can successfully capture and transport the target object, revealing the actual loading ability. The in situ generated crystalline domains in the monolayer not only lead to the anisotropic swelling and subsequent bending behavior, but also render the bilayer device with excellent mechanical properties. It is believed that this strategy can be generally suitable for both synthetic and natural polymers, providing more possibilities of fabricating fascinating supramolecular devices in soft actuator fields.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Dr Qy Liu gratefully acknowledges the financial support from the Natural Science Foundation of China (51603195). Prof. Xj Xu gratefully acknowledges the financial support from the National Natural Science Foundation of China (21875167 and 21574102).
References
- C. Ma, W. Lu, X. Yang, J. He and X. Xiao, Adv. Funct. Mater., 2018, 28, 1704568 CrossRef.
- H. Xu, C. Han, S. Liu, X. Hao and Z. Sun, Ionics, 2020, 26, 6371 CrossRef CAS.
- F. Cheng, H. Chen and H. Li, J. Mater. Chem. B, 2021, 9, 1762 RSC.
- C. Jiang, Y. Li, H. Wang, D. Chen and Y. Wen, Sens. Actuators, B, 2020, 307, 127625 CrossRef.
- Y. Zhang, Y. Tao, K. Wang, S. Zhao and H. Cheng, J. Appl. Polym. Sci., 2021, 138, 50628 CrossRef CAS.
- J. A. Rui, A. Fw, A. Ql, Y. A. Xu, A. Ml, C. A. Yue, A. Wz, G. B. Hao, S. A. Peng and A. Gl, Sens. Actuators, B, 2020, 327, 128992 Search PubMed.
- M. S. Kalairaj, H. Banerjee, K. G. Lopez and H. Ren, Transport Porous Med., 2021 DOI:10.1007/s11242-021-01625-y.
- H. Li, Y. Liang, G. Gao, S. Wei and T. Chen, Chem. Eng. J., 2021, 415, 128988 CrossRef CAS.
- S. Kanai, Y. Watanabe, N. I. Shiblee, A. Khosla, J. Ogawa, M. Kawakami and H. Furukawa, ECS Trans., 2020, 98, 23 CrossRef CAS.
- Z. Han, P. Wang, G. Mao, T. Yin, D. Zhong, B. Yiming, X. Hu, Z. Jia, G. Nian, S. Qu and W. Yang, ACS Appl. Mater. Interfaces, 2020, 12, 12010 CrossRef CAS PubMed.
- D. Boso, E. Carraro, E. Maghin, S. Todros and M. Piccoli, Biomedicines, 2021, 9, 709 CrossRef CAS PubMed.
- J. Wang, J. Wang, Z. Chen, S. Fang, Y. Zhu, R. H. Baughman and L. Jiang, Chem. Mater., 2017, 29, 9793 CrossRef CAS.
- J. Yin, D. Zhang, Z. Xu, W. Fan and K. Sui, ACS Appl. Mater. Interfaces, 2020, 12, 49042 CrossRef CAS PubMed.
- M. Li, X. Wang, B. Dong and M. Sitti, Nat. Commun., 2020, 11, 3988 CrossRef CAS PubMed.
- J. Duan, X. Liang, K. Zhu, J. Guo and L. Zhang, Soft Matter, 2017, 13, 345 RSC.
- Q. Liu, Z. Dong, Z. Ding, Z. Hu, Y. Dan, H. Yang, N. Abidi and L. Wei, ACS Sustainable Chem. Eng., 2018, 6, 7052 CrossRef CAS.
- C. Yang, W. Wang, C. Yao, R. Xie, X. J. Ju, Z. Liu and L. Y. Chu, Sci. Rep., 2015, 5, 13622 CrossRef CAS PubMed.
- L. Chao, X. Hong, S. Qing, W. Gong, Y. Wang, Q. Chen, Y. Zhang, L. Liu and H. Sun, Adv. Mater. Interfaces, 2017, 4, 1601002 CrossRef.
- S. Zhou, B. Wu, Q. Zhou, Y. Jian, X. Le, H. Lu, D. Zhang, J. Zhang, Z. Zhang and T. Chen, Macromol. Rapid Commun., 2020, 41, 1900543 CrossRef CAS PubMed.
- K. T. Huang, K. Ishihara and C. J. Huang, Biomacromolecules, 2019, 20, 3524 CrossRef CAS PubMed.
- A. Gevorkian, S. M. Morozova, S. Kheiri, N. Khuu, H. Chen, E. Young, N. Yan and E. Kumacheva, Adv. Funct. Mater., 2021, 31, 2010743 CrossRef CAS.
- X. Le, W. Lu, J. Zhang and T. Chen, Adv. Sci., 2019, 6, 1801584 CrossRef PubMed.
- S. Xiao, M. Zhang, X. He, L. Huang, Y. Zhang, B. Ren, M. Zhong, Y. Chang, J. Yang and J. Zheng, ACS Appl. Mater. Interfaces, 2018, 10, 21642–21653 CrossRef CAS PubMed.
- X. X. Le, Y. C. Zhang, W. Lu, L. Wang, J. Zheng, I. Ali, J. W. Zhang, Y. J. Huang, M. J. Serpe and X. T. Yang, Macromol. Rapid Commun., 2018, 39, 1800019 CrossRef PubMed.
- L. Huang, R. Jiang, J. Wu, J. Song, H. Bai, B. Li, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1605390 CrossRef PubMed.
- H. Thérien-Aubin, Z. L. Wu, Z. Nie and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 4834 CrossRef PubMed.
- M. Liu, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima, M. Takata and T. Aida, Nature, 2015, 517, 68 CrossRef CAS PubMed.
- Y. Liu, M. Takafuji, H. Ihara, M. Zhu, M. Yang, K. Gu and W. Guo, Soft Matter, 2012, 8, 3295 RSC.
- A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413 CrossRef PubMed.
- C. Yao, Z. Liu, C. Yang, W. Wang, X. J. Ju, R. Xie and L. Y. Chu, Adv. Funct. Mater., 2015, 25, 2980 CrossRef CAS.
- D. Niu, W. Jiang, H. Liu, T. Zhao, B. Lei, Y. Li, L. Yin, Y. Shi, B. Chen and B. Lu, Sci. Rep., 2016, 6, 1 CrossRef PubMed.
- T. Chung, J. Han and Y. S. Kim, Nano Convergence, 2019, 6, 1 CrossRef PubMed.
- Q. Zhao, Y. Liang, L. Ren, Z. Yu, Z. Zhang, F. Qiu and L. Ren, J. Mater. Chem. B, 2018, 6, 1260 RSC.
- Z. Sun, C. Wei, W. Liu, H. Liu, J. Liu, R. Hao, M. Huang and S. He, ACS Appl. Mater. Interfaces, 2021, 13, 33404 CrossRef CAS PubMed.
- F. Zhang, J. Fan, P. Zhang, M. Liu, J. Meng, L. Jiang and S. Wang, NPG Asia Mater., 2017, 9, e380 CrossRef CAS.
- L. Zhao, J. Huang, Y. Zhang, T. Wang, W. Sun and Z. Tong, ACS Appl. Mater. Interfaces, 2017, 9, 11866 CrossRef CAS PubMed.
- W. J. Zheng, N. An, J. H. Yang, J. Zhou and Y. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 1758 CrossRef CAS PubMed.
- K. M. Lee, H. J. Kim, D. Jung, Y. Oh, H. Lee, C. Han, J. Y. Chang and H. Kim, ACS Omega, 2018, 3, 3096 CrossRef CAS PubMed.
- J. L. Holloway, A. M. Lowman and G. R. Palmese, Soft Matter, 2012, 9, 826 RSC.
- C. Dong, J. Zhou, D. Shi, Y. Song, X. Yu, W. Dong, M. Chen and D. Kaneko, Chem. Commun., 2021, 57, 3789 RSC.
- C. M. Hassan and N. A. Peppas, Macromolecules, 2000, 33, 2472 CrossRef CAS.
- M. He, Z. Wang, Y. Cao, Y. Zhao, B. Duan, Y. Chen, M. Xu and L. Zhang, Biomacromolecules, 2014, 15, 3358 CrossRef CAS PubMed.
- J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706 CrossRef CAS.
- H. Bing, Z. Yi and Y. Lin, Chem. Eng. J., 2014, 235, 207 CrossRef.
- M. Darabi, A. Khosrozadeh, Y. Wang, N. Ashammakhi, H. Alem, A. Erdem, Q. Chang, K. Xu, Y. Liu, G. Luo, A. Khademhosseini and M. Xing, Adv. Sci., 2020, 7, 1902740 CrossRef CAS PubMed.
- L. Xu, S. Gao, Q. Guo, C. Wang, Y. Qiao and D. Qiu, Adv. Mater., 2020, 32, 2004579 CrossRef PubMed.
- W. Chen, N. Li, Y. Ma, M. L. Minus and H. Zhu, Biomacromolecules, 2019, 20, 4476 CrossRef CAS PubMed.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279 CrossRef CAS.
- Q. Yu, L. A. Jauregui, W. Wu, R. Colby and J. Tian, Nat. Mater., 2011, 10, 443 CrossRef CAS PubMed.
- A. M. Hubbard, W. Cui, Y. Huang, R. Takahashi and J. P. Gong, Matter, 2019, 1, 674 CrossRef CAS.
- A. Sharkawy, M. F. Barreiro and A. E. Rodrigues, Carbohydr. Polym., 2020, 250, 116885 CrossRef CAS PubMed.
- Y. Shu, K. Khare and P. Lin, Adv. Funct. Mater., 2010, 20, 2550 CrossRef.
- H. Chen, Y. Liu, B. Ren, Y. Zhang, J. Ma, L. Xu, Q. Chen and J. Zheng, Adv. Funct. Mater., 2017, 27, 1703086 CrossRef.
- K. Li, Y. Wang, X. Chen, B. Sun and Y. Liu, Biomacromolecules, 2021, 22, 2684 CrossRef CAS PubMed.
- K. Li, X. Chen, Y. Wang, B. Sun, Z. Yuan and Y. Liu, ACS Appl. Polym. Mater., 2021, 3, 4101 CrossRef CAS.
- S. Lin, X. Liu and X. Zhao, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10244 CrossRef CAS PubMed.
- G. Li, Q. Yan, H. Xia and Y. Zhao, ACS Appl. Mater. Interfaces, 2015, 7, 12067 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b and
Qingye
Liu
*b and
Qingye
Liu