Less phagocytosis of viral vectors by tethering with CD47 ectodomain
Received
19th August 2021
, Accepted 24th November 2021
First published on 30th November 2021
Abstract
Many viral vectors, which are effective when administrated in situ, lack efficacy when delivered intravenously. The key reason for this is the rapid clearance of the viruses from the blood circulation via the immune system before they reach target sites. Therefore, avoiding their clearance by the immune system is essential. In this study, lentiviral vectors were tethered with the ectodomain of self-marker protein CD47 to suppress phagocytosis via interacting with SIRPα on the outer membrane of macrophage cells. CD47 ectodomain and core-streptavidin fusion gene (CD47ED-coreSA) was constructed into pET-30a(+) plasmid and transformed into Lemo21 (DE3) competent E. coli cells. The expressed CD47ED-coreSA chimeric protein was purified by cobalt-nitrilotriacetate affinity column and characterized by SDS-PAGE and western blot. The purified chimeric protein was anchored on biotinylated lentivirus via biotin–streptavidin binding. The CD47ED-capped lentiviruses encoding GFP were used to infect J774A.1 macrophage cells to assess the impact on phagocytosis. Our results showed that the overexpressed CD47ED-coreSA chimeric protein was purified and bound on the surface of biotinylated lentivirus which was confirmed via immunoblotting assay. The process to produce biotinylated lentivirus did not affect native viral infectivity. It was shown that the level of GFP expression in J774A.1 macrophages transduced with CD47ED-lentiviruses was threefold lower in comparison to control lentiviruses, indicating an antiphagocytic effect triggered by the interaction of CD47ED and SIRPα. Through the test of blocking antibodies against CD47ED and/or SIRPα, it was confirmed that the phagocytosis inhibition was mediated through the CD47ED-SIRPα axis signaling. In conclusion, surface immobilization of CD47ED on lentiviral vectors inhibits their phagocytosis by macrophages. The chimeric protein of CD47 ectodomain and core-streptavidin is effective in mediating the surface binding and endowing the lentiviral nanoparticles with the antiphagocytic property.
1. Introduction
Viral vectors are at the forefront of gene delivery vector technology with a large usage in clinical trials.1 Despite their potential, many viral vectors are highly vulnerable to the immune system of the host when administrated intravenously.2–4 Studies of viral therapeutics in murine models have revealed that liver and spleen uptake is a major impediment to systemic delivery, where the vast majority of the virus is immediately taken up from the circulation by resident macrophages of the mononuclear phagocyte system following intravenous infusion.5,6 To overcome the obstacles that will allow us to move toward efficacious viral gene therapy, several attempts have been made to improve the circulatory persistence and efficacy of viral vectors. An intriguing strategy to overcome the host immune response is to hide the virus from immune scrutiny. This approach relies upon the use of an immunologically inert cell carrier (e.g., mesenchymal stem cells, tumor-infiltrating lymphocytes)7,8 that is infected ex vivo followed by the administration to mice. Another immune evasive approach is to chemically shield viruses with polymers. Chemical modification of viral coat proteins by conjugation of biocompatible hydrophilic polymers, such as N-(2-hydroxypropyl)methacrylamide and polyethylene glycol masks viral surface epitopes, and therefore provides viruses the ability to evade the humoral and cellular immune response and extend the circulation times to facilitate targeting.9–16
An alternative strategy is to disguise foreign viral particles as “self” using a transmembrane protein CD47. This molecule has been suggested to function as a “marker of self” on red blood cells since in the absence of CD47 such cells are cleared rapidly from the bloodstream by splenic red pulp macrophages.17 This self-marker protein protects red cells from elimination by binding to a cognate receptor, signal regulatory protein α (SIRPα) on macrophage cell outer membranes.18,19 It was also shown that evasion of phagocytosis through up-regulation of the antiphagocytic antigen CD47 is the mechanism by which tumor cells escape immune surveillance.20–22 In fact, CD47 analogues have also been encoded by myxoma virus23 to escape this type of immune surveillance. Therefore, CD47-SIRPα interaction may represent a viable approach for the design of immune-evading viral vectors. Sosale et al.24 used HEK 293T cells transduced with a lentivector encoding CD47-GFP and puromycin resistance transgenes as a CD47-overexpressing producer cell line to generate CD47-lentivectors. It was found that CD47-lentivectors showed prolonged circulation in lung cancer xenografts and the macrophages transduced with CD47-lentiviruses were threefold lower comparing to the ones done with lentiviruses without CD47 functionalization. Another published study by Milani et al.25 using a multiple-plasmid co-transfection method to generate CD47-shielded lentiviral vectors from transfected HEK 293T cells. It showed that CD47-shielded lentiviral vectors improved gene therapy in non-human primates with diminished phagocytosis by liver macrophages and consequent immune sensing. The enhanced transduction contributed by the prolonged circulation of the lentiviral particles augmented hepatocyte gene transfer efficiency. Both of the aforementioned “inside-out” approaches generate CD47-expressing lentiviral particles involving overexpressing the full length of CD47 on the lentiviral producer cell line (i.e., HEK 293T). Thus, when viral particles bud out of producer cells, they pinch off the cell membrane already expressing CD47 protein. The level of CD47 protein generated via such cell machinery of virus is difficult to tweak than having the protein of interest tethered from outside. Moreover, since HEK 293T cells are limited to two-dimensional space for cultivation, large-scale production of CD47-lentivirus using suspension HEK 293F cells needs to re-examine all of the operating conditions which are time-consuming and costly.
Since the diminution of macrophage phagocytosis via CD47-SIRPα interaction mainly relies on the ectodomain of CD47 rather than the full-length protein, the “outside-in” approach was attempted in the present study by tethering VSV-G pseudotyped lentiviral vectors with CD47 ectodomain via biotin–streptavidin affinity. Ectodomain of mouse CD47 fused with core streptavidin protein (CD47ED-coreSA) was constructed, expressed, and purified with some modifications of a previously reported paper.26 The CD47ED-coreSA chimeric protein was further tethered on the biotinylated lentivirus obtained from HEK 293T cells pre-treated with biotin-lipid. The immunoblotting assay demonstrated the presence of CD47ED on the surface of the biotinylated virus through biotin–streptavidin binding. Our results showed that the infectivity of CD47ED-tagged lentivirus to murine J774A.1 macrophage was much less in comparison with mock lentivirus, indicating the signal created via the interaction of CD47ED tethered on the surface of lentivirus and SIRPα on macrophage cell outer membrane led to the suppression of phagocytosis. Moreover, the levels of secreted proinflammatory cytokines (IL-6 and TNF-α) were decreased if the lentiviral particles were anchored with CD47ED.
2. Materials
High Fidelity Phusion polymerase, Quick-Load Purple 2-log DNA ladder, Fast digest restriction enzymes (XhoI, EcoRI, and BamHI), T4 DNA ligase, T7 Express Lemo21(DE3) competent E. coli, L-rhamnose, and NEB 5-alpha competent E. coli were all purchased from New England Biolabs (Ipswich, MA, USA). Plasmid Maxi Kit was purchased from QIAGEN (Valencia, CA, USA). Dulbecco's modified Eagles’ medium (DMEM) culture media, penicillin–streptomycin, 0.25% trypsin–EDTA, B-PER bacterial protein extraction reagent, HaltTM protease inhibitor EDTA-free, streptavidin–fluorescein isothiocyanate (SA-FITC), fetal bovine serum (FBS), HisPur™ cobalt resin, Coomassie brilliant blue R 250, ECL western blotting substrate, Tween-20, bicinchoninic acid (BCA) protein assay kit including bovine serum albumin (BSA) standard, mouse TNF-α ELISA kit and IL-6 ELISA kit, streptavidin monoclonal antibody, protein concentrator PES MWCO = 10 kDa, and methanol were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). Biotin-Cap-PE was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Mouse IgG HRP (horseradish peroxidase)-conjugated antibody was bought from R&D Systems (Minneapolis, MN, USA). Anti-mouse CD47 monoclonal antibody was purchased from BioLegend (San Diego, CA, USA). Phosphate buffer saline (PBS) was purchased from Boston BioProducts (Ashland, MA, USA). Maxi columns were purchased from G-Biosciences (St. Louis, MO, USA). Ethanol, 2-(4-hydroxyazobenzene) benzoic acid (HABA), hexadimethrine bromide (Polybrene), isopropyl β-D-1-thiogalactopyranoside (IPTG), lysogeny broth (LB) media, imidazole, sodium hydroxide, polyethyleneimine (PEI), and potassium phosphate were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2-(N-Morpholino) ethanesulfonic acid (MES) was purchased from TCI (Portland, OR, USA). Kanamycin sulfate and dimethyl sulfoxide (DMSO) were purchased from Santa Cruz Biotech (Dallas, TX, USA). Tris/glycine/SDS buffer, Laemmli sample buffer, Tris-buffer saline (TBS), TGX FastCast acrylamide solutions, and polyvinylidene difluoride (PVDF) membrane (0.2 μm) were purchased from Bio-Rad (Hercules, CA, USA). 2-Mercaptoethanol was purchased from Gibco (Grand Island, NY, USA). Tris-hydrochloride (Tris-HCl) was bought from Promega (Madison, WI, USA). Acetic acid was bought from J. T. Baker (Philipsburg, NJ, USA). Trypan blue was purchased from BTC Beantown chemical (Hudson, NH, USA). J774A.1 macrophage cell line was obtained from ATCC (Manassas, VA, USA).
3. Methods
3.1 Construction of mouse CD47ED-coreSA encoding plasmid
Core-streptavidin (coreSA) was amplified from pSTE2-215 (yol)27 by polymerase chain reaction (PCR) using forward 5′-AGATCCGAATTCGGTGCTGCTGAAGCAGGT-3′’ and reverse primers 5′-ATTATACTCGAGGGAGGCGGCGGACGGCTT-3′. A commercially available clone of mouse CD47 in pCMV-SPORT6 vector was procured from Harvard Medical School (Boston, MA, USA). The ectodomain of mouse CD47 (CD47ED) was PCR amplified using forward primer 5′-GCGATGGCCATGGATATCATGCTACTGTTTAGTAACGTCA-3′ and reverse primer 5′-GCTCGAATTCGGATCCTCCTTTCTCCTCCTCGTAAGAACAG-3′. A 50 μL PCR was set up for each DNA by mixing the following components: 1× Phusion HF buffer, 200 mM dNTPs, 0.5 μM forward primer, 0.5 μM reverse primer, 1 μg DNA, 1.0 unit of Phusion DNA polymerase, and reaction volume was made up with nuclease-free water (New England Biolabs). The PCR for both genes were carried out using a T-100 thermocycler (Bio-Rad) with an initial denaturation temperature of 98 °C for 30 s, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 15 s, and extension at 72 °C for 15 s. The final extension was set for 5 min at 72 °C and PCR products were purified using a Monarch PCR & DNA cleanup kit (New England Biolab). Finally, PCR products were confirmed by 1% agarose gel electrophoresis. CoreSA was digested and cloned into pET-30a(+) between XhoI and EcoRI sites to yield plasmid pME005. Further, mouse CD47 ectodomain was inserted into pME005 between the EcoRI and BamHI sites yielding plasmid pME006.
3.2 Expression of CD47ED-coreSA chimeric protein
The expression of CD47ED-coreSA fusion gene encoded in pET-30a(+) plasmid transformed into Lemo21(DE3) was performed using previously reported procedures.26 Briefly, Lemo21 (DE3) competent E. coli cells carrying pME006 plasmid were grown in 1 liter of LB media containing 50 μg mL−1 kanamycin and 0.5 mM of L-rhamnose at 37 °C with shaking at 225 rpm. When optical density at 600 nm (OD600) reached to 0.6–0.8, culture was induced with 0.4 mM IPTG and kept shaking overnight at 18 °C. Then, cells were harvested by centrifugation at 4500 × g for 15 min and cell pellets were resuspended in a lysis buffer containing proprietary non-ionic detergent B-PER supplemented with 50 mM Tris–HCl, and 1× EDTA-free protease inhibitor (pH 7.5). The lysate was incubated at room temperature (RT) for 20 min and sonicated under cold conditions for 30 min with power output set at 5 of a tip sonicator (Misonix, Farmingdale, NY, USA). The bacterial lysate was centrifuged for 20 min at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, and the supernatant was collected and purified (i.e., soluble crude protein).
000 × g, and the supernatant was collected and purified (i.e., soluble crude protein).
3.3 Purification of CD47ED-coreSA chimeric protein
The purification of CD47ED-coreSA chimeric protein was carried out by immobilized metal affinity chromatography (IMAC) using HisPur™ cobalt nitrilotriacetic acid (Co-NTA) resin according to the manufacturer's instructions and protocol. In brief, the Co-NTA resin was equilibrated with two resin-bed volumes of equilibrium buffer (10 mM imidazole, 50 mM Na2HPO4, 300 mM NaCl, pH 7.4) in the affinity column. Mixture of crude protein and equilibrium buffer (1:1 ratio) was prepared and then added to the equilibrated Co-NTA resin. Polyhistidine tagged CD47ED-coreSA chimeric protein in the crude protein was bound to the resin by gentle shaking at 4 °C for 2 hours. After collecting the non-binding protein flow-through, the resin was washed with five resin-bed volumes of wash buffer (10 mM imidazole, 50 mM Na2HPO4, 300 mM NaCl, pH 7.4) or until the absorbance of wash-fraction at OD280 approached the minimum baseline. The target protein was eluted out from the resin with five resin-bed volumes of the elution buffer (250 mM imidazole, 50 mM Na2HPO4, 300 mM NaCl, pH 7.4) or until the absorbance of elution–fraction at OD280 approached the minimum baseline. The eluted protein fractions were concentrated using polyethersulfone (PES) membrane with a molecular weight cut off (MWCO) equal to 10 kDa. The protein solution was aliquoted and stored at −80 °C until further use. The coreSA protein used for the control group was expressed and purified from plasmid pME005 (pET-30a(+)-coreSA) with the same procedures mentioned above.
3.4 Characterization of CD47ED-coreSA chimeric protein
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the expressed recombinant histag-chimeric protein was carried out on a vertical slab assembly using polyacrylamide gels run under denaturing conditions. The 12% polyacrylamide gels prepared by Mini-PROTEAN Tetra handcast systems as guided by Bio-Rad were used. 15μL of each sample was diluted to 1:1 ratio with 2× Laemmli sample loading buffer supplemented with 5% (v/v) of 2-mercaptoethanol and heated to 95 °C for 5 min prior to loading. Electrophoresis was performed with Tris-glycine electrode buffer (1×), pH 8.3 at 200 V for 40 min; the gels were stained for three hours with Coomassie brilliant blue R-250. For western blot analysis, three electrophoresed samples were transferred to separate nitrocellulose membranes 0.2 μm (Bio-Rad) by Trans-Blot® semi-dry system (Bio-Rad) conducted at 20 V for 1 hour. Next, membranes were immersed in blocking buffer (5% BSA in TBST) for 1 hour at RT. The blots were washed with TBST buffer thrice for 5 min each and then incubated separately with primary antibodies solutions of anti-histag monoclonal antibody, anti-streptavidin monoclonal antibody, and anti-mouse CD47 monoclonal antibody at 4 °C overnight. After incubation, the blots were washed thrice again and incubated separately with secondary antibody solutions (1:1000 dilution of HRP-labeled mouse IgG in blocking buffer) at RT for 2 hours. Finally, after gentle washing, the blots were developed using ECL substrate and imaged using a chemiluminescence imager (PXi Syngene, Frederick, MD, USA). BCA protein assay was performed to determine the concentration of CD47ED-coreSA chimeric protein. According to the manufacturer's instructions, serial dilution of BSA solution (2 mg mL−1) was initially prepared as protein standards. Next, working reagent was prepared by mixing BCA reagents A and B at 50:1 ratio, respectively. 30 μL of BSA proteins standards and chimeric protein were mixed with 200 μL of working reagent and maintained at 37 °C for 30 min. Finally, the absorbance of all samples was measured at 562 nm by a microplate reader (Spectramax M2e, Molecular Devices, Sunnyvale, CA, USA) and concentration was calculated using a calibration curve of the BSA standard curve.
3.5 Preparation of PEI/DNA complexes
A pH neutralized solution of PEI (200 μg mL−1) was prepared and filter sterilized. Next, 6 μg of each DNA plasmid [all purchased from Addgene (Watertown, MA, USA)]: the packaging plasmid (pCMV delta R8.2, ID#12263), the virus envelope plasmid (pMD2.G, cat#12259), and the tracker EGFP-encoding plasmid (pLJM1-EGFP, cat#19319) were mixed with the PEI solution at N/P ratio of 5:1 (PEI:DNA). The complex was then incubated at RT for 30 min before being added to cultured cells.
3.6 Native lentivirus and biotinylated lentivirus production
HEK 293T cells were cultured in two T-75 flasks containing Dulbecco's modified eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37 °C in a 5% CO2 and humidified incubator. One of the flasks was additionally supplemented with 0.02 mg mL−1 biotin–lipid (Biotin-Cap-PE). Upon 80% confluency, cells in both flasks were transfected with the PEI/DNA complexes. Lentiviruses from both flasks were collected every 24 hours and replaced with fresh culture media supplemented with and without biotin–lipid. Native lentivirus and biotinylated lentivirus were separately filtered through a 0.45 μm syringe filter and stored at −80 °C for later use. Culture supernatant containing lentivirus collected on the third day post-transfection was concentrated using a previously reported method.28 Briefly, the supernatant was mixed with an equal volume of 320 μg mL−1 of a polybrene solution and incubated for 30 min at 37 °C. After incubation, the lentivirus was pelleted by centrifugation at 10000 × g for 10 min at 4 °C. The pellet was resuspended in 1 mL PBS and stored at −80 °C till further use.
3.7 Quantification of biotin on the lentivirus surface
Biotin amount on lentivirus surface was estimated with HABA assay according to a previously published report.29 Lentivirus culture was diluted 10-fold using the HABA/Avidin solution (6 μmol of HABA and 5 mg of avidin). To do so, a cuvette containing 0.9 mL of the HABA/Avidin solution was set to measure optical density at 500 nm (OD500) using a microplate reader. The biotinylated virus (0.1 mL) was added to the cuvette and then measured spectrophotometrically at OD500. The decrease in absorbance compared to the HABA/Avidin was recorded and the amount of biotin per lentivirus was calculated.
3.8 Infectivity of biotinylated and native lentivirus
HEK 293T cells were seeded in 6-well tissue culture plates. Upon reaching confluency of 50%, cells were infected with biotinylated and native lentivirus, respectively at a multiplicity of infection (MOI) of 12. After incubating for 3 hours, the media were replaced and the cells were imaged at 24, 48 and 72 hours post-infection, using fluorescence microscopy (DMi8 equipped with Leica EC3 camera, Leica Microsystems, Wetzlar, Germany). Post-completion, cells were harvested to measure GFP expression using a flow cytometer (Accuri C6, BD Biosciences, San Jose, CA, USA). For flow cytometry, cells were trypsinized and centrifuged. The cell pellets were washed with PBS, harvested by centrifugation, resuspended again in PBS, and incubated in ice. Fluorescence was determined for at least 100000 events. To define the timing of viral entrance, biotinylated lentivirus was concentrated and mixed with 0.02 mg mL−1 of streptavidin–FITC with gentle shaking at 4 °C for an hour. Unbound streptavidin–FITC was removed by centrifugation and washing of pellet with PBS. The streptavidin–FITC labeled virus particles were used to infect the J774A.1 macrophages, which were cultured in a 6-well tissue culture plate. Phagocytosis was monitored at an hourly interval up until 4 hours post-infection using fluorescence microscopy.
3.9 Biotinylated lentivirus immobilized with CD47ED-coreSA chimeric protein
The biotinylated viral suspension (500 μL) was mixed with 1 mL of CD47ED-coreSA chimeric protein and rotated gently for 1 hour at 4 °C to allow streptavidin–biotin binding. Unbound protein was removed by washing twice with PBS through centrifugation. The pellet was finally resuspended in PBS. Attachment of CD47ED-coreSA on the surface of biotinylated lentivirus was characterized using the dot blot assay. Five μL of the sample (CD47ED-capped virus) was spotted on the nitrocellulose membrane. After the sample was air-dried, the membrane was immersed in a blocking buffer for 1 hour at RT. The membrane was washed thrice for 5 min each using TBST buffer and allowed to react with primary antibody solution (1:1000 dilution of anti-mouse CD47 antibody in blocking buffer) for 1 hour at RT. The membrane was again washed thrice followed by incubation with secondary antibody solution (1:1000 dilution of HRP-conjugated mouse IgG antibody in blocking buffer) for 1 hour at RT. The image was detected using ECL substrate. Uncoated biotinylated lentivirus and CD47ED-coreSA protein were used as the negative and positive control, respectively.
3.10 Phagocytosis of biotinylated lentivirus immobilized with CD47ED-coreSA
J774A.1 macrophage cells were seeded on a 6-well tissue culture plate according to protocols reported previously. Upon 60% confluency, cells were infected with biotinylated lentivirus, coreSA-tagged lentivirus, and CD47ED-tagged lentivirus. Three hours post-infection, the cells were washed and replenished with culture media. Cells were imaged and analyzed for the percentage of phagocytosis (i.e., rate of infectivity) using fluorescence microscopy. HEK 293T cells were used as the non-phagocytic cell control and infected with biotinylated lentivirus tagged and non-tagged with CD47ED.
3.11 Blocking CD47ED/SIRPα interaction
An inhibition assay was performed to assess the impact of blocking CD47ED and/or SIRPα on the uptake of lentivirus by J774A.1 cells. Before the infection, CD47ED-modified lentivirus was treated with anti-mouse CD47 antibodies under gentle shaking conditions at 4 °C for an hour. Similarly, macrophages were treated with anti-SIRPα antibody for an hour before being infected with CD47ED-modified lentivirus. CD47ED-modified lentivirus and J774A.1 macrophages were both blocked with anti-mouse CD47 and anti-SIRPα antibodies, respectively, before incubating together. Unblocked CD47ED-modified lentivirus and macrophages were used as controls.
3.12 Effect of CD47ED on proinflammatory cytokine secretion
J774A.1 macrophages were seeded at a density of 5 × 104 cells cm−2 in 6-well plates, before treating with either CD47ED-modified lentivirus or native lentivirus for a period of 18 hours. The secretion of IL-6 and TNF-α in the culture media was determined by commercial ELISA assays. In short, the 96 well ELISA plate was coated overnight at 4 °C with 100 μL coating buffer containing either IL-6 or TNF-α capture antibody. The coating solution was then removed, and the wells were washed three times with a wash buffer (PBS + 0.05% Tween 20), followed by blocking with 200 μL 1 × ELISPOT diluent per well for 1 hour at RT. The wells were washed three times and 100 μL of culture media or appropriate standards and controls were added to the test plate for 2 hours at RT. Following removal of samples, the plate was again washed three times, and the IL-6 and TNF-α detection antibodies prepared in ELISPOT diluent were added in 100 μL per well for 1 hour at RT. The detection antibodies were removed, and the wells were washed three times. 100 μL per streptavidin-HRP in ELISPOT diluent was added and incubated at room temperature for 30 min. Following removal of the conjugate, the wells were washed five times, and 100 μL of 1 × tetramethyl benzidine solution was added to each well for 15 min at RT. Addition of 50 μL per well of 1 M phosphoric acid stopped the reaction. The resultant absorbance at 450 nm was determined on a microplate reader. The concentrations of IL-6 and TNF-α in the culture media were determined using the standard curves.
3.13 Statistical analysis
All data are presented as mean ± standard deviation from three independent experiments and analyzed using unpaired Student's t-test. P value less than0.05 is considered statistically significant.
4. Results
4.1 Construction of plasmid encoding CD47ED-coreSA chimeric gene
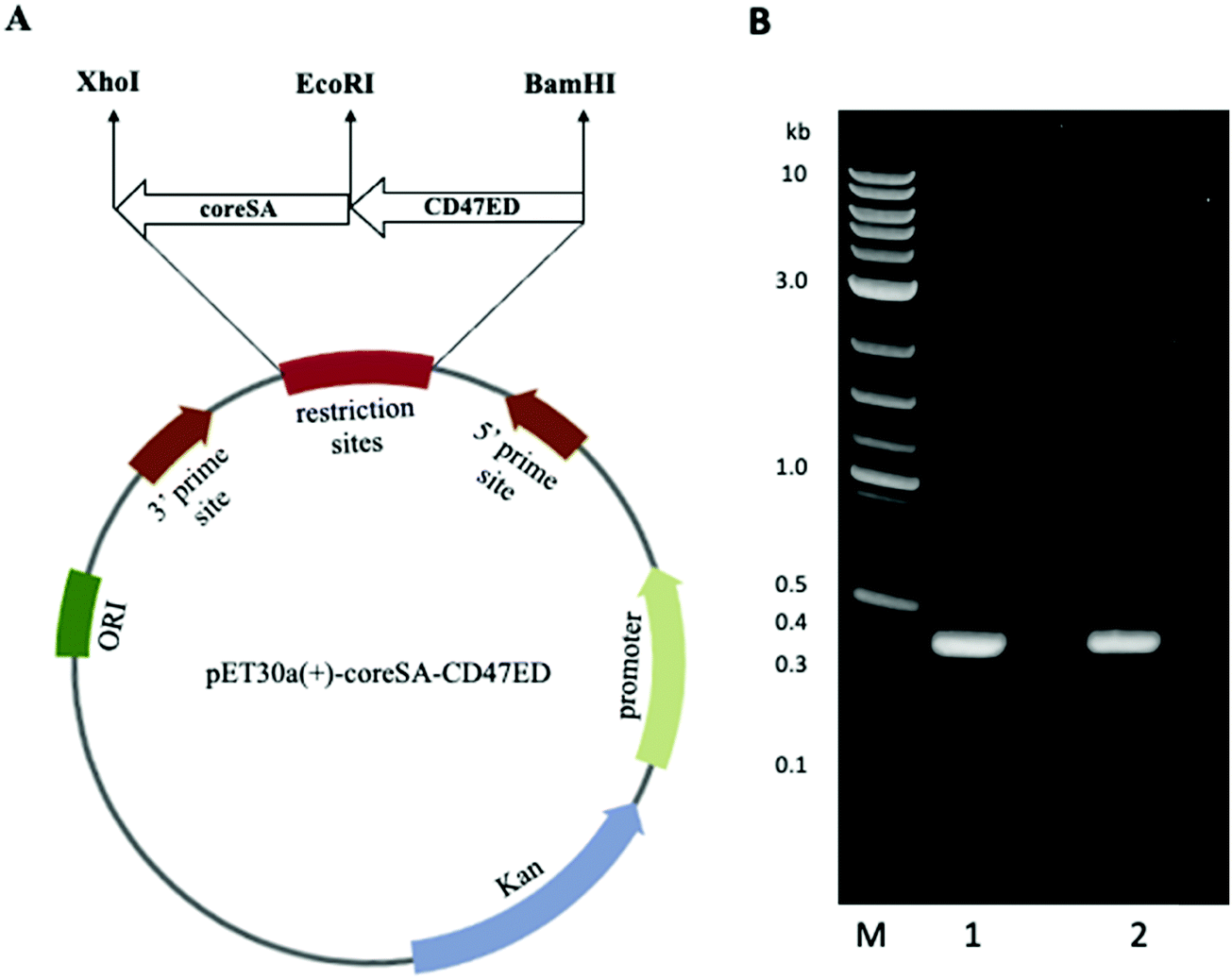
PCR products of CD47ED ∼400 bp and coreSA ∼387 bp were obtained by PCR. The pME006 was constructed by cloning PCR product of CD47ED and coreSA in between EcoRI/BamHI and XhoI/EcoRI respectively of pET-30a(+) as shown in Fig. 1A. The gene coding sequence of CD47ED (∼400 bp) and coreSA (∼387 bp) in pME006 were verified by PCR and running on 1% agarose gel shown in Fig. 1B. Given the sequence of CD47ED 400 bp (∼14.6 kDa) and coreSA 387 bp (∼14.2 kDa), the molecular weight of CD47ED-coreSA chimeric protein was estimated around 29 kDa which is consistent with the SDS-PAGE analysis shown in Fig. 2B.
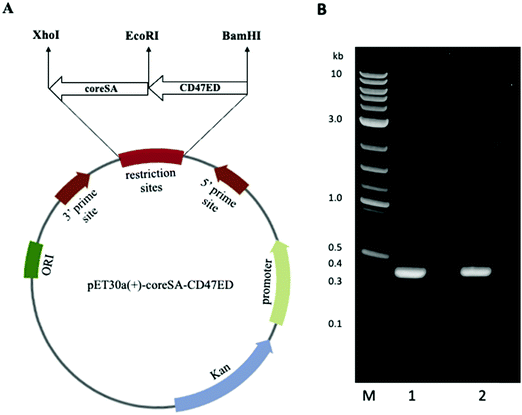 |
| | Fig. 1 Plasmid map and PCR amplification of genes of interest. (A) Vector map of plasmid pET-30a(+) showing cloning site of both genes of interest; ectodomain of mouse CD47 (CD47ED) and core streptavidin (coreSA) in between respective restriction sites; (B) DNA gel electrophoresis of PCR products of CD47ED in lane 1, coreSA in lane 2; M reflects the DNA ladder. | |
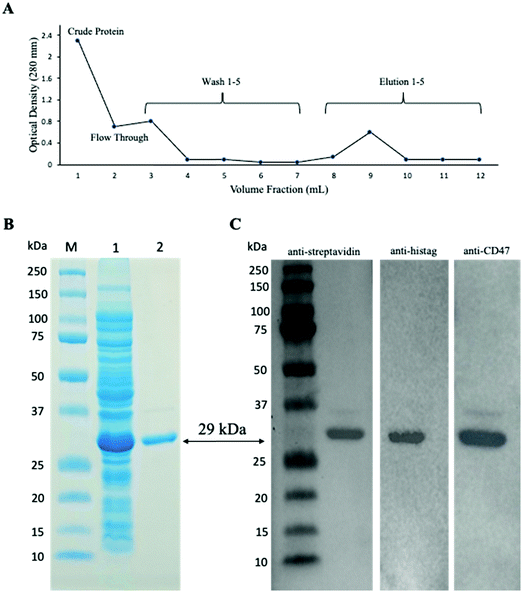 |
| | Fig. 2 Characterization of CD47ED-coreSA chimeric protein. (A) Elution profile of the protein purification process showing that most of the target protein eluted at the fraction 9; (B) SDS-PAGE image showing the protein marker (lane M), soluble crude protein with overexpressed chimeric protein around 29 kDa (lane 1), and purified concentrated chimeric protein ∼29 kDa (lane 2); (C) western blot analysis revealing reactivity of anti-streptavidin antibody, anti-histag antibody and anti-CD47 antibody against the purified chimeric protein. | |
4.2 Expression and purification of CD47ED-coreSA chimeric protein
The constructed pME006 vector was transformed into Lemo21(DE3) competent E. coli cells and screened using a kanamycin agar plate. A single colony was picked and cultured in LB media supplemented with 500 μM L-rhamnose and induced expression with inoculation of 400 μM IPTG. The target chimeric protein was purified from lysates using IMAC columns packed with Co-NTA resin, and the elution profile was plotted by measuring absorbance at OD280 of each wash and elution fraction (Fig. 2A). The soluble crude fraction along with purified chimeric protein was run on 12% SDS-PAGE followed by Coomassie staining, showing 29 kDa predominant band as predicted for the CD47ED-coreSA chimeric protein in the soluble crude extracts of the E. coli cells (Fig. 2B lane 2). The purified chimeric protein appeared as a monomeric band of 29 kDa (Fig. 2B lane 3). The elution fractions were concentrated using a protein concentrator (MWCO = 10 kDa) and the concentration was measured by BCA to be 383 ± 23 μg mL−1. The purified protein was further characterized by western blot analysis against three commercially procured antibodies: anti-histag antibody, anti-streptavidin antibody, and anti-mouse CD47 antibody, which bind the purified chimeric protein. Each antibody exhibits a single band at the target protein size of MW ∼ 29 kDa (Fig. 2C).
4.3 Infectivity of biotinylated lentivirus
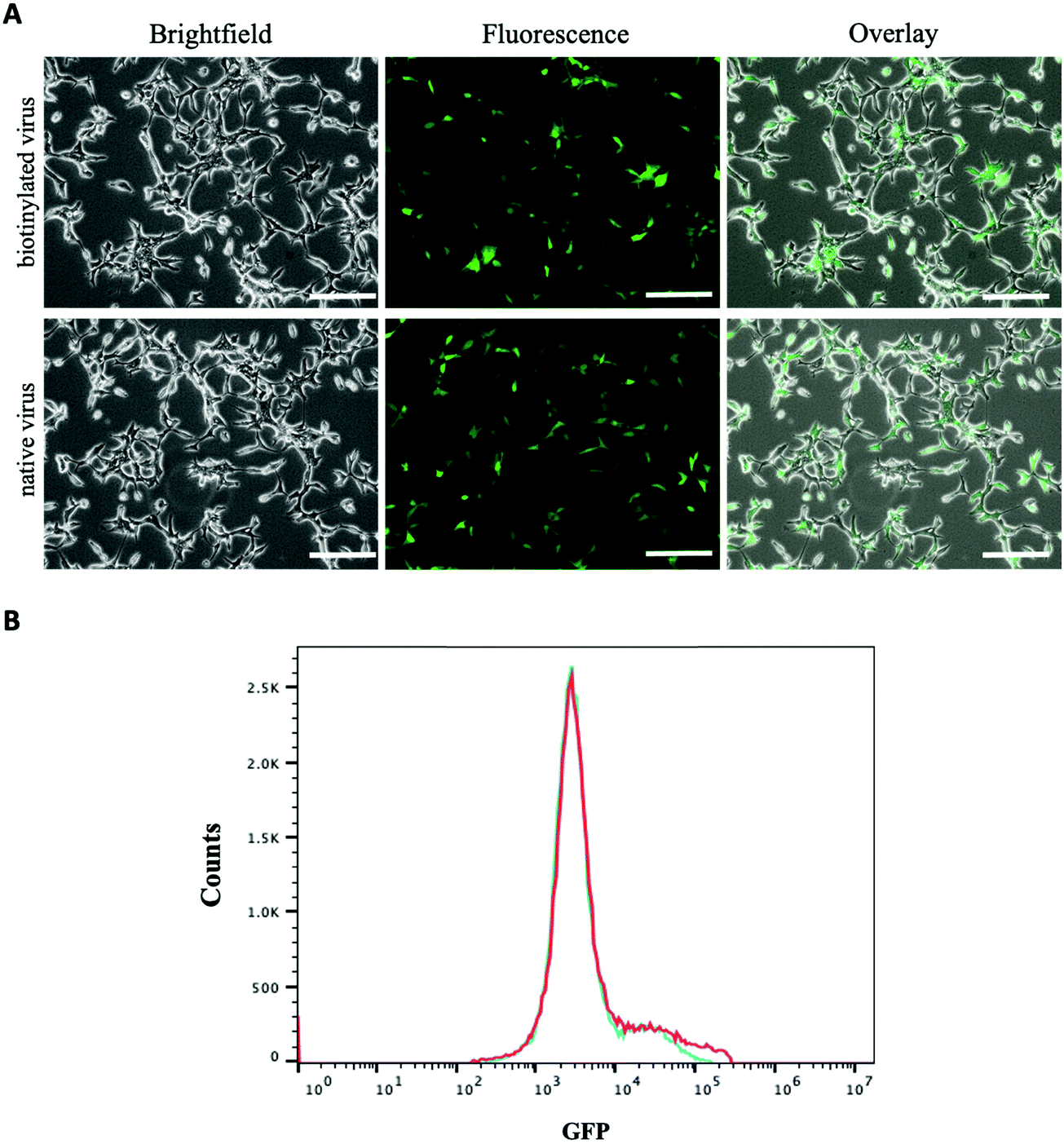
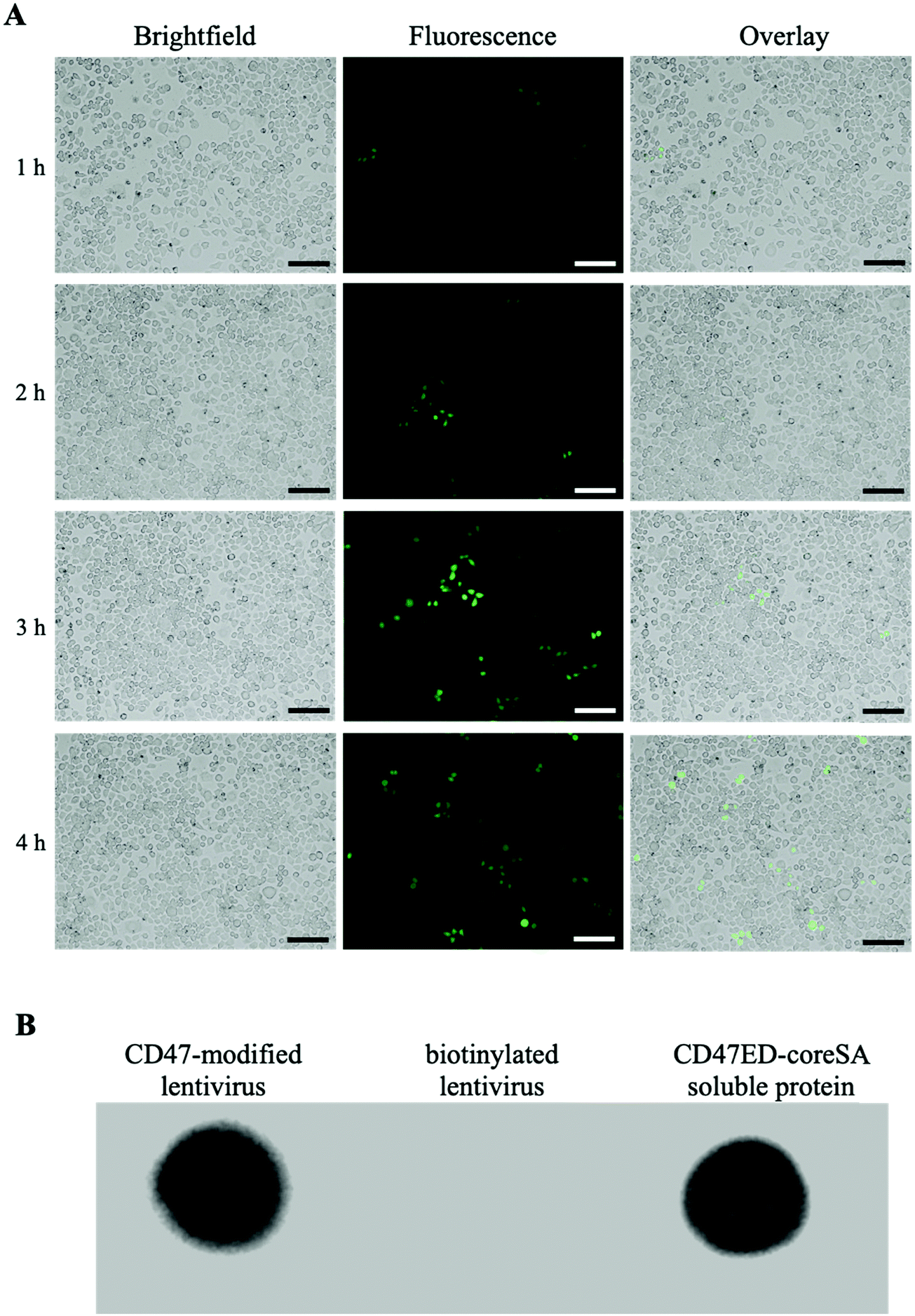
The amount of biotin bound on the lentivirus surface was quantitated using the HABA/Avidin assay. It was estimated about 1.27 × 10−7 μmoles of biotin per virion particle (equivalent to 7.65 × 1010 molecules). The effect of biotinylation on infectivity of lentiviral vectors was qualitatively or quantitatively assessed against HEK 293T cells using fluorescence microscopy and flow cytometry. Fig. 3A illustrates the levels of GFP expression in HEK 293T cells infected with native and biotinylated lentivirus are similar. The flow cytometry histograms further quantitatively confirmed that with mean fluorescent intensity (MFI) = 7087 for biotinylated lentivirus and MFI = 6935 for native lentivirus (Fig. 3B). As a result, the infectivity of biotinylated lentivirus remains intact after biotinylation. In addition, biotinylated lentiviruses were tagged with streptavidin-FITC before infecting J774A.1 macrophage cells to visualize cell entry of dye-labeled viruses. The fluorescence intensity of FITC confirmed the early time entry of the virus into macrophages within 1 hour. The fluorescence intensity was found to increase with the passage of time, which was observed on an hourly basis for up to 4 hours. Fig. 4A demonstrated the virus entering the macrophages within 4 hours post-infection was much higher comparing to 1 hour treatment, but similar to 3 hours post-infection. Therefore, the viral treatment for three hours was chosen as the time period for macrophage phagocytosis of lentiviruses in this study.
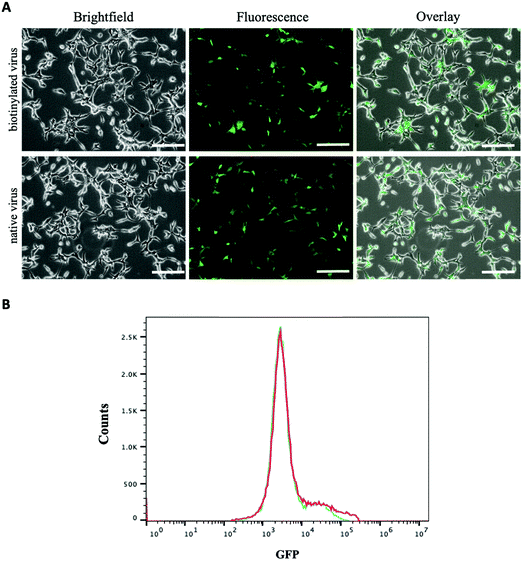 |
| | Fig. 3 Characterization of biotinylated and native lentivirus. (A) Showing phase contrast, fluorescence, and overlay images of HEK 293T cells treated with biotinylated and native lentivirus for 72 hours (scale bar denotes 100 μm); (B) representing the histograms of GFP intensity in HEK 293T cells treated with biotinylated (red, MFI = 7087) and native lentivirus (blue, MFI = 6935), respectively. | |
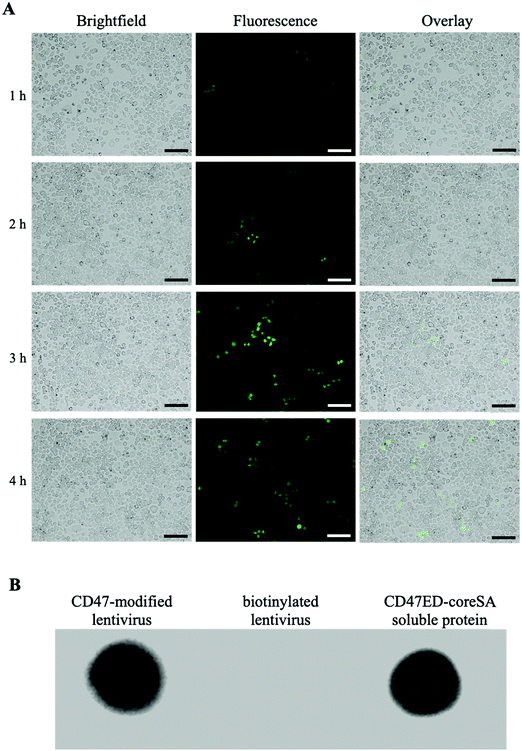 |
| | Fig. 4 Cell entrance of biotinylated lentivirus. (A) Biotinylated lentivirus conjugated to streptavidin-FITC was loaded onto J774A.1 macrophages. According to the level of green FITC fluorescence, lentiviruses entered macrophages 1 hour post-infection and continued the process at the end of 4 h observation (scale bar denotes 100 μm); (B) dot blot assay showing biotinylated lentiviral particles can bind CD47ED-coreSA via biotin–streptavidin and gotten detected by anti-CD47 monoclonal antibody, while biotinylated lentivirus only (negative control) revealing no CD47ED signal. Here, soluble CD47ED-coreSA protein was used as a positive control. | |
4.4 Characterization of biotinylated lentivirus tagged with CD47ED-coreSA
Five μL of biotinylated lentivirus tagged with CD47ED-coreSA was added to the PVDF membrane along with controls. Anti-mouse CD47 antibodies bound to biotinylated lentiviruses tagged with CD47ED-coreSA (shown in Fig. 4B) and soluble CD47ED-coreSA protein (positive control, shown in Fig. 4B), whereas the antibodies were not able to bind to biotinylated lentiviruses (shown in Fig. 4B). The presence of the CD47ED-coreSA protein and the CD47ED-modified virus were clearly expressed, while there was no expression of CD47ED-coreSA protein on the biotinylated lentivirus surface. Judging by the dot blot assay, the binding of CD47ED-coreSA with biotinylated lentivirus was successful.
4.5 Antiphagocytic effect of CD47ED-coreSA immobilized on biotinylated lentivirus
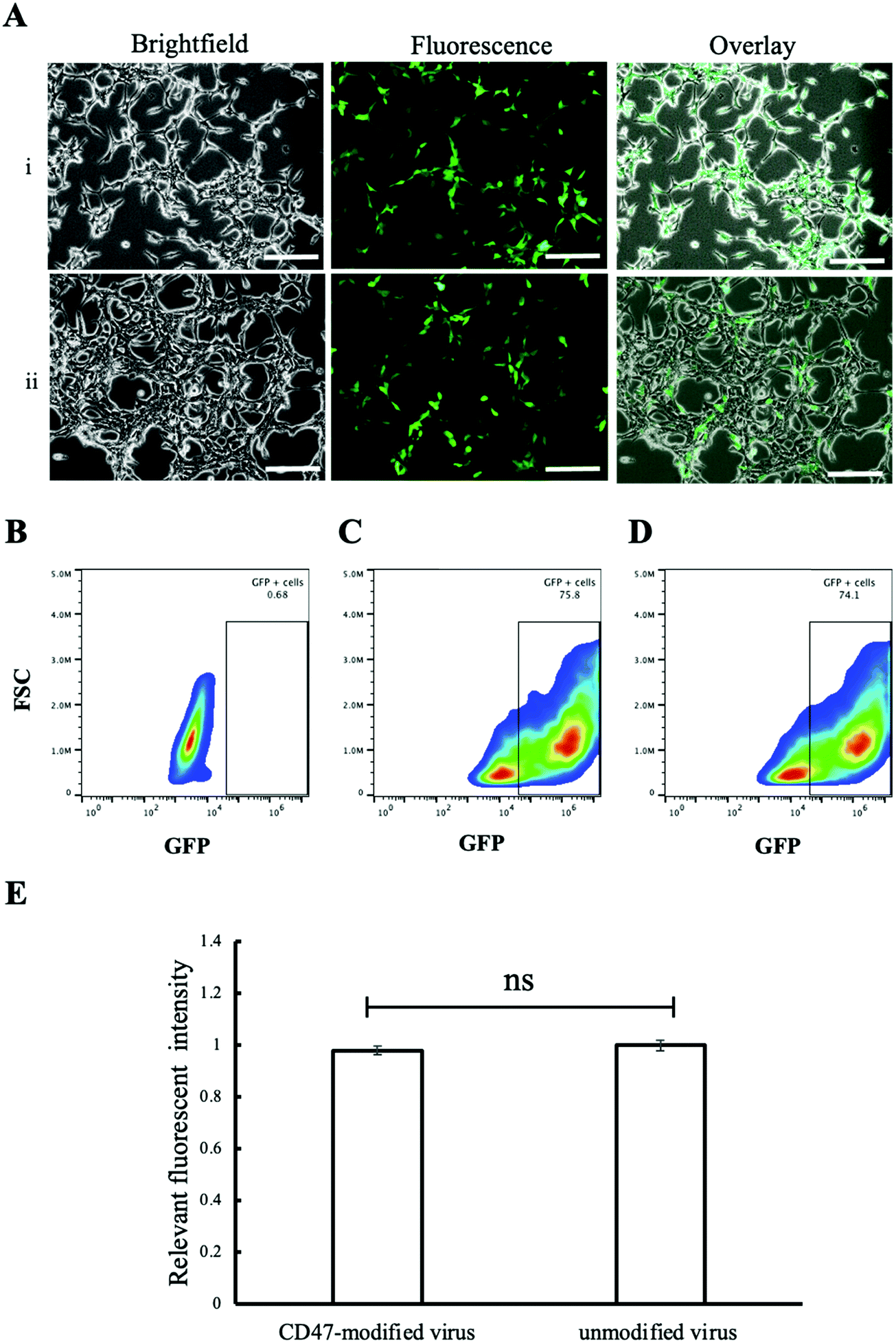
In vitro study was performed using two cell lines, phagocytic J774A.1 and non-phagocytic HEK 293T cells, to study the effect of CD47ED protein on macrophage activation. Phagocytic J774A.1 cells were treated with biotinylated lentivirus, coreSA-modified lentivirus, and CD47ED-modified lentivirus. After 72 hours of infection, the level of GFP expression in phagocytic J774A.1 macrophages treated with biotinylated lentivirus (Fig. 5A(i)) and coreSA-modified lentivirus (Fig. 5A(ii)) was way much higher than the macrophages treated with CD47ED-modified lentivirus (Fig. 5A(iii)). According to density plot analysis of GFP expression, the percentages of GFP expression are 0.18% (Fig. 5B), 62.2% (Fig. 5C), 55.9% (Fig. 5D), and 34.3% (Fig. 5E) for J77A.1 cells untreated with biotinylated lentivirus, treated with the biotinylated virus, coreSA-modified lentivirus, and CD47ED-modified lentivirus, respectively. The decrease of GFP expression level for the CD47ED-capped virus group was about 40–45% compared to the biotinylated virus and coreSA-tethered virus. The finding was furthered confirmed by fluorescence plate reading, the macrophage cells expressed 3-fold less GFP in the group treated with CD47ED-modified lentivirus in comparison with biotinylated lentivirus and coreSA-modified lentivirus (Fig. 5F). The decreasing of the phagocytosis activity illustrates that CD47ED-modified virus could interact with SIRPα receptor located on the surface of macrophages and inhibit their activation to prevent the virus entry, therefore GFP expression (i.e., virus infectivity) decreased. Unlike macrophages, HEK 293T cells treated with CD47ED-modified lentivirus could not inhibit phagocytosis due to the very low expression of SIRPα24 on their surfaces (Fig. 6A). Therefore, the percentage of GFP expression for HEK 293T cells treated with the biotinylated virus (75.8%, Fig. 6C) was similar to the ones infected with CD47ED-modified virus (74.1%, Fig. 6D). These findings were consistent with the fluorescence plate reading data (Fig. 6E) which demonstrated that no statistical significance between CD47-modified lentivirus and unmodified lentivirus.
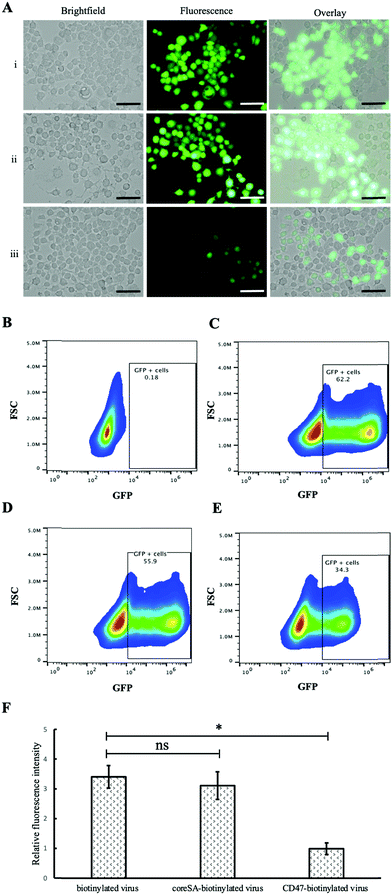 |
| | Fig. 5 Reduced macrophage phagocytosis of lentivirus tethered with CD47ED. (A) Showing brightfield, fluorescent, and overlay images of 774A.1 macrophage cells ingested (i) biotinylated lentivirus, (ii) coreSA-biotinylated lentivirus, and (iii) CD47ED-biotinylated lentivirus, respectively; (B–E) density plot of macrophages expressing GFP using flow cytometer – (B) untreated J774A.1 macrophages (negative control), (C) J774A.1 macrophages treated with unmodified biotinylated lentivirus (positive control), (D) 774A.1 macrophages treated with coreSA-modified biotinylated lentivirus, and (E) J774A.1 macrophages treated with CD47ED-modified biotinylated lentivirus; (F) measurement of relative fluorescence intensity of J774A.1 macrophages treated with biotinylated lentivirus, coreSA-biotinylated lentivirus, and CD47ED-biotinylated lentivirus at 72 hours post-treatment. Scale bar denotes 100 μm and data were presented as mean ± standard deviation (n = 3). *Denotes P < 0.05. | |
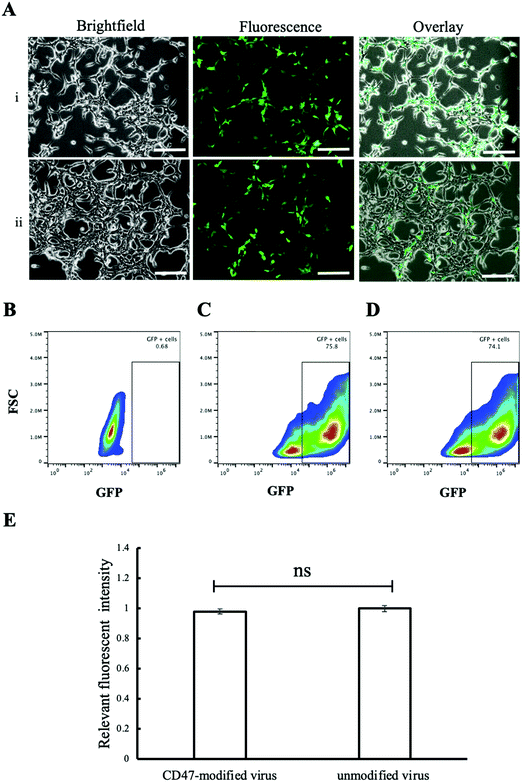 |
| | Fig. 6 Non-phagocytic cells treated with CD47ED-capped virus. (A) (i) HEK-293T cells treated with CD47ED-modified and (ii) unmodified biotinylated lentivirus for a period of 72 hours culture; (B-D) density plot of HEK-293T cells expressing GFP using flow cytometer – (B) untreated HEK-293T cells (negative control), (C) HEK-293T cells treated with unmodified biotinylated lentivirus (positive control), (D) HEK-293T cells treated with CD47ED-modified biotinylated lentivirus, and (E) measurement of relative fluorescence intensity of HEK-293T cells treated with CD47ED-modified lentivirus and unmodified lentivirus at 72 hours post-treatment. Scale bar denotes 100 μm and data were presented as mean ± standard deviation (n = 3); ns denotes not significant. | |
4.6 Inhibition of phagocytosis by blocking of CD47-SIRPα interaction
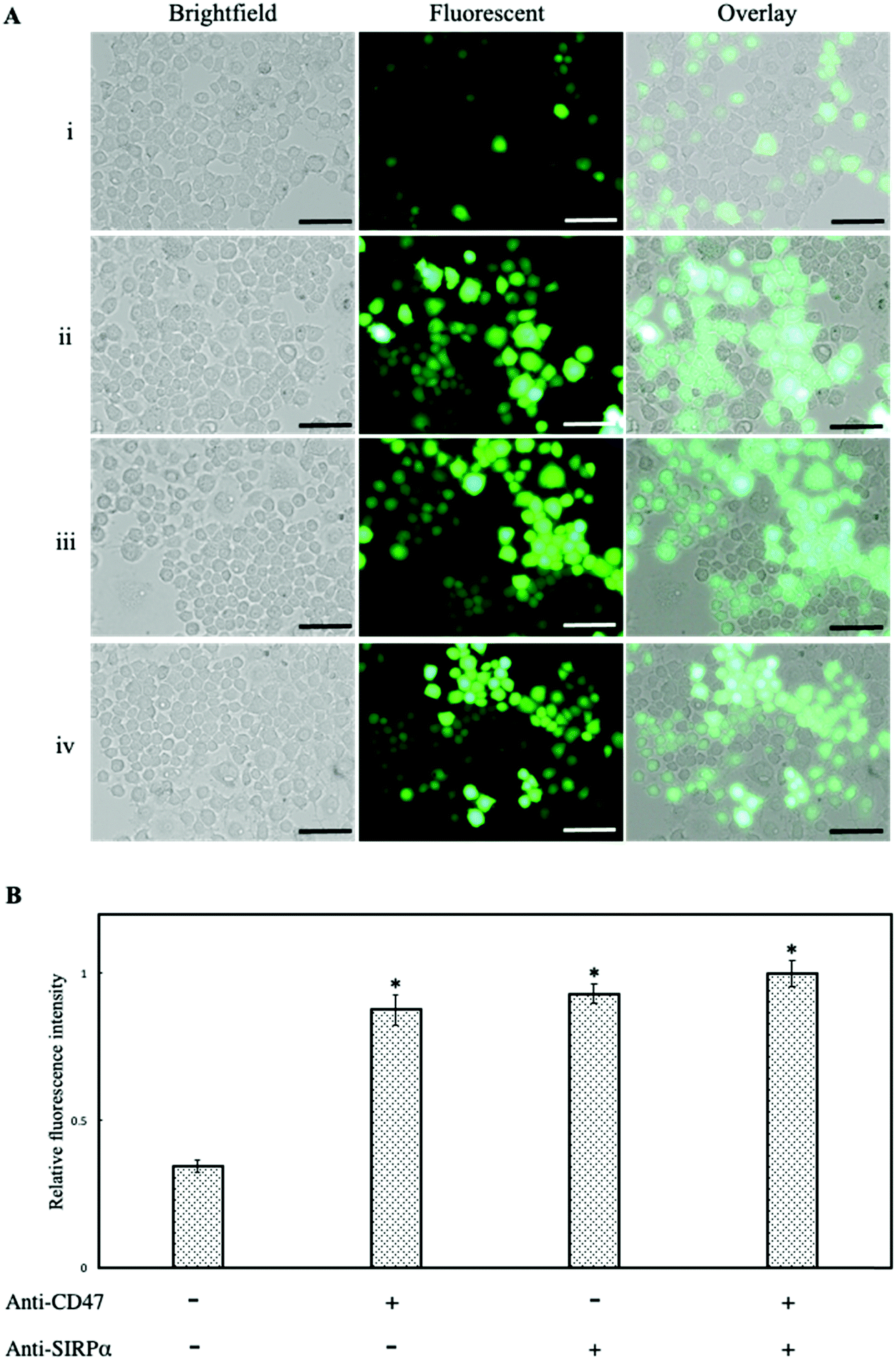
It has been previously reported that CD47 activates the SIRPα receptor on the macrophage surface suppressing phagocytic engulfment.30 To confirm the findings, anti-CD47 and anti-SIRPα monoclonal antibodies were used separately to block CD47ED on lentivirus and SIRPα on the macrophage cell membrane. Infection of macrophages was mediated with blocked CD47 but unblocked SIRPα, blocked SIRPα but unblocked CD47ED and blocked both CD47ED and SIRPα. Fig. 7A showed the blocking of CD47ED protein or the SIRPα receptor on the macrophage surface increased phagocytosis of virus engulfment by macrophages due to the disruption of CD47-SIRPα axis signaling, therefore increasing GFP expression. The relative change in fluorescence intensity was measured using a microplate reader, with respect to unblocked CD47ED-modified virus control, demonstrated a statistically significant increase in the phagocytosis of viral particles upon the blockage of either CD47ED, SIRPα, or both (Fig. 7B).
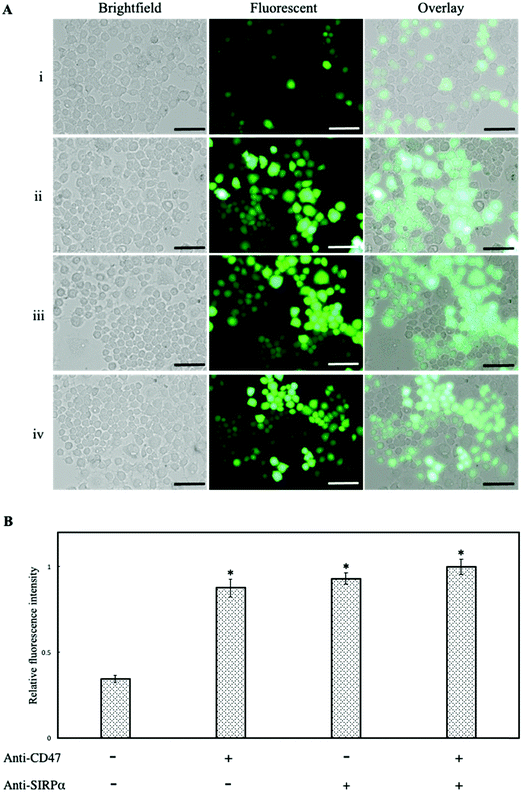 |
| | Fig. 7 Inhibition of macrophage phagocytosis via CD47ED-SIRPα axis signaling. (A) Showing brightfield, fluorescence, and overlay images of J774A.1 macrophages infected with mouse CD47ED-modified lentivirus with (i) unblocked, (ii) blocking CD47ED protein, (iii) blocking SIRPα receptor, and (iv) blocking CD47ED protein and SIRPα receptor together; (B) relative fluorescence intensity of J774A.1 macrophages incubated with and without antibodies blocking either CD47ED, SIRPα, or both, then treated with CD47ED-tethered lentivirus. Fluorescence intensity was read using a microplate reader. Scale bar denotes 100 μm and data were presented as mean ± standard deviation (n = 3). *P < 0.05 compared with the CD47ED-modified virus. | |
4.7 Effect of CD47ED on proinflammatory cytokine secretion
The secretion of proinflammatory cytokines (IL-6 and TNF-α) from macrophages in response to the treatment of CD47ED-modified lentivirus, biotinylated lentivirus, or untreated control was evaluated using ELISA. The amount of cytokine's secretion from untreated J774A.1 macrophages was 403 ± 84 pg mL−1 for TNF-α and 17 ± 3 pg mL−1 for IL-6. Fig. 8A showed J774A.1 macrophages treated with biotinylated lentivirus secreted about 1216 ± 121 pg mL−1 for TNF-α. However, the level of TNF-α secretion from macrophages treated with CD47ED-coated lentivirus was dropped to 883 ± 104 pg mL−1, which is about 27% less than the one treated with biotinylated lentivirus. Similarly, the amount of IL-6 secreted from cells treated with biotinylated lentivirus group was 48 ± 6 pg mL−1, which was decreased about 35.4% for the cells treated with CD47ED-modified lentivirus (32 ± 5 pg mL−1) (Fig. 8B).
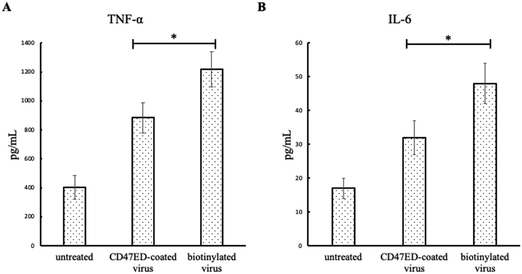 |
| | Fig. 8 IL-6 and TNF-α secreted by J774A.1 macrophages. Proinflammatory cytokines secretion of TNF-α (A) and IL-6 (B) from macrophages challenged with untreated, CD47ED-capped lentivirus, and biotinylated lentivirus for 18 hours. Data were presented as mean ± standard deviation (n = 3); *P < 0.05. | |
5. Discussion
The soluble and immobilized CD47 ectodomain have been previously reported in our studies and found there was inhibition of the phagocytosis activity due to the CD47-SIRPα interaction, resulting in fewer particles taken up by macrophages.31,32 However, the structure of virus and the components that make up the viral genome are much more complicated comparing to nonviral particles. Virus immunogenicity has become a huge challenge for gene therapy.33 The immune system within a body is able to recognize foreign bodies including vectors carrying transgenes and will generate antibodies to fight them, therefore, transgenes will be cleared by the mononuclear phagocyte system before arriving at their target and expressing the genes of interest.34 The present study investigated the efficacy of surface immobilization of CD47ED on lentiviral nanoparticles for overcoming macrophage-mediated phagocytosis. CD47-mediated stealth functionalization is an important biomimetic strategy used for the treatment of cancer and coating of implant surfaces.35,36 While its importance in therapeutic drug delivery has been discussed, its utility in preparing viral nanoparticles for gene delivery is less explored. In this study, we used an in vitro set up with J774A.1 macrophages and infected them with biotinylated lentiviral vectors immobilized with the CD47ED-coreSA chimeric protein and found a reduced uptake of particles in comparison to the mock group. It has been reported previously, the presence of CD47 on lentiviral vectors, inhibits phagocytosis of the virus.24,25 However, while all studies had the full length of CD47 expressed through the viral membrane,24,25 we have demonstrated that the viral particles can reduce phagocytosis through surface immobilization of CD47ED. This also indicates that the ectodomain of CD47 is functionally capable and immobilization has not resulted in structural vis-à-vis functional changes in the chimeric protein. To further confirm that the inhibition of phagocytosis is mediated through the CD47-SIRPα pathway,37,38 we simultaneously blocked the CD47ED protein on the lentivirus, the SIRPα protein on the macrophage membrane, and both to assess the impact on phagocytosis. Expectedly, inhibition of either or both protein moieties resulted in more virus engulfment by macrophages. Furthermore, macrophages are known to secrete proinflammatory cytokines in response to pathogens including viruses.39–41 When J774A.1 macrophages were treated with biotinylated lentivirus, the amounts of secreted TNF-α and IL-6 were increased; while macrophages that were not exposed to inflammatory stimuli showed low levels of TNF-α and IL-6. In this study, cytokines secretion from J774A.1 macrophages treated with CD47ED-capped lentivirus was significantly less than uncoated lentivirus, indicating phagocytic resistance attributed to CD47ED/SIRPα interaction which could downregulate proinflammatory cytokine secretion.
We have genetically engineered a chimera that combines the ectodomain of mouse CD47 with the affinity domain of core-streptavidin. It has been previously reported that full-length streptavidin (SA) forms an aggregate of higher-order and thus displays poor solubility. Contrastingly, core-streptavidin is found to be more soluble than full-length SA.42 Further, the combination of L-rhamnose and IPTG to induce protein overexpression along with native conditions, permitted threefold overexpression of the soluble CD47ED-coreSA chimeric protein. The method permitted overcoming the formation of insoluble proteins in inclusion bodies, which needs a higher recovery time and reduces protein yield.32,43 The use of the ectodomain is justified by the functional significance of the domain in regulating several immune responses through multiple cellular processes.44,45 Ultracentrifugation is a commonly used method for the collection of viral particles. However, it is a time-consuming process and the envelope proteins are not robust under high shear stress.46 For the separation of biotinylated viruses, conventionally used techniques involve the use of avidin-based affinity columns28 or avidin-coated paramagnetic particles.47 These methods necessitate the recovery of viral particles from the column thereby increasing the time of viral recovery. Here, we constructed biotinylated lentiviral vectors, concentrated and purified them using a simple flocculation method described previously by Landazuri et al.28 It is a simple, cost-effective, and efficient method including flocculation and disaggregation.
Our dot blotting assay confirmed the active binding of the chimeric protein on the biotinylated lentiviral surface. Moreover, their infectivity was not reduced as demonstrated by similar infectivity of both biotinylated and native lentivirus to HEK 293T cells. It should be noted that the percentage of GFP expression by J774A.1 macrophages (shown in Fig. 5) was less than that expressed by HEK 293T cells (shown in Fig. 6). This is because that HEK 293T cell expresses viral SV40 large T antigen. So, the transfected plasmid that has SV40 origin of replication (i.e., GFP plasmid used for this study) can replicate and express more and maintain high copy number. Independently, the ability of the lentiviral vectors tagged with CD47ED protein was also assessed and no significant difference was observed in the ability of the CD47ED-tagged virus to infect the cells. It is believed that the immobilization via biotin–streptavidin affinity has not introduced any surface changes on the virus thus enabling their uptake by the HEK 293T cells. This also indicates that despite the presence of the chimeric protein, the virus can infect the cells, thus mediating gene transfer, which is the preliminary function of lentivirus-based vectors.
While existing chemical biotinylation technologies have been reported to increase viral transduction of target cells,48–51 they are limited by the fact that the chemical biotin labeling methods involve random covalent attachment to virion amino acids and require prior purification of the labeled substrate. Moreover, one must play a balancing act of adding sufficient numbers of biotin for the expected functionality, while avoiding over-alkylation with biotin derivatives that may lead to inactivation of the biotinylated virus.52,53 In this study, an alternative approach – producing biotinylated viruses directly from living cells pre-fed with biotinylated lipid.54 Due to membrane fluidity, biotin-lipids are distributed in various viral budding sites. Subsequently, viruses are spontaneously labeled with biotin during viral natural assembly and budding processes. Since the whole viral biotinylation performed spontaneously in the viral replication cycle, operations such as incubation with conjugate chemicals (e.g., Sulfo-NHS-LC-Biotin) and additional purification before and/or after conjugation can be avoided. Thus, biotinylated viral bioactivity can be kept to the greatest extent compared with traditional chemical biotinylation of viruses. It should be noted that for free biotin, one coreSA binds four biotins. However, since one coreSA probably can only bind up to two biotins immobilized on the viral surface, the number of CD47ED molecules tagged on a virus via biotin-SA affinity should be no more than half of the number of biotin molecules per virion. Based on the biotin molecules calculated from the HABA/Avidin assay (i.e., 7.65 × 1010 molecules) and lentivirus diameter of ∼100 nm, the surface density of CD47ED tethered on the virion is estimated to be 1.22 × 1012 molecules μm−2. This value probably is overestimated due to the inaccuracy of HABA/Avidin assay.55 Nonetheless, the surface density of CD47ED capped on the viral surface is way above the minimum level of CD47 density on red blood cells (∼1000 molecules μm−2)56 and cancer cells (∼600 molecules μm−2).30 Hence, the amount of CD47ED coated on viruses should be sufficient over the threshold value to engage with SIRPα receptors on J774A.1 macrophages and lead to reduced phagocytosis. With that being said, since the estimated surface density of CD47ED tethered on the virion surface (i.e., 1.22 × 1012 molecules μm−2) is way higher than the reported threshold values, it is probably not feasible to achieve the desired range of thousand molecules μm−2 of CD47ED by just shortening the biotin-lipid treatment time from currently used 24 hours to 3 hours or even less. Instead of using in situ host cell biotinylation approach harnessed in this study, rapid biotinylation of virus using the lipid tail of function-spacer-lipid biotin (i.e., FSL–biotin) incorporating into lentiviral surface is speculated to be able to fine-tune the level of CD47ED on the virion surface for investigating the effect of immobilized amount of CD47ED-coreSA. It should also be noted that the results and discussion of this study were made merely based on in vitro assay. Further study is needed to examine if “outside-in” approach will provide expected outcomes using in vivo animal model.
6. Conclusion
The study, to the best of our knowledge, is a preliminary report on immobilization of ectodomain of mouse CD47 on the lentiviral membrane to assess evasion of macrophage phagocytosis. The study using recombineering to functionalize lentivirus presents a novel approach to biomimicry that does not involve genetic engineering of lentivirus. The surface immobilization of viral vectors with CD47 ectodomain can provide an alternative approach to endow therapeutic viruses with stealthy functionalization and achieve prolonged blood circulation for enhanced drug targeting.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the financial support from the National Cancer Institute (Grant No. 1 R03 CA216179-01).
References
- K. Lundstrom, Diseases, 2018, 6, 42 CrossRef PubMed.
- Y. K. Sung and S. W. Kim, Biomater. Res., 2019, 23, 1–7 CrossRef PubMed.
- K. Morizono and I. S. Y. Chen, Cell Cycle, 2005, 4, 854–856 CrossRef CAS PubMed.
- D. A. Carbonaro-Sarracino, A. F. Tarantal, C. C. I. Lee, M. L. Kaufman, S. Wandro, X. Jin, M. Martinez, D. N. Clark, K. Chun, C. Koziol, C. L. Hardee, X. Wang and D. B. Kohn, Mol. Ther.--Methods Clin. Dev., 2020, 16, 78–93 CrossRef CAS PubMed.
- S. Worgall, G. Wolff, E. Falck-Pedersen and R. G. Crystal, Hum. Gene Ther., 1997, 8, 37–44 CrossRef CAS PubMed.
- X. Ye, M. Jerebtsova and P. E. Ray, Hum. Gene Ther., 2000, 11, 621–627 CrossRef CAS PubMed.
- J. García-Castro, R. Alemany, M. Cascalló, J. Martínez-Quintanilla, M. del Mar Arriero, Á. Lassaletta, L. Madero and M. Ramírez, Cancer Gene Ther., 2010, 17, 476–483 CrossRef.
- P. Mayor, K. Starbuck and E. Zsiros, Gynecol. Oncol., 2018, 150, 361–369 CrossRef CAS.
- M. Z. Tesfay, A. C. Kirk, E. M. Hadac, G. E. Griesmann, M. J. Federspiel, G. N. Barber, S. M. Henry, K.-W. Peng and S. J. Russell, J. Virology, 2013, 87, 3752 LP–3759 CrossRef PubMed.
- M. A. Croyle, S. M. Callahan, A. Auricchio, G. Schumer, K. D. Linse, J. M. Wilson, L. J. Brunner and G. P. Kobinger, J. Virol., 2004, 78, 912–921 CrossRef CAS PubMed.
- K. D. Fisher and L. W. Seymour, Adv. Drug Delivery Rev., 2010, 62, 240–245 CrossRef CAS PubMed.
- J. Morrison, S. S. Briggs, N. Green, K. Fisher, V. Subr, K. Ulbrich, S. Kehoe and L. W. Seymour, Mol. Ther., 2008, 16, 244–251 CrossRef CAS PubMed.
- Y. Eto, Y. Yoshioka, Y. Mukai, N. Okada and S. Nakagawa, Int. J. Pharm., 2008, 354, 3–8 CrossRef CAS PubMed.
- K. Doronin, E. V. Shashkova, S. M. May, S. E. Hofherr and M. A. Barry, Hum. Gene Ther., 2009, 20, 975–988 CrossRef CAS PubMed.
- N. K. Green, C. W. Herbert, S. J. Hale, A. B. Hale, V. Mautner, R. Harkins, T. Hermiston, K. Ulbrich, K. D. Fisher and L. W. Seymour, Gene Ther., 2004, 11, 1256–1263 CrossRef CAS PubMed.
- R. Alemany, K. Suzuki and D. T. Curiel, J. Gen. Virol., 2000, 81, 2605–2609 CrossRef CAS PubMed.
- P. A. Oldenborg, A. Zheleznyak, Y. F. Fang, C. F. Lagenaur, H. D. Gresham and F. P. Lindberg, Science, 2000, 288, 2051–2054 CrossRef CAS.
- H. Wang, J. VerHalen, M. L. Madariaga, S. Xiang, S. Wang, P. Lan, P.-A. Oldenborg, M. Sykes and Y.-G. Yang, Blood, 2006, 109, 836–842 CrossRef PubMed.
- T. Matozaki, Y. Murata, H. Okazawa and H. Ohnishi, Trends Cell Biol., 2009, 19, 72–80 CrossRef CAS.
- S. Jaiswal, M. P. Chao, R. Majeti and I. L. Weissman, Trends Immunol., 2010, 31, 212–219 CrossRef CAS PubMed.
- M. P. Chao, A. A. Alizadeh, C. Tang, J. H. Myklebust, B. Varghese, S. Gill, M. Jan, A. C. Cha, C. K. Chan, B. T. Tan, C. Y. Park, F. Zhao, H. E. Kohrt, R. Malumbres, J. Briones, R. D. Gascoyne, I. S. Lossos, R. Levy, I. L. Weissman and R. Majeti, Cell, 2010, 142, 699–713 CrossRef CAS PubMed.
- X. W. Zhao, E. M. van Beek, K. Schornagel, H. Van der Maaden, M. Van Houdt, M. A. Otten, P. Finetti, M. Van Egmond, T. Matozaki, G. Kraal, D. Birnbaum, A. van Elsas, T. W. Kuijpers, F. Bertucci and T. K. van den Berg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18342–18347 CrossRef CAS.
- C. M. Cameron, J. W. Barrett, M. Mann, A. Lucas and G. McFadden, Virology, 2005, 337, 55–67 CrossRef CAS PubMed.
- N. G. Sosale, I. I. Ivanovska, R. K. Tsai, J. Swift, J. W. Hsu, C. M. Alvey, P. W. Zoltick and D. E. Discher, Mol. Ther.--Methods Clin. Dev., 2016, 3, 16080 CrossRef PubMed.
- M. Milani, A. Annoni, F. Moalli, T. Liu, D. Cesana, A. Calabria, S. Bartolaccini, M. Biffi, F. Russo, I. Visigalli, A. Raimondi, S. Patarroyo-White, D. Drager, P. Cristofori, E. Ayuso, E. Montini, R. Peters, M. Iannacone, A. Cantore and L. Naldini, Sci. Transl. Med., 2019, 11, 7325 CrossRef PubMed.
- A. Tarar, E. M. Alyami and C.-A. Peng, Methods Protocols, 2020, 3, 82 CrossRef CAS.
- S. Dübel, F. Breitling, R. Kontermann, T. Schmidt, A. Skerra and M. Little, J. Immunol. Methods, 1995, 178, 201–209 CrossRef.
- N. Landázuri, M. Gupta and J. M. Le Doux, J. Biotechnol., 2006, 125, 529–539 CrossRef.
- W.-W. Hu, M. W. Lang and P. H. Krebsbach, J. Gene Med., 2008, 10, 1102–1112 CrossRef CAS PubMed.
- M. A. Morrissey and R. D. Vale, bioRxiv, 2019, 752311 Search PubMed.
- Y. C. Hsu, M. Acuña, S. M. Tahara and C. A. Peng, Pharm. Res., 2003, 20, 1539–1542 CrossRef CAS.
- N. Salehi and C.-A. Peng, Biotechnol. Prog., 2016, 32, 949–958 CrossRef CAS.
- A. D. Miller, Hum. Gene Ther., 1990, 1, 5–14 CrossRef CAS PubMed.
- P. U. Emeagi, C. Goyvaerts, S. Maenhout, J. Pen, K. Thielemans and K. Breckpot, Curr. Mol. Med., 2013, 13, 602–625 CrossRef CAS PubMed.
- S. J. Stachelek, M. J. Finley, I. S. Alferiev, F. Wang, R. K. Tsai, E. C. Eckells, N. Tomczyk, J. M. Connolly, D. E. Discher, D. M. Eckmann and R. J. Levy, Biomaterials, 2011, 32, 4317–4326 CrossRef CAS.
- C. Guido, G. Maiorano, B. Cortese, S. D’Amone and I. E. Palamà, Bioengineering, 2020, 7, 111 CrossRef CAS PubMed.
- Y. Murata, T. Kotani, H. Ohnishi and T. Matozaki, J. Biochem., 2014, 155, 335–344 CrossRef CAS.
- B. Oronsky, C. Carter, T. Reid, F. Brinkhaus and S. J. Knox, Semin. Oncol., 2020, 47, 117–124 CrossRef CAS.
- R. Z. Murray, J. G. Kay, D. G. Sangermani and J. L. Stow, Science, 2005, 310, 1492 LP–1495 CrossRef.
- A. Aderem, J. Infect. Dis., 2003, 187, S340–S345 CrossRef CAS.
- A. P. Manderson, J. G. Kay, L. A. Hammond, D. L. Brown and J. L. Stow, J. Cell Biol., 2007, 178, 57–69 CrossRef CAS.
- T. Sano, M. W. Pandori, X. Chen, C. L. Smith and C. R. Cantor, J. Biol. Chem., 1995, 270, 28204–28209 CrossRef CAS.
- A. Singh, V. Upadhyay, A. K. Upadhyay, S. M. Singh and A. K. Panda, Microb. Cell Fact., 2015, 14, 41 CrossRef PubMed.
- E. J. Brown and W. A. Frazier, Trends Cell Biol., 2001, 11, 130–135 CrossRef CAS.
- K. Hayashida, A. H. Bartlett, Y. Chen and P. W. Park, Anatomical Record (Hoboken, N.J.: 2007), 2010, 293, 925–937 CrossRef CAS PubMed.
- J. C. Burns, T. Friedmann, W. Driever, M. Burrascano and J. K. Yee, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 8033–8037 CrossRef CAS PubMed.
- L. Chan, D. Nesbeth, T. MacKey, J. Galea-Lauri, J. Gäken, F. Martin, M. Collins, G. Mufti, F. Farzaneh and D. Darling, J. Virol., 2005, 79, 13190–13194 CrossRef CAS.
- J. S. Smith, J. R. Keller, N. C. Lohrey, C. S. McCauslin, M. Ortiz, K. Cowan and S. E. Spence, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8855–8860 CrossRef CAS.
- Q. Zhong, J. K. Kolls and P. Schwarzenberger, J. Biol. Chem., 2001, 276, 24601–24607 CrossRef CAS.
- P. Selvarangan, M. Gandham, K. Sanjay, J. A. Thompson and M. Castillas, J. Virol., 2002, 76, 12900–12907 CrossRef.
- B. Purow and K. Staveley-O’Carroll, J. Surg. Res., 2005, 123, 49–54 CrossRef CAS PubMed.
- M. A. Barry, S. K. Campos, D. Ghosh, K. E. Adams, H. Mok, G. T. Mercier and M. B. Parrott, Expert Opin. Biol. Ther., 2003, 3, 925–940 CrossRef CAS PubMed.
- M. B. Parrott, K. E. Adams, G. T. Mercier, H. Mok, S. K. Campos and M. A. Barry, Mol. Ther., 2003, 8, 688–700 CrossRef CAS PubMed.
- B. H. Huang, Y. Lin, Z. L. Zhang, F. Zhuan, A. A. Liu, M. Xie, Z. Q. Tian, Z. Zhang, H. Wang and D. W. Pang, ACS Chem. Biol., 2012, 7, 683–688 CrossRef CAS PubMed.
- K.-J. Sung, E. A. Miller and H. D. Sikes, Mol. Syst. Des. Eng., 2018, 3, 877–882 RSC.
- I. Mouro-Chanteloup, J. Delaunay, P. Gane, V. Nicolas, M. Johansen, E. J. Brown, L. L. Peters, C. Le Van Kim, J. P. Cartron and Y. Colin, Blood, 2003, 101, 338–344 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Ammar
Tarar
,
Ammar
Tarar