
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One step electrochemical fabrication of high performance Ni@Fe-doped Ni(oxy)hydroxide anode for practical alkaline water electrolysis†
Tao
Jiang
 *ae,
Xinge
Jiang
b,
Jaromír
Hnát
c,
Alena
Michalcova
d,
Indro
Biswas
a,
Regine
Reissner
a,
Vasileios
Kyriakou
e,
Fatemeh
Razmjooei
a,
Hanlin
Liao
b,
Karel
Bouzek
c and
Syed-asif
Ansar
a
*ae,
Xinge
Jiang
b,
Jaromír
Hnát
c,
Alena
Michalcova
d,
Indro
Biswas
a,
Regine
Reissner
a,
Vasileios
Kyriakou
e,
Fatemeh
Razmjooei
a,
Hanlin
Liao
b,
Karel
Bouzek
c and
Syed-asif
Ansar
a
aInstitute of Technical Thermodynamics, German Aerospace Center (DLR), Stuttgart 70569, Germany. E-mail: taojiang0510@gmail.com
bUBFC, ICB-PMDM-LERMPS UMR6303, Sevenans 90010, France
cDepartment of Inorganic Technology, University of Chemistry and Technology Prague, 166 28 Prague 6, Czech Republic
dDepartment of Metals and Corrosion Engineering, University of Chemistry and Technology Prague, 166 28 Prague 6, Czech Republic
eEnergy Systems & Conversions, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG, Groningen, The Netherlands
First published on 28th October 2022
Abstract
Oxygen evolution reaction (OER) is a rate-determining process in alkaline water electrolysis (AWE). Herein, we report a novel one-step oxidation–electrodeposition (OSOE) approach to generate core@shell nanoarrays-based AWE electrode with outstanding OER performances: an overpotential of 245 mV at 10 mA cm−2 (Tafel slope: 37 mV dec−1), and excellent stability under huge current densities. Moreover, the alkaline (AEL) cell equipped with NM-OSOE-23 anode recorded significant performance improvement of 200 mV lower voltage (2 A cm−1) compared with a similar cell used bare Ni mesh as an anode, which was contributed by notable enhancements of interface contact, anodic charge transfer, and mass transfer. These promising results are attributed to the constructed specific core@shell Ni@Fe-doped Ni(oxy)hydroxide nanoarray architecture on commercial nickel mesh. This study demonstrates this first reported OSOE can be commercialized to make highly efficient anodes enabling next-generation AWE.
Introduction
One of the grand challenges of this century is the transition to renewable energy systems, which are environmentally friendly, scalable, and highly efficient. In this context, the role of green hydrogen (H2) as an energy carrier is becoming successively a solid option for the deep decarbonization of global energy systems.1,2 Although various technologies are available for H2 production, water electrolysis technology powered by renewable energies, such as wind, solar, and hydro, is suggested to come out as a low-emission way to reserve excess electricity and produce high-quality green hydrogen.3 Water electrolysis to produce green H2 may be carried out under either acidic conditions, called polymer electrolyte membrane electrolyzer (PEMEL), or alkaline conditions, called alkaline electrolyzer (AEL). However, besides the efficiency, the lifetime and the cost govern whether PEMEL or AEL is the most promising system design for the scale-up application.4 Unlike PEMEL, in which highly expensive precious metal catalysts are used, AEL allows the use of less expensive catalysts for the hydrogen evolution reaction (HER) in the cathode and the oxygen evolution reaction (OER) in the anode.4,5 In addition, the AEL system has an expected higher operation lifetime of over 100 years than the PEMEL systems.6 Therefore, efficiency, durability, and cost are the main factors determining the electrolyzer technology for a large-scale application. Among various types of electrolyzer technologies, the alkaline electrolyzer is the most mature, durable, and affordable electrolyzer technology to produce hydrogen.7However, it has to be noted that the anodic reaction is sluggish owing to the complicated four-electron transfer process, which is the bottleneck in realizing efficient water electrolysis.8–11 To date, materials based on precious metals, such as IrO2 and RuO2, are still considered state-of-the-art OER electrocatalysts, but the shortage, high cost, and insufficient stability of these materials severely limit their scale-up application in industry.12–14 As a consequence, it is imperative to develop substitute electrocatalysts for OER based on cost-effective earth-abundant metals such as Fe, Ni, and Co.11,15,16 Particularly, Ni/Fe-based electrocatalysts have been examined as efficient catalysts for OER in alkaline electrolytes because of their synergistic effects between Fe and Ni.17–19 Concretely, the doping of the Fe element could tune Ni-based electrocatalysts' electronic structure and accelerate the evolution of the high-valence of Ni species, which could conversely lead to OER performance improvement.20–22 It is noteworthy that the most recently reported catalysts are mostly operated at low current densities (typically ≤100 mA cm−2). However, to compete with PEMEL, which is capable of operating at high current densities, industrial alkaline water electrolysis is highly required to be operated at huge current densities (e.g. ≥500 mA cm−2).19 This brings out big challenges for developing electrocatalysts. Although there are already some electrocatalysts that can drive water electrolysis with large current densities, the operation time is generally far from the need of the industry.19,23 Developing such electrocatalysts encounters a lot of obstacles. First of all, the gas production, at high current densities, is so vigorous that could break the binding between catalysts and current collectors. In this situation, self-supported catalysts are highly recommended.19,21,24–32 In addition, at high current densities, the catalysts suffer serious surface and structure reconstructions due to intense reactions, and eventually might essentially lose catalytic activity. Meanwhile, the high reaction rate also leads to extra demands on the charge and mass transfer efficiency. To efficiently deal with these problems, OER catalysts should be designed with fast mass transport, high efficiency of electron transfer, and abundant active sites. Hence, in situ generation of NiFe-based core@shell (CS) nanoarrays on collector skeleton would be one of the most feasible answers.19
On the other hand, the costs of both catalyst materials and the responding synthesis approaches should also be considered, in the case of the development of practical AWE electrodes. Electrodeposition is simple and cost-effective in preparing practical electrodes with nano-structured catalysts, compared with other usually used techniques such as hydro-/solvothermal procedures, co-precipitation, and physical or chemical vapor depositions. Moreover, this method allows nano-structured catalysts to be in situ grown on conductive substrates with excellent mechanical adhesion, thus avoiding the drawbacks of using additional binders. The structure, morphology, and loading of the catalysts can be easily regulated by controlling the parameters, such as applied potential/current density, additives, stirring speed, and deposition time. More importantly, this kind of method can be potentially scaled up for the production of large-scale practical electrodes for AWE.33
Based on the above discussion, herein a facile one-step oxidation-electrodeposition (OSOE) approach, for the first time, was developed to construct CS Ni@Fe-doped Ni(oxy)hydroxide, which can dramatically promote the kinetics of O2 evolution. Due to the inherent oxidizing solution environment, a trace Fe-doped Ni(oxy)hydroxide layer was in situ formed on the surface of the metallic Ni nanoarray growing on the commercial perforated nickel mesh (NM, 69% effective area) cathode, decreasing the electron transfer resistance within catalysts and improving the charge transferability. Notably, trace Fe was incorporated into the Ni(oxy)hydroxide shell, targeting to enhance its intrinsic catalytic activity. Specifically, the Ni metallic core facilitates the charge transport to the surface of the Fe-doped Ni(oxy)hydroxide shell, whereas the nanoarray structure enables fast ion diffusion and provides abundant active sites, which would not only lead to an excellent catalytic OER performance but also guarantee a significant improvement of AEL cell. Consequently, an outstanding OER performance, that is, an overpotential of 245 mV is obtained under a current density of 10 mA cm−2. The corresponding Tafel slope is 37 mV dec−1. Strikingly, it delivers a total catalyst loading mass activity of 1336 A g−1 and a high turnover frequency of 0.285 s−1, which is over 7-fold higher than state-of-the-art IrO2 and almost 30-fold higher than the without Fe-free CS Ni@Ni(oxy)hydroxide. Moreover, the electrode can also survive at 500 mA cm−2 for over 500 h without apparent degradation. To further validate the catalyst activity for the real application, the optimized catalyst was tested as the anode in the 4 cm2 AEL cell where bare NM was used as the cathode. Significant performance improvements were revealed, which led to a 200 mV lower voltage at 2 A cm−1 compared with a similar AEL cell but with commercial Ni mesh as an anode, which was contributed by notable enhancements of interface contact, anodic charge transfer, and mass transfer. These promising results are attributed to the constructed specific core@shell Ni@Fe-doped Ni(oxy)hydroxide nanoarray architecture on commercial nickel mesh. This work suggests that the developed OSOE is a very promising approach to not only engineering transition metal compounds (TMCs)-based CS nanostructures with the TM core and catalytically active (oxy)hydroxide substance as the shell but also constructing nanoarrays architecture with superior interface contact and mass transfer, directing the merits of two parts and synergistically proceeding alkaline water electrolysis. The approach we employed is highly reproducible, and the electrode size can be easily controlled, making it a promising strategy to advance the next generation of AWE technology.
Results and discussion
Synthesis and characterization of the NM/CS Ni@Fe–Ni(oxy)hydroxide electrode
CS Ni@Fe–Ni(oxy)hydroxide was prepared, for the first time, by a facile OSOE, which was a modified electrodeposition method (see Experimental section, ESI†). The electrochemical and concomitant chemical reactions involved in the synthesis of CS Ni@Fe–Ni(oxy)hydroxide can be probably expressed as follows:| NH4F ↔ NH4+ + F− | (1) |
| Fe(III) + 6F− → [FeF6]3− | (2) |
Anode:
| Ni → Ni(II) + 2e− | (3) |
Cathode:
| 2H2O + 2e− → 2OH− + H2↑ | (4) |
| Ni(II) + 2e− → Ni | (5) |
| [FeF6]3− → Fe(III) + 6F− | (6) |
| 4OH− → O2↑ + 2H2O + 4e− | (7) |
| Fe(III) + 3e− → Fe | (8) |
In the first stage: Fe(III) is fixed in the [FeF6]3− complex (reaction (2)) and dispersed in the electrolyte; for the anode, the nickel atom is transformed into Ni(II) due to the loss of electrons (reaction (3)), and moves quickly to the vicinity of the cathode driven by the electric field force (20 V); for the cathode, at the same time, the H2O near cathode becomes an electron acceptor, and turns into OH− accompanied by release of H2 gas (reaction (4)), immediately afterward, the strongly drawn Ni(II) rapidly reaches the surface of the cathode, loses electrons instantly and deposits on NM surface (reaction (5)), which results in the formation of metallic Ni nanoarray. Fe(III) is slowly electrochemically reduced to metallic Fe at the cathode through reaction (8). In the second stage: Fe(III) is released from [FeF6]3− in the vicinity of the cathode (reaction (6)) due to its consumption in Fe electrodeposition (reaction (8)); for the anode, O2 gas is gradually released based on reaction (7), which leads to an increase of the O2 concentration in the electrolyte; for the cathode, due to the oxidizing environment of the electrolyte, most of the later deposited Ni/Fe (trace) has become the (oxy)hydroxide shell over the metallic Ni core nanoarray. Different from conventional electrodeposition, organic solvent instead of water as the main component of the electrolyte could reduce the ion concentration of the electrolyte so that the current density was not too large even when a huge voltage was applied. Applying a much higher voltage (20 V) than in conventional electrodeposition was very advantageous for driving Ni(II)/Fe(III) and inducing the construction of a metallic Ni nanoarray core at the first stage. In the later stage, Fe was also electrodeposited at the cathode, while the oxidizing environment in the electrolyte, caused by the production of by-product oxygen gas at the anode, brought about the in situ formation of the Fe–Ni(oxy)hydroxide layer over the already formed metallic Ni nanoarray core, resulting in the formation of CS Ni@Fe–Ni(oxy)hydroxide-23 nanoarrays deposited on commercial NM (Fig. 1a). To optimize catalytic activities, different electrodes were prepared by simply tuning the deposition voltage of 10 V, 20 V, and 40 V, which were named CS Ni@Fe–Ni(oxy)hydroxide-13, CS Ni@Fe–Ni(oxy)hydroxide-23, and CS Ni@Fe–Ni(oxy)hydroxide-43, respectively. X-Ray diffraction (XRD) analysis of the prepared NM/CS Ni@Fe–Ni(oxy)hydroxide-23 electrode (Fig. S1†) displayed strong metallic Ni (JCPDS 04-0850) reflections from NM substrate and weak deposit reflection corresponding to (003) of Ni(oxy)hydroxide (JCPDS 89-7111). These weak XRD peaks are due to the very thin deposit layer and tiny loading (0.1 mg cm−2) of Ni(oxy)hydroxides.34
 | ||
| Fig. 1 Morphological and structural characterization of CS Ni@Fe–Ni(oxy)hydroxide-23. (a) Schematic illustration for the synthesis of CS Ni@Fe–Ni(oxy)hydroxide-23 electrode via OSOE and its catalytic function for the OER. (b) SEM image of the vertical standing architecture of nanoarrays; the inset is an optical image of the prepared 4 cm2 squared NM supported Ni@Fe–Ni(oxy)hydroxide-23 electrode. (c) HR-SEM of the CS Ni@Fe–Ni(oxy)hydroxide-23 nanoarrays; (d) HAADF-STEM image of the single nanoarray and corresponding EELS elemental mapping of Ni, Fe, and O. (e) SAED pattern of the CS Ni@Fe–Ni(oxy)hydroxide-23. (f) HR-TEM image of the CS heterostructure. | ||
Scanning electron microscopy (SEM) images exhibit that numerous CS Ni@Fe–Ni(oxy)hydroxide-23 nanoarrays are highly oriented and vertically rooted on the NM with typical gaps between nanoarrays (Fig. 1b, c, and S2†). The top surface of the NM substrate was shown in Fig. S3.† Additionally, the top surface SEM images of the Ni@Fe–Ni(oxy)hydroxide-13 and -43 are shown in Fig. S4.† The Ni@Fe–Ni(oxy)hydroxide-13 sample (Fig. S4a†) shows big spherical particle accumulation because of a lower voltage (10 V) applied in OSOE. In contrast, the coarsening and overlapping of the nanoarrays are shown in the Ni@Fe–Ni(oxy)hydroxide-43 sample (Fig. S4b†), mainly due to a higher voltage (40 V) applied during OSOE. The thickness of the Ni@Fe–Ni(oxy)hydroxide samples is about 1–2 μm, which can be roughly estimated from the surface topography. A low-resolution High-Angle Annular Dark-Field Scanning TEM (HAADF-STEM) image shows that CS Ni@Fe–Ni(oxy)hydroxide-23 nanoarrays feature a clear contrast between the center and periphery (Fig. S5†), manifesting the emergence of core@shell structure. The growth of such a core@shell structure is related to the special OSOE synthesis protocol, which involves electrochemical and chemical reactions mediated growth of nanoarrays with Ni core and Fe-doped Ni(oxy)hydroxide shell. Such a nanoarray structure will absorb electrolytes onto the surface of the electrode on account of strong capillary forces. Fig. S6† showed the hydrophilicity of the samples. The Ni@Fe-doped Ni(oxy)hydroxide-23 sample displayed the smallest contact angle with a value of 4.29 ± 0.21°, compare with those of the bare NM sample (85.97 ± 0.26°) and Ni@Ni(oxy)hydroxide sample (20.81 ± 0.31°). The results further reveal the OSOE would promote the wettability of the samples and the Ni@Fe-doped Ni(oxy)hydroxide-23 has the best wettability. Consequently, it decreases interface friction between gas and solid and enhances oxygen bubbles desorption from the electrocatalyst surface, which is critical for the catalytic OER performance even applying large current densities.19,35–37 Last but not least, the well-aligned feature of CS Ni@Fe–Ni(oxy)hydroxide-23 decreases the pile-up of electrocatalysts, ensuring a sufficient amount of catalytic active site exposure. HAADF-STEM image (Fig. 1d) further exhibits the heterostructure of the nanoarray with the features of a brighter center (a metallic element) and darker edges (nonmetallic element doping). The images of corresponding electron energy loss spectroscopy (EELS) elemental mapping corroborate such heterostructure with metallic Ni uniformly distributed in the whole region while Fe and O reside mainly on the shell, manifesting the formation of metallic Ni core and Fe/Ni oxide/(oxy)hydroxide shell structure. Moreover, the corresponding EELS elements content analysis also reveals that approximately 4.1 at% Fe was incorporated into Ni(oxy)hydroxide shell for the CS Ni@Fe–Ni(oxy)hydroxide-23 sample (52 ± 3 at% Ni, 44 ± 3 at% O; Table S1 and Fig. S7†) while no Fe doping was found for the CS Ni@Ni(oxy)hydroxide-23 sample. EELS elemental mapping images of the Ni@Ni(oxy)hydroxide-23 sample also showed that its nanoarray structure was constituted by a metallic Ni core and Ni(oxy)hydroxide shell with the contents of Ni (51 ± 3 at%) and O (48 ± 3 at%) (Fig. S8, S9, and Table S1†). In addition, the selected area electron diffraction (SAED) pattern was carried out to verify the crystal structure of CS Ni@Fe–Ni(oxy)hydroxide-23. All the well-defined diffraction rings can be indexed into the (111), (200), and (311) planes of metallic Ni (JCPDS 04-0850) with perfect crystallinity; and all the moon halo with a few bright spots can be indexed into (104), (110), (205) planes of crystalline/amorphous Ni(oxy)hydroxide (JCPDS 89-711), as shown in Fig. 1e. As shown in Fig. 1f, the HR-TEM image of CS Ni@Ni(oxy)hydroxide-23 nanoarray shell demonstrated lattice fringes with distances of 0.242 nm and 0.227 nm, corresponding to (104) and (015) planes of Ni(oxy)hydroxide with the measured angle very consistent with the actual value. The core of the CS Ni@Ni(oxy)hydroxide-23 nanoarray was detected to be much darker than its shell, indicating its metallic features. Noteworthy, the nanoarray core part consists of the metallic phase with well-crystalline features, ensuring good electron conductivity. Meanwhile, on the surface part of the nanoarray, the metal atoms in both crystalline/amorphous boundaries and amorphous domains are favorable for accelerating the catalytic activity. Consequently, the ultrathin amorphous layer ensures high efficiency of surface-core electron transfer.38
X-Ray photoelectron spectroscopy (XPS) was performed to further reveal the chemical composition and oxidation state of the shell parts for CS Ni@Fe–Ni(oxy)hydroxide-23 and CS Ni@Ni(oxy)hydroxide-23. Concretely, the wide scanning XPS spectrum of the CS Ni@Fe–Ni(oxy)hydroxide-23 sample exhibits the co-existence of O, Ni, and Fe elements on the surface of the nanoarrays (Fig. S10†), while the CS Ni@Ni(oxy)hydroxide-23 only displays the co-existence of O, and Ni elements (Fig. S11†), suggesting the Fe was successfully doped into the shell part of CS Ni@Fe–Ni(oxy)hydroxide-23 sample. Fig. 2a showed that the fitting peaks of Ni 2p3/2/2p1/2 at 855.7 and 873.8 eV are characteristic of Ni(II), and those at 862.9 and 880.8 eV were Ni(II) satellite peaks (abbreviated as “sat.”); the peaks at 857.5 and 875.1 eV correspond to Ni(III), while the minor peak at 853.1 eV in the Ni 2p3/2 spectrum is assigned to metallic Ni, further confirming that Ni(oxy)hydroxide mainly constitutes the surface of CS Ni@Fe–Ni(oxy)hydroxide-23 sample.34,39,40 The deconvoluted Fe 2p spectrum exhibited a peak at 706.3 eV (Fig. 2b), which is the pre-peak of Fe 2p3/2.41 The binding energies at 711.7 eV and 724.4 eV correspond to Fe 2p3/2 and Fe 2p1/2 peaks, respectively, which are typical for Fe(III).42,43 The missing shakeup satellites at 719 eV and 732 eV precluded the emergence of a separate Fe oxide/(Oxy)hydroxide,42 implying the doping of Fe(III) into the Ni(oxy)hydroxide lattice.22 The O 1s spectrum (Fig. 2c) presented three deconvoluted peaks at 529.9, 531.9, and 533.4 eV, which can be attributed to the typical metal–oxygen bonds, oxygen in the hydroxide group, and chemisorbed water molecules, respectively.19,44,45 The results suggest the presence of oxidized nickel and iron, and (oxy)hydroxide species on the nanoarray surface. Noteworthy, the Ni 2p peeks of CS Ni@Fe–Ni(oxy)hydroxide-23 nanoarray are positively shifted compared to those of CS Ni@Ni(oxy)hydroxide-23 nanoarray, further revealing that Fe was incorporated into the Ni(oxy)hydroxide.46
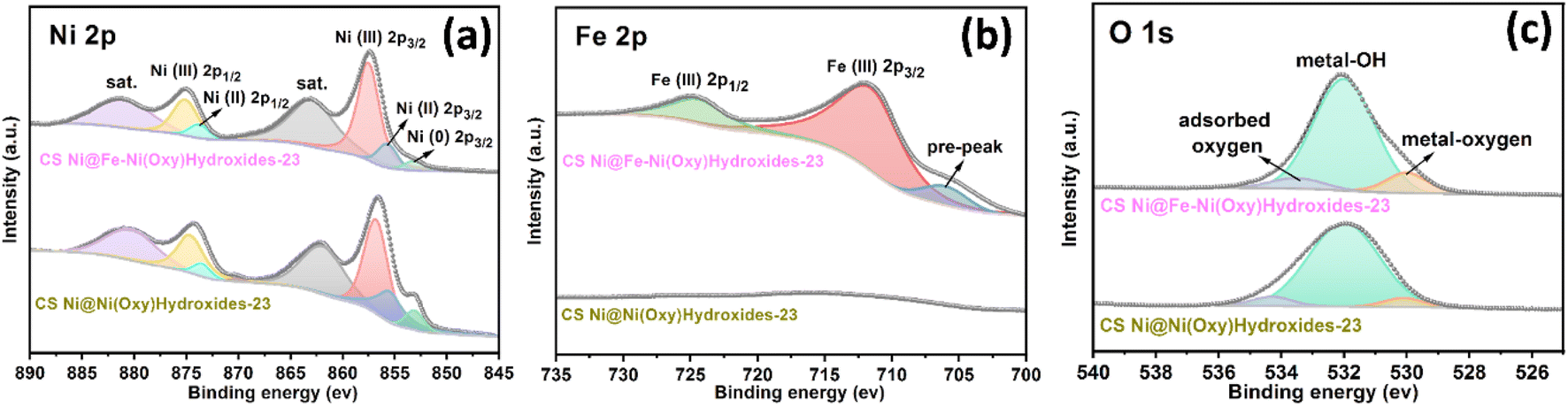 | ||
| Fig. 2 The high-resolution XPS spectra of the as-achieved CS Ni@Fe–Ni(oxy)hydroxide-23 and CS Ni@Ni(oxy)hydroxide-23. (a) Ni 2p. (b) Fe 2p. (c) O 1s. | ||
Electrocatalytic properties of the CS Ni@Fe–Ni(oxy)hydroxide-23 electrodes for the OER (three-electrode cells)
The developed electrodes were directly measured for OER electrocatalysis in a homemade three-electrode cell with an electrolyte of 1 M KOH. The linear sweep voltammogram (LSV, Fig. 3a) curves (details see Experiment section, ESI†) of all as-prepared Ni(oxy)hydroxide based catalysts at a scan rate of 5 mV s−1 show the conversion of Ni(II) to Ni(III) within the potential range from 1.30 to 1.40 V vs. RHE (reversible hydrogen electrode), which is considered to be the active substance for OER.47,48 NM with commercial IrO2 catalyst loaded (IrO2/NM) and bare NM samples were also recorded as controls. Overpotentials at 10 and 100 mA cmgeometric−2 were investigated to evaluate total electrode activity. As illustrated in Fig. 3a and S12,† the Fe–Ni(oxy)hydroxide-based catalysts show remarkably promoted OER activity compared to control electrodes that were employed. Notably, CS Ni@Fe–Ni(oxy)hydroxide-23 sample shows the lowest overpotential of 245 mV to reach 10 mA cmgeometric−2, which is 51, 68, and 114 mV smaller than that of IrO2/NM (296 mV), CS Ni@Ni(oxy)hydroxide (313 mV), the bare NM (359 mV). Additionally, CS Ni@Fe–Ni(oxy)hydroxide-23 sample shows the lowest overpotential of 288 mV to reach 100 mA cmgeometric−2, which is 68, 94, and 258 mV smaller than that of IrO2/NM (356 mV), CS Ni@Ni(oxy)hydroxide (382 mV), the bare NM (546 mV). The growth of CS Ni@Fe–Ni(oxy)hydroxide on NM significantly reduced the overpotential and boosted the total activity of the electrode, as Fe-doped Ni(oxy)hydroxide is considered to be much more advantageous for adsorbing OER intermediate of OH species than the metallic nickel surface on NM sample.21 Moreover, all Fe-doped Ni(oxy)hydroxide samples exhibit similar lower overpotential from 245 to 250 mV when reaching 10 mA cmgeometric−2, which is superior to CS Ni@Ni(oxy)hydroxide without Fe doping and state-of-the-art IrO2 electrode. Furthermore, the overpotential gap further increased when all the electrodes were loading a much higher current density (100 mA cmgeometric−2). The Tafel slopes of those three samples with Fe-doped Ni(oxy)hydroxide have similar values from 37 to 46 mV dec−1, which are much lower than the 55 mV dec−1 obtained for the IrO2/NM sample, 58 mV dec−1 for Ni(oxy)hydroxide sample and 138 mV dec−1 for bare NF sample, as displayed in Fig. 3b. The Tafel slope is employed to investigate the kinetics of the rate-determining step in OER: the smaller Tafel slopes of Fe-doped Ni(oxy)hydroxide samples suggest that Fe-doped surface promotes hydroxyl species adsorption (OH− = OHads + e−), thus accelerating the subsequent rate-determining step of OHads deprotonation (OHads + OH− → Oads + H2O + e−).21,49 Electrochemical impedance spectroscopy (EIS) measurement investigations were run for Fe-doped Ni(oxy)hydroxide samples and other control samples (Fig. S13†). Based on the equivalent circuit, all EIS results are well-fitted (Fig. S13,† inset), and Table S2† lists the values of solution resistance (Rs) and charge-transfer resistance (Rct). As intended, CS Ni@Fe–Ni(oxy)hydroxide-23 has the smallest Rct of 0.58 Ω cm2, which is significantly smaller than those of CS Ni@Fe–Ni(oxy)hydroxide-13 (1.08 Ω cm2), CS Ni@Fe–Ni(oxy)hydroxide-43 (0.89 Ω cm2), CS Ni@Ni(oxy)hydroxide-23 (1.99 Ω cm2), and IrO2/NM (1.76 Ω cm2) respectively. The bare NM has the largest Rct of 17.66 Ω cm2 due to insufficient OER activity. Remarkably, all Ni(oxy)hydroxide samples have slightly smaller values of Rs than that of bare NM. Nevertheless, this difference does not impact significantly efficient charge transport through the system. | ||
| Fig. 3 Electrocatalytic oxygen evolution reaction. (a) The LSV curves of different electrodes. (b) The corresponding Tafel plots. (c) LSV of CS Ni@Ni(oxy)hydroxide sample and CS Ni@Fe–Ni(oxy)hydroxide samples based on ECSA. (d) Comparison of TOF and a total catalyst loading mass activity of the samples at the overpotential of 300 mV. (e) The long-term durability of the CS Ni@Fe–Ni(oxy)hydroxides catalyst under 500 mA cmgeometric−2 in 30 wt% KOH at 70 °C. | ||
To define the intrinsic activity of the developed samples, the OER current was normalized to the electrochemically active surface area (ECSA; named jECSA). The ECSA values were approximated by using a strategy of electrochemical capacitance measurement (Fig. S14 and S15†). As shown in Fig. 3c, CS Ni@Fe–Ni(oxy)hydroxide-23 reaches jECSA of 10 mA cm−2 with a very lower overpotential of 316 mV, while CS Ni@Ni(oxy)hydroxide-23, CS Ni@Fe–Ni(oxy)hydroxide-13, and CS Ni@Fe–Ni(oxy)hydroxide-43 exhibit significantly lower jECSA of 0.16, 5.62, and 2.44 mA cm−2, respectively, at the same overpotential. Obviously, the intrinsic activity of CS Ni@Fe–Ni(oxy)hydroxide-23 is outstandingly better than the CS Ni@Ni(oxy)hydroxide-23 without Fe doping, and the introduction of the right OSOE process parameter can further advance its OER intrinsic activity. Additionally, turnover frequency (TOF) and mass activity at an overpotential of 300 mV were estimated to exhibit the intrinsic property of the catalysts (Fig. 3d). The TOF is a very important kinetic parameter for OER, which is essential for evaluating the performance of the catalysts.25 Note that in our work all the Ni/Fe atoms in the Ni(oxy)hydroxide of CC Ni@Ni(oxy)hydroxide sample and CC Ni@Fe–Ni(oxy)hydroxide samples were assumed to be accessible for catalyzing water splitting. The CS Ni@Fe–Ni(oxy)hydroxide-23 sample displays the highest TOF of 0.285 s−1, which is nearly 26-fold higher than the CS Ni@Ni(oxy)hydroxide-23 at 0.011 s−1 and 7-fold higher than the IrO2/NM at 0.036 s−1. Moreover, CS Ni@Fe–Ni(oxy)hydroxide-23 delivers a mass activity of 1336.0 A g−1 (based on the total catalyst loading mass), which is about 10, and 30 folds higher than that of IrO2/NM, and CS Ni@Ni(oxy)hydroxide-23, respectively. Significantly, the results verified that the outstanding catalytic activity of CS Ni@Fe–Ni(oxy)hydroxide-23 is due to the huge enhanced intrinsic activity of each active site in transforming hydroxyl species into oxygen bubbles. The intrinsic activity of CS Ni@Fe–Ni(oxy)hydroxide-23 is appreciably better than many other state-of-the-art OER catalysts (Table S3†). The combination of electrochemical performance, TOF values, and mass activity demonstrate that the growth of the Fe-doped Ni(oxy)hydroxide with core@shell nanoarray structure on the commercial nickel mesh can significantly improve the electrocatalytic activities. This also proves that the OSOE is a simple and effective method to construct a highly active catalyst with a heterogeneous structure.
The electrochemical stability of the catalyst is also a key item to evaluate catalytic OER performance. To characterize the performance stability of CS Ni@Fe–Ni(oxy)hydroxide-23, we carried out a prolonged chronoamperometric experiment at 10 mA cm−2 (Fig. S16†). Notably, CS Ni@Fe–Ni(oxy)hydroxide-23 withstood accelerated degradation tests and showed excellent stability for 12 h with a small overpotential of approximate 246 mV, which is much smaller than that of CS Ni@Ni(oxy)hydroxide-23 at 315 mV, corroborating that the catalytic activity can be sustained upon OER testing. Additionally, the faradaic efficiency (FE) for H2 and O2 evolution by two-electrode electrolyzer with the CS Ni@Fe–Ni(oxy)hydroxide-23 as both electrodes was also evaluated (Fig. S17†). Hydrogen and oxygen with a volume ratio close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 can be recorded. Meanwhile, the measured hydrogen and oxygen amounts match very well with the calculated ones, demonstrating a nearly 100% FE for both the HER and OER processes during water electrolysis.
:1 can be recorded. Meanwhile, the measured hydrogen and oxygen amounts match very well with the calculated ones, demonstrating a nearly 100% FE for both the HER and OER processes during water electrolysis.
To further reveal the durability of the prepared CS Ni@Fe–Ni(oxy)hydroxide-23 catalyst, we employed the intermittent power supply mode (every 30 minutes), mimicking the condition, in which the catalyst functions under transient operation in the electrolysis cell driven by intermittent renewable energy such as wind and solar energy in the future. Herein, we investigated the long-term electrochemical stability of the CS Ni@Ni(oxy)hydroxide-23 catalyst as the anode (bare NM as the cathode) by testing in 30 wt% KOH at 70 °C, and we found that the electrode can survive at 500 mA cmgeometric−2 for a total of 500 hours without apparent degradation (Fig. 3e), suggesting its outstanding durability. These results verify the exceptional durability of the CS Ni@Ni(oxy)hydroxide-23 catalyst towards OER activity in AWE. The overall surface of the post-OER sample was kept well compared to the fresh one, as shown in Fig. S18.† Moreover, SEM (Fig. S19†) and TEM (Fig. S20†) analyses were employed to probe the post-OER sample. Its core@shell nanoarray structural and morphological integrity was well preserved, which further proves the morphology and structure stability of CS Ni@Ni(oxy)hydroxide-23 after the durability test. Besides, XPS was performed to clarify the chemical states of the post-OER sample after a long-term chronopotentiometric test (50 mA cm−2 for 10 h in 1 M KOH; Fig. S21†). The Ni 2p peaks of the post-OER sample shift to higher binding energy compared to the fresh sample (Fig. S21b†), confirming the formation of the high valence Ni species. The Fe(III) was still doped into the Ni(oxy)hydroxide lattice after a long-term OER test, as shown in Fig. S21c.† Additionally, in the O 1s spectra of the post-OER sample (Fig. S21d†), the peak located at 533.4 eV confirms the increased chemisorbed water molecules, due to the long-term OER test. These results further suggest the potential of CS Ni@Ni(oxy)hydroxide-23 as an OER electrocatalyst in practical alkaline water electrolysis.
Effect of NM@OSOE-23 electrode as an anode on the performance of 4 cm2 AEL cell (full cell, 6 M KOH at 70 °C)
As schematized in Fig. 4a and b, squared NM@OSOE-23 (i.e. CS Ni@Fe–Ni(oxy)hydroxide-23 catalyst deposited on commercial 4 cm2 perforated NM; similarly, NM@OSOE-23(Fe free)(+) refers to Ni@Ni(oxy)hydroxide-23 sample) and bare NM electrodes were implemented in a 4 cm2 AEL cell in 6 M KOH at 70 °C to explore their effect on AEL cell performance. Three different cells were constructed, with two bare NM electrodes as both cathode and anode (NM(−)||NM(+), Fig. 4a), bare NM as the cathode and NM@OSOE-23(Fe free) as the anode (NM(−)||NM@OSOE-23(Fe free)(+)), and bare NM as the cathode and NM@OSOE-23 as the anode (NM(−)||NM@OSOE-23(+), Fig. 4b). In all cases, the anode and cathode were separated by a membrane (Zirfon PERL, 500 μm, Agfa). Nickel foams (NF, 2 mm thickness) and nickel plates were employed as gas diffusion layers (GDL) and bipolar plates (BPP), respectively, for both anode and cathode sides in all cell configurations. Fig. 4c shows the polarization curves of AEL cells with the specified cell configurations, operated at atmospheric pressure. The NM(−)||NM(+) and NM(−)||NM@OSOE-23(Fe free)(+) cells implemented the high cell voltages of 2.256 V and 2.192 V at a current density of 2 A cm−2, while the NM(−)||NM@OSOE-23(+) cell delivered a significantly lower cell voltage of 2.055 V. Thus, a considerable amelioration of more than 200 mV for the cell construction of NM(−)||NM@OSOE-23(+) at 2 A cm−2 compared with that of the control cell with the bare NM as both electrodes was obtained. This result suggests that the introduction of the NM@OSOE-23 anode into the cell is favorable for reducing the voltage at high current densities since the NM@OSOE-23 anode provides excellent OER activity and desirable interfacial contacts, which is attributed to the specific catalytic nanoarrays of the core@shell Fe-doped Ni(oxy)hydroxide. The amelioration of NM(−)||NM@OSOE-23(Fe free)(+) cell was only 64 mV, mainly due to the insufficient OER activity of the Fe-free Ni@Ni(oxy)hydroxide-23 nanoarrays. | ||
| Fig. 4 Impact of NM@OSOE-23 anode on the performance of 4 cm2 AEL cell with bare NM as a cathode. (a and b) Schematic illustration of two different AEL cells with bare NM as cathode: (a) bare NM as anode and (b) NM@OSOE-23 as an anode. (c) Polarization curves for different cell configurations: NM (−)||NM (+), NM (−)||NM@OSOE-23(Fe free)(+), and NM (−)||NM@OSOE-23 (+) in 6 M KOH at 70 °C. (d) Nyquist plots from the electrochemical impedance spectroscopy (EIS) measurements for different configurations: NM (−)||NM (+), NM (−)||NM@OSOE-23(Fe free)(+), and NM (−)||NM@OSOE-23 (+) at 0.5 A cm−2 (from 50 kHz to 100 mHz); the applied equivalent circuit is shown in the inset. (e) Nyquist plot parameters obtained from (d). (f) Chronopotentiometric measurement at a constant current density of 1 A cm−2 for the NM (−)||NM@OSOE-23 (+) cell in 6 M KOH at 70 °C. | ||
The positive effect of the NM@OSOE-23 anode in the AEL cell was also established by conducting the EIS investigation, which was systematically tracked during the in situ AEL measurements. To inspect the EIS plots, a fitting process was handled with an equivalent electric circuit model, which was fixed based on physical processes and their interactivities in the system of the AEL cell. Ohmic, cathodic charge transfer (HER), anodic charge transfer (OER), and mass-transfer resistances were included.50Fig. 4d displays the investigation and comparison of the Nyquist plots with model fits for the NM(−)||NM(+) cell, NM(−)||NM@OSOE-23(Fe free)(+) cell, and NM(−)||NM@OSOE-23(+) cell by applying a current density of 0.5 A cm−2 and the insert shows a suggested equivalent circuit model, exhibiting the contribution of each resistance loss. From the high to low-frequency range, three arc loops are detected, indicating three resistor-constant phase element (CPE) pairs. Specifically, the equivalent circuit comprises an ohmic resistance (R) in a series with three circuits, each involving a resistance (R1, cathodic charge transfer; R2, anodic charge transfer; and R3, mass-transfer resistances) and a CPE (CPE1, CPE2, and CPE3) in parallel to each other. The inductor (L) in series with the R indicates probable inductive parts from cables and other components. The R or high-frequency resistance (HFR), which emerges as the x-axis intercepts on the left side of the Nyquist plot, represents the internal ohmic resistance of the cell.51 The R denotes total ohmic resistance contributions including the diaphragm, electrodes, GDLs, BPPs, and contact resistances.52 The middle section in the equivalent circuit model depicts the activation losses, specifically because of the cathodic charge transfer (R1) kinetic and anodic charge transfer (R2) kinetic. They are noticeable as two arcs at high- and middle-frequency ranges.53 The CPE1 and CPE2 are relevant to the double-layer capacitances of the HER cathode and OER anode, respectively. The charge-transfer resistances from two electrodes and the related CPEs are separated and should be considered independently because two different arcs have been noticed, which is highly favorable to defining each side of charge-transfer resistances. The last arc denotes the mass-transport resistance and limitation (R3), which is noticeable at low frequencies of the EIS plot. Moreover, in terms of AWE, the generated bubbles can influence every process: dissolved gases accumulate and ultimately arrive at a concentration above the supersaturation threshold for bubble generation when the cell is in operation. Consequently, electrodes with appropriate catalyst morphologies are required for favorable bubble evolution and desorption, thus improving the total performance and efficiency of water electrolysis.4
Fig. 4e exhibits the results of the fitted equivalent circuit model for NM(−)||NM(+) cell, NM(−)||NM@OSOE-23(Fe free)(+) cell, and NM(−)||NM@OSOE-23(+) cell. Notably, the ohmic resistance (R) of the NM(−)||NM@OSOE-23(+) cell is the lowest, which can be only attributed to the employed NM@OSOE-23 anode because the three cells use the same diaphragm, cathode, GDLs, and BPPs. By incorporating the NM@OSOE-23 as an anode, the HFR drops to 73.8 mΩ cm2 for the NM(−)||NM@OSOE-23(+) cell compared to 88.4 mΩ cm2 for the NM(−)||NM(+) cell. The decrement in the HFR upon incorporating the NM@OSOE-23 anode in the cell can be ascribed to the smaller contact resistance of the NM@OSOE-23 electrode compared with the bare NM electrode. The HFR for NM(−)||NM@OSOE-23(Fe free)(+) cell (83.1 mΩ cm2) was slightly decreased, compared to that for NM(−)||NM(+) cell. The NM@OSOE-23 electrode with appropriate core–shell nanoarray distribution (Fig. 1b and c) can increase the contact areas with the porous GDL and reduce the trapped-bubble concentrations between them, which leads to an increment in overall cell performance. It can be found that from high to middle frequencies, the cathodic charge transfer resistances (R1) show almost the same values with 45.6, 46.1, and 46.8 mΩ cm2 of NM(−)||NM(+) cell, NM(−)||NM@OSOE-23(Fe free)(+) cell, and NM(−)||NM@OSOE-23(+) cell, respectively, which is due to the same bare NM as a cathode in all cells; while the anodic charge transfer resistances (R2) show a sharp decrease from 100.4 mΩ cm2 of the NM(−)||NM(+) cell to 66.1 mΩ cm2 of NM(−)||NM@OSOE-23(+) cell as expected, which is mainly attributable to the employed NM@OSOE-23 anode with very high OER activity. Meanwhile, this also could be since the bare NM in contact with the GDL is not favorable enough for supporting bubble desorption, which causes bubble coalescence and augmentation that lead to covering the surface of the active electrode and finally to increasing the anode charge transfer resistance of the NM(−)||NM(+) cell. The NM(−)||NM@OSOE-23(Fe free)(+) cell also displayed a measurable enhancement (78.1 mΩ cm2) because of the good OER activity of the Ni@Ni(oxy)hydroxide nanoarrays. Therefore, the implementation of NM@OSOE-23 anode with both superior OER performance and appropriate nanoarray distribution in the cell might not only enhance intrinsic OER activity but also decrease bubble coalescence. Lastly, mass-transport limitations for both the NM(−)||NM@OSOE-23(+) cell (60.2 mΩ cm2) and NM(−)||NM@OSOE-23(Fe free)(+) cell (66.7 mΩ cm2) dropped compared to that of the NM(−)||NM(+) cell (75.2 mΩ cm2). It is detectable that the mechanisms of mass-transport loss change with the employment of the NM@OSOE-23 and NM@OSOE-23(Fe free) anodes with uniform distribution of nanoarrays on their surface. This can be caused by the innate transmission channel between every two nanoarrays, which can bring much more effective contact with the nearby porous GDL. In other words, nanoarrays on the electrode surface, like numberless bridges, extend into porous GDL, and in turn contact two sides, which is favorable enough for optimizing liquid/gas flow. The durability of NM(−)||NM@OSOE-23(+) cell in 6 M KOH was investigated under 1 A cm−2 at 70 °C for 150 h, and the cell-voltage changes with the test time are shown in Fig. 4f. After 150 h of operation, the cell voltage shows a 22 mV increment from 1.925 V to 1.946 V with negligible degradation of 1.2%. The black surface of the post NM@OSOE-23 electrode revealed that the catalyst layer remained at the surface, as shown in Fig. S22.† These results show that the introduction of the NM@OSOE-23 anode into the AEL cell successfully brings a great improvement in cell performance, due to the excellent OER intrinsic activity and favorable mass transport of the specific core@shell Fe-doped Ni(oxy)hydroxide nanoarrays.
Conclusions and outlook
This work demonstrates an exceptionally high OER performance of electrodes based on non-precious transition metal (Ni, Fe) oxyhydroxides that were designed by the newly developed OSOE approach, which can potentially meet the industrial standard of AEL. A low overpotential of 245 mV is obtained at 10 mA cm−2 together with a corresponding low Tafel slope of 37 mV dec−1 for the optimum Ni@Fe–Ni(oxy)hydroxide-23 anode. Strikingly, it delivers a high turnover frequency of 0.285 s−1 and a total catalyst loading mass activity of 1336 A g−1, which are over 7-fold higher than state-of-the-art IrO2 and almost 30-fold higher than the Fe-free CS Ni@Ni(oxy)hydroxide. Moreover, the electrode can also survive at 500 mA cm−2 for over 500 h without apparent degradation. Furthermore, the optimized catalyst was tested as the anode in a 4 cm2 AEL cell where bare NM was used as the cathode. Significant performance improvements were revealed, which led to a 200 mV lower voltage at 2 A cm−1 compared with a similar AEL cell but with commercial Ni mesh as the anode. This was attributed to notable enhancements of interface contact, anodic charge transfer, and mass transfer originating from the constructed specific core@shell Ni@Fe-doped Ni(oxy)hydroxide nanoarray architecture on commercial nickel mesh.This work suggests that the developed OSOE is a very promising approach to not only engineering TMC-based CS nanostructures with the TM core and catalytically active (oxy)hydroxide substance as the shell but also constructing nanoarrays architecture with superior interface contact and mass transfer, directing the merits of two parts and synergistically proceeding AWE. The approach we employed is highly reproducible, and the electrode size can be easily controlled, making it a promising strategy to advance the next generation of AWE technology. We hope the performance of the electrode can be further promoted with a more favorable charge and mass transfer through engineering the electronic structure by single atom doping extra elements (such as Mo, W, Pt, Ir, and Ru), and optimizing morphology features. Last but not least, the developed high-performance electrodes will also be extended and assembled in the state-of-the-art anion exchange membrane water electrolyzer (AEMWEL) cell. Our work is essential for accelerating the development of the next generation of AWE technology.
Author contributions
T. Jiang conceived and conducted the experiments and carried out the electrochemical tests along with analysis of the electrochemical and physical results and writing of the manuscript. X. G. Jiang provided SEM and XRD data. J. Hnat and A. Michalcova contributed to the TEM/STEM characterizations. I. Biswas provided support on XPS measurement. R. Reisser provided support during the long-term half-cell test. V. Kyriakou provided support on contact angle measurement. F. Razmjooei provided support during the full-cell test. H. L. Liao provided helpful suggestions. K. Bouzek provided helpful suggestions and edited the manuscript. S. A. Ansar provided support at each stage of the tests. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 862509 (“NEXTAEC”). We gratefully acknowledge financial support from DLR/DAAD Research Fellowships (No. 57540124). We acknowledge the NEWELY project. We also acknowledge CzechNanoLab Research Infrastructure supported by MEYS CR (LM2018110) and Dr Jan Duchon for performing the STEM/EELS analysis. The authors thank Mr Franz Egert, Mr Sven Marx, and Mr Günter Roth for their assistance during the work.References
- S. Mitchell and J. Pérez-Ramírez, Nat. Rev. Mater., 2021, 6, 969–985 CrossRef
.
- S. A. Chala, M. C. Tsai, B. W. Olbasa, K. Lakshmanan, W. H. Huang, W. N. Su, Y. F. Liao, J. F. Lee, H. J. Dai and B. J. Hwang, ACS Nano, 2021, 15, 14996–15006 CrossRef CAS
- Y. Zhou, Z. Wang, Z. Pan, L. Liu, J. Xi, X. Luo and Y. Shen, Adv. Mater., 2019, 31, 1806769 CrossRef
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle and K. A. Friedrich, Joule, 2021, 5, 1776–1799 CrossRef CAS
- L. Wang, V. A. Saveleva, M. J. Eslamibidgoli, D. Antipin, C. Bouillet, I. Biswas and K. A. Friedrich, ACS Appl. Energy Mater., 2022, 5, 2221–2230 CrossRef CAS
- G. Calado and R. Castro, Appl. Sci., 2021, 11, 5561 CrossRef CAS
- F. Qureshi, M. Yusuf, H. Kamyab, D. V. N. Vo, S. Chelliapan, S. W. Joo and Y. Vasseghian, Renewable Sustainable Energy Rev., 2022, 168, 112916 CrossRef CAS
- B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing and R. Wu, Adv. Energy Mater., 2020, 10, 2001963 CrossRef CAS
- Q. Zhou, Y. Chen, G. Zhao, Y. Lin, Z. Yu, X. Xu and S. X. Dou, ACS Catal., 2018, 8, 5382–5390 CrossRef CAS
- Y. Tang, Q. Liu, L. Dong, H. B. Wu and X. Y. Yu, Appl. Catal., B, 2020, 266, 118627 CrossRef CAS
- S. F. Hung, Y. Y. Hsu, C. J. Chang, C. S. Hsu, N. T. Suen, T. S. Chan and H. M. Chen, Adv. Energy Mater., 2018, 8, 1701686 CrossRef
- N. K. Oh, J. Seo, S. Lee, H. J. Kim, U. Kim, J. Lee and H. Park, Nat. Commun., 2021, 12, 1–12 CrossRef
- H. Yang, L. Gong, H. Wang, C. Dong, J. Wang, K. Qi and B. Y. Xia, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed
- Z. Gong, R. Liu, H. Gong, G. Ye, J. Liu, J. Dong and H. Fei, ACS Catal., 2021, 11, 12284–12292 CrossRef CAS
- R. Yang, Y. Zhou, Y. Xing, D. Li, D. Jiang, M. Chen and S. Yuan, Appl. Catal., B, 2019, 253, 131–139 CrossRef CAS
- W. Hao, R. Wu, H. Huang, X. Ou, L. Wang, D. Sun, X. Ma and Y. Guo, Energy Environ. Sci., 2020, 13, 102–110 RSC
- H. Sun, Y. Min, W. Yang, Y. Lian, L. Lin, K. Feng and Y. Peng, ACS Catal., 2019, 9, 8882–8892 CrossRef CAS
- R. Gao and D. Yan, Adv. Energy Mater., 2020, 10, 1900954 CrossRef CAS
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC
- Q. Cao, M. Luo, Y. Huang, Q. Liu, X. Kong, J. Lei and J. Wang, Sustainable Energy Fuels, 2020, 4, 1522–1531 RSC
- T. Kou, S. Wang, J. L. Hauser, M. Chen, S. R. J. Oliver, Y. Ye, J. Guo and Y. Li, ACS Energy Lett., 2019, 4, 622–628 CrossRef CAS
- Z. Wu, Z. Zou, J. Huang and F. Gao, J. Catal., 2018, 358, 243–252 CrossRef CAS
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC
- W. Zhu, W. Chen, H. Yu, Y. Zeng, F. Ming, H. Liang and Z. Wang, Appl. Catal., B, 2020, 278, 119326 CrossRef CAS
- Y. Lyu, R. Wang, L. Tao, Y. Zou, H. Zhou, T. Liu, Y. Zhou, J. Huo, S. P. Jiang, J. Zheng and S. Wang, Appl. Catal., B, 2019, 248, 277–285 CrossRef CAS
- W. Zhu, T. Zhang, Y. Zhang, Z. Yue, Y. Li, R. Wang, Y. Ji, X. Sun and J. Wang, Appl. Catal., B, 2019, 244, 844–852 CrossRef CAS
- Y. Li, S. Guo, T. Jin, Y. Wang, F. Cheng and L. Jiao, Nano Energy, 2019, 63, 103821 CrossRef CAS
- L. Peng, J. Shen, X. Zheng, R. Xiang, M. Deng, Z. Mao, Z. Feng, L. Zhang, L. Li and Z. Wei, J. Catal., 2019, 369, 345–351 CrossRef CAS
- Z. Xia, H. Sun, X. He, Z. Sun, C. Lu, J. Li, Y. Peng, S. Dou, J. Sun and Z. Liu, Nano Energy, 2019, 60, 385–393 CrossRef CAS
- K. Xiao, L. Zhou, M. Shao and M. Wei, J. Mater. Chem. A, 2018, 6, 7585–7591 RSC
- Q. Xu, H. Jiang, H. Zhang, Y. Hu and C. Li, Appl. Catal., B, 2019, 242, 60–66 CrossRef CAS
- H. Yang, Z. Chen, W. Hao, H. Xu, Y. Guo and R. Wu, Appl. Catal., B, 2019, 252, 214–221 CrossRef CAS
- T. Jiang,
et al.
, ACS Appl. Energy Mater., 2019, 2, 8809–8817 CrossRef CAS
- T. Wang, G. Nam, Y. Jin, X. Wang, P. Ren, M. G. Kim, J. Liang, X. Wen, H. Jang, J. Han, Y. Huang, Q. Li and J. Cho, Adv. Mater., 2018, 30, 1800757 CrossRef
- Y. Li, Z. Wang, J. Hu, S. Li, Y. Du, X. Han and P. Xu, Adv. Funct. Mater., 2020, 30, 1910498 CrossRef CAS
- Y. Luo, L. Tang, U. Khan, Q. Yu, H. M. Cheng, X. Zou and B. Liu, Nat. Commun., 2019, 10, 1–9 CrossRef
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang and X. Wang, Nat. Commun., 2018, 9, 1–12 CrossRef
- F. Guo, Y. Wu, H. Chen, Y. Liu, L. Yang, X. Ai and X. Zou, Energy Environ. Sci., 2019, 12, 684–692 RSC
- Y. Jia, L. Zhang, G. Gao, H. Chen, B. Wang, J. Zhou and X. Yao, Adv. Mater., 2017, 29, 1700017 CrossRef
- Z. Wu, Z. Zou, J. Huang and F. Gao, ACS Appl. Mater. Interfaces, 2018, 10, 26283–26292 CrossRef CAS
- G. Dong, M. Fang, J. Zhang, R. Wei, L. Shu, X. Liang and J. C. Ho, J. Mater. Chem. A, 2017, 5, 11009–11015 RSC
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef CAS
- K. Fominykh, P. Chernev, I. Zaharieva, J. Sicklinger, G. Stefanic, M. Döblinger and D. Fattakhova-Rohlfing, ACS Nano, 2015, 9, 5180–5188 CrossRef CAS
- Y. Wang, C. Xie, Z. Zhang, D. Liu, R. Chen and S. Wang, Adv. Funct. Mater., 2018, 28, 1703363 CrossRef
- C. Dong, T. Kou, H. Gao, Z. Peng and Z. Zhang, Adv. Energy Mater., 2018, 8, 1701347 CrossRef
- Y. Li, H. Zhang, M. Jiang, Q. Zhang, P. He and X. Sun, Adv. Funct. Mater., 2017, 27, 1702513 CrossRef
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong and M. Wang, Nat. Commun., 2018, 9, 1–12 CrossRef
- Y. F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS
- D. A. García-Osorio, R. Jaimes, J. Vazquez-Arenas, R. H. Lara and J. Alvarez-Ramirez, J. Electrochem. Soc., 2017, 164, 3321 CrossRef
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2, 7903–7912 CrossRef CAS
- S. H. Frensch, A. C. Olesen, S. S. Araya and S. K. Kær, Electrochim. Acta, 2018, 263, 228–236 CrossRef CAS
- J. Rodríguez, S. Palmas, M. Sánchez-Molina, E. Amores, L. Mais and R. Campana, Membranes, 2019, 9, 129 CrossRef
- S. Siracusano, S. Trocino, N. Briguglio, V. Baglio and A. S. Aricò, Materials, 2018, 11, 1368 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06813c |
| This journal is © The Royal Society of Chemistry 2022 |