
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upcycling waste poly(ethylene terephthalate) into polymer electrolytes†
Ming Yan
Tan
a,
Leonard
Goh
a,
Dorsasadat
Safanama
a,
Wei Wei
Loh
a,
Ning
Ding
a,
Sheau Wei
Chien
a,
Shermin S.
Goh
 a,
Warintorn
Thitsartarn
a,
Jason Y. C.
Lim
*ab and
Derrick W. H.
Fam
*acd
a,
Warintorn
Thitsartarn
a,
Jason Y. C.
Lim
*ab and
Derrick W. H.
Fam
*acd
aInstitute of Materials Research and Engineering (IMRE), 2 Fusionopolis Way, Singapore 138634, Singapore. E-mail: jason_lim@imre.a-star.edu.sg; derrickfamwh@imre.a-star.edu.sg
bDepartment of Materials Science and Engineering, National University of Singapore (NUS), 9 Engineering Drive 1, Singapore 117576, Singapore
cSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Ave, Singapore 639798, Singapore
dDepartment of Mechanical Engineering, College of Design and Engineering, National University of Singapore, 9 Engineering Drive 1, Block EA #07-08, Singapore 117575, Singapore
First published on 15th November 2022
Abstract
Lithium-ion batteries (LiB) play an important role in energy storage in our increasingly-electrified modern world, with polymer electrolyte (PE) materials poised to revolutionise battery design by eliminating the most critical safety hazards associated with liquid electrolytes in use today. Although there is growing focus on sustainable PE designs, the use of abundant waste commodity plastics as an alternative feedstock for PE production remains surprisingly overlooked. Herein, we report the first examples of PEs obtained by chemical upcycling of waste poly(ethylene terephthalate) (PET) bottles, exploiting the susceptibility of PET's ester linkages for chemical solvolysis and the structural rigidity of the terephthalate aromatic components to allow for free-standing conductive film formation. Our PET-derived polyurethane PEs show promising ionic conductivity when used as both solid and gel polymer electrolytes, and can be assembled into a working lithium-ion battery. This sets a precedent for designing future sustainable PE materials from waste plastics and contributing towards a circular materials economy.
Plastics are ubiquitous materials in modern society due to their low cost, durability and versatility for diverse applications. With soaring demand worldwide, the quantity of post-consumer waste plastics produced is also increasing rapidly. Of the 460 million tons of plastics produced globally in 2019, only 9% is recycled, with the remainder being either incinerated, disposed of in landfills or irresponsibly discarded in the environment.1 Polyethylene terephthalate (PET, SPI code 1) is amongst the most abundant plastics produced today (∼31 million tonnes in 2019).2 Being mechanically tough, ductile and mouldable, PET is commonly used as bottles and jars, synthetic fibres for clothing, carpets and ropes, protective packaging and even construction materials. Due to its relatively uncomplicated waste stream,3,4 PET is also the most-recycled plastic worldwide.5 However, with rapid degradation of physical properties during repeated mechanical recycling due to polymer chain cleavage, only a minority of recycled PET is suitable for use in its original applications.5 While chemical recycling offers the possibility of reforming high molecular weight PET polymers after first breaking them down into monomers,6 this process is disadvantaged by its high energy demands and need for monomer purification.7 Moreover, recycled PET can cost more than virgin polymers,8,9 reducing the economic viability of recycling.
In contrast, plastic upcycling transforms low-value waste polymers into chemicals and materials of higher value.10–15 PET has been upcycled into small molecules which can be used as solvents and reagents for the chemical industry,16 including the production of new polymers such as polyhydroxyalkanoates (PHA),17 as well as porous materials for carbon dioxide (CO2) capture18 and utilisation.19 Recently, with growing interest in developing sustainable materials for energy storage applications to meet ever-increasing global energy demands,20 upcycling of post-consumer plastics into energy storage materials is expected to be of growing importance. In this aspect, waste plastics have traditionally been converted pyrolytically into carbon materials for supercapacitors and batteries.21–23 Thus far, other than Sardon's recent report of upcycling bisphenol A polycarbonate plastics into aliphatic polycarbonates useful as polymer electrolytes (PEs) for solid-state lithium ion batteries (LiBs),24 upcycling of other readily-available, abundant waste commodity plastics for this lucrative application has been largely overlooked.25 Indeed, compared to conventional liquid electrolytes currently used in LiBs, PEs offer numerous practical advantages such as reducing the risks of electrolyte leakage, thermal runaway, uncontrolled volume expansion, dendrite growth, and unwanted electrolyte side reactions.26
Herein, we report the first examples of PEs derived from waste PET beverage bottles. PET offers numerous desirable ready-made features for upcycling to solid polymer electrolytes (SPEs): other than its susceptibility towards chemolysis into polymer building blocks,6 the rigid aromatic terephthalate components which are responsible for PET's excellent mechanical properties27 can also be exploited for enhancing the mechanical robustness of SPEs – facilitating device integration and fabrication, suppressing dendrite formation28 and potentially, for use in structural power devices.29 After assessing the viability of our PET-derived polymers as solid polymer electrolytes, we further evaluated their ionic conductivity and cycling performance when used as gel polymer electrolytes for LiBs.
The design of our PET-derived polymer electrolytes is summarised in Fig. 1. The structural rigidity conferred by the “hard” aromatic units from PET is balanced by “soft” poly(ethylene glycol) (PEG) segments which provide the requisite flexibility for Li+ transport by segmental chain motion.28 Urethane linkages were chosen to covalently link the different “soft” and “hard” segments together to form the target polymer electrolytes, due to their well-established synthesis by polyaddition of alcohols with isocyanates.30–33 Indeed, polyurethanes (PUs) are gaining popularity as polymer electrolyte materials due to their often excellent mechanical strength, desirable thermal and electrochemical stability.34 They also offer a vast design space for manipulation of polymer structure and properties using different combinations of easily-accessible diol and diisocyanate starting materials, allowing PUs to be used as polymer and gel electrolytes for batteries and supercapacitors.34–36 In addition, extensive hydrogen bonding from the abundant amide and urethane groups present can enhance polymer–polymer interactions for better mechanical robustness. These hydrogen bond donors also facilitate interphase compatibility with additives such as LiTFSI and ionic liquids.34
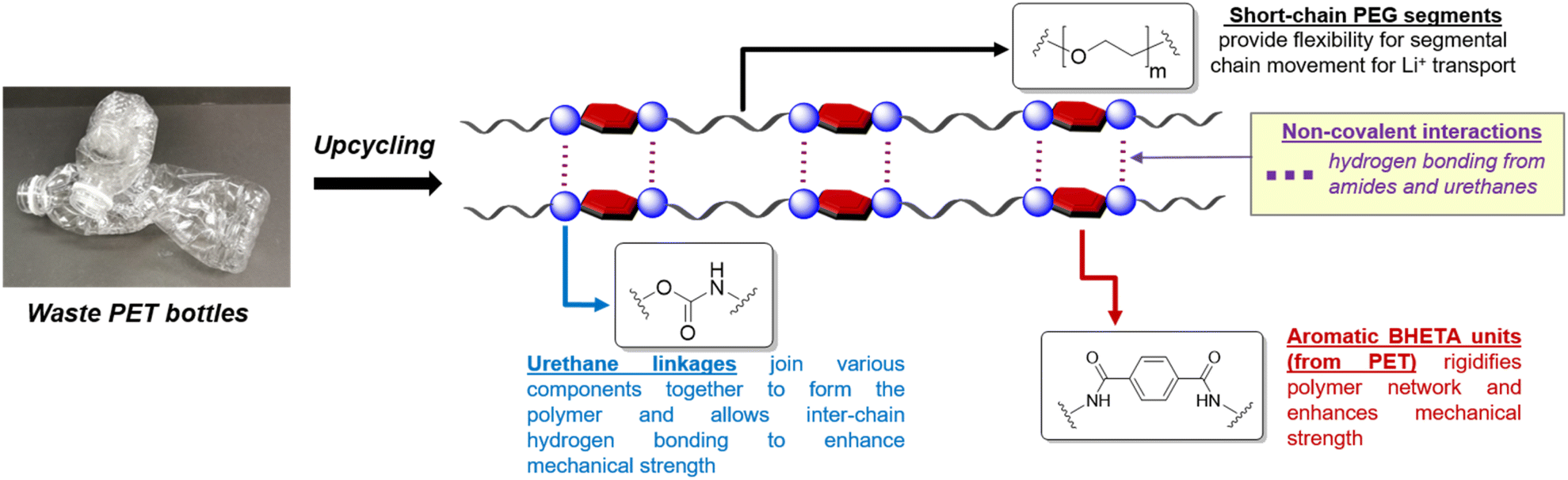 | ||
| Fig. 1 Design of polyurethane polymer electrolytes derived from waste PET with hard (aromatic) and soft (PEG) components. | ||
The synthesis of the polyurethane electrolytes from waste PET bottles is summarised in Scheme 1. Clean waste PET bottles were first depolymerised by aminolysis using neat ethanolamine to form bis(2-hydroxyethyl)terephthalamide (BHETA) in the presence of the 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) organocatalyst (Fig. S1†).37 Compared with synthesising BHETA directly from terephthalic acid, production of terephthalamides from PET aminolysis avoids the usage of hazardous acid chloride reagents traditionally required,38 potentially offering a more sustainable synthesis route.14 Thereafter, the easily-purified BHETA monomer was mixed with PEG (Mn 17.5 kDa) in different mass ratios, and reacted with hexamethylene diisocyanate (HMDI) to form a family of polyurethanes containing different ratios of hard (BHETA): soft (PEG) blocks in generally excellent yields (Table 1).‡ To better appreciate the roles of the BHETA component in our polymer electrolyte design, we also synthesised a polyurethane control (named PEG-PU) made up solely of PEG and HMDI components (Fig. S2†) for comparison.
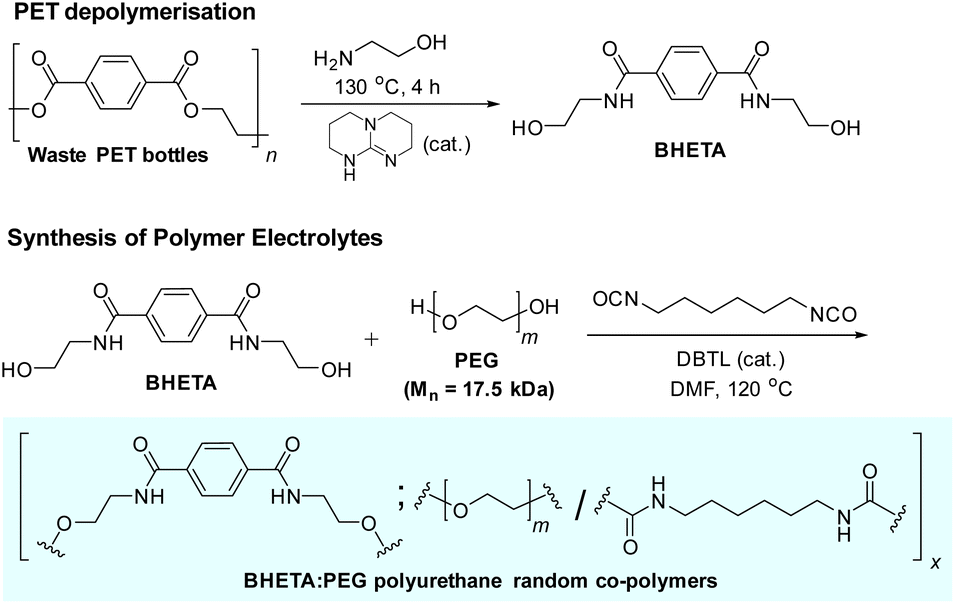 | ||
| Scheme 1 Synthesis of BHETA:PEG polymer electrolytes from waste PET bottles. | ||
| Polymera | Mass percentages of each polymer componentb/% | T g /°C | T c /°C | Polymer yields/% | ||
|---|---|---|---|---|---|---|
| BHETA | PEG | HMDI linkers | ||||
| a Polymers are named according to their approximate BHETA:PEG mass percentage compositions. b Determined by integration of the aromatic BHETA (7.92 ppm), PEG (3.51 ppm) and HMDI (2.93 ppm) signals in the polymers 1H NMR spectra (d6-DMSO). c Determined from DSC measurements during the second heating scan (heating rate = 20 °C min−1; cooling rate = 20 °C min−1). | ||||||
| 1BHETA:2PEG | 26.9 | 54.8 | 18.3 | −59.0 | 21.8 | 91.2 |
| 1BHETA:4PEG | 16.8 | 71.4 | 11.8 | −36.9 | 26.3 | 95.8 |
| 1BHETA:6PEG | 13 | 74.8 | 12.2 | −55.8 | 32.1 | 71.4 |
| 1BHETA:7PEG | 10.9 | 79.8 | 9.3 | −22.0 | 35.0 | 96.6 |
| PU w/o BHETA | 0 | 99.8 | 0.23 | −44.3 | 30.7 | 91.8 |
1H NMR spectroscopy showed unequivocal evidence of the formation of polymers containing PET-derived BHETA and PEG, evident from diagnostic resonances of the different components (Fig. 2). The presence of amide and urethane bonds on the polymer were further verified by their characteristic 13C NMR signals at 165.8 and 156.1 ppm respectively (Fig. S3†). In addition, the absence of 13C NMR signals at ca. 120 ppm and FTIR absorbance peaks between 2250–2300 cm−1 further indicates that all isocyanate groups from the HMDI linker have been fully consumed to form urethanes.39 FTIR spectroscopy provides further evidence of extensive hydrogen bonding between polymers: compared to free urethane C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches with typical absorbance at 1710–1730 cm−1, the lower-than-expected urethane CO stretch of the 1BHETA-2PEG polymer at 1689 cm−1 (Fig. S4†) is characteristic of hydrogen-bonded urethane groups in ordered hard polymeric domains.40 The compositions of the BHETA-PEG series of polymers were determined by integrating the 1H NMR signals at 7.9, 3.5 and 1.9 ppm to obtain the percentage masses of the BHETA, PEG and HMDI components respectively for each polymer (Table 1).
O stretches with typical absorbance at 1710–1730 cm−1, the lower-than-expected urethane CO stretch of the 1BHETA-2PEG polymer at 1689 cm−1 (Fig. S4†) is characteristic of hydrogen-bonded urethane groups in ordered hard polymeric domains.40 The compositions of the BHETA-PEG series of polymers were determined by integrating the 1H NMR signals at 7.9, 3.5 and 1.9 ppm to obtain the percentage masses of the BHETA, PEG and HMDI components respectively for each polymer (Table 1).
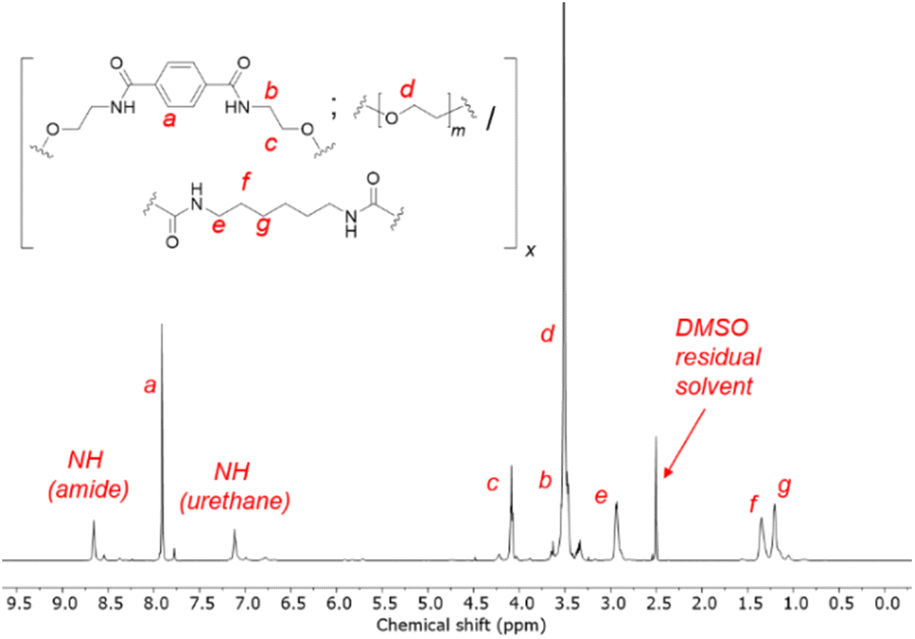 | ||
| Fig. 2 1H NMR of the 1BHETA:2PEG polyurethane in d6-DMSO. | ||
The interactions between 1BHETA:2PEG polymers were further probed by 1H NMR dilution experiments. At gradually lower polymer concentrations in d6-DMSO, the polymer's aromatic and aliphatic NMR signals became increasingly sharper and more well-defined (Fig. S5†). This suggested that at high polymer concentrations, considerable interactions between the polymers occurred, restricting their conformational and translational freedom in solution. Primarily, this could be due to the extensive hydrogen bonding from the amide and urethane groups present, with insignificant contributions from π–π stacking as observed from the lack of significant chemical shift perturbations of the BHETA aromatic signals upon dilution.
The thermal properties of the PET-derived BHETA-PEG family of polymers were probed using TGA and DSC to determine their thermal stability, glass transition (Tg) and crystallisation temperatures (Tc).§ TGA studies showed that all polymers were stable up to 250 °C (Fig. S6†), well beyond the working temperatures for battery applications. Derivative thermogravimetry (DTG) (Fig. S7†) reveals that the BHETA-containing polymers show a first thermal degradation peak centred around ∼300 °C, which likely resulted from decomposition of the BHETA component as this peak became less pronounced with polymers containing smaller percentages of BHETA. DSC studies (Table 1 and Fig. S8†) showed that generally, lower Tc was observed with the polymers containing the highest percentages of BHETA (1BHETA:2PEG and 1BHETA:4PEG). These findings suggested that BHETA was able to reduce the interchain interactions and crystallinity of the PEG components, potentially facilitating segmental chain motion and Li+ ion transport.
The PET-derived BHETA-PEG polyurethanes were first studied as SPEs. The polyurethane films were cast as a 10 wt% solution in anhydrous DMSO at 120 °C with LiTFSI salt (10 mol% w.r.t. PEG repeating units) in an Ar-filled glovebox, before the DMSO was evaporated to obtain a solid film (see Fig. 3A inset). As LiTFSI can act as a plasticiser,41 only 1BHETA:2PEG and 1BHETA:4PEG samples were able to form free-standing films under such conditions. Indeed, the presence of LiTFSI reduced the elastic modulus of the 1BHETA:2PEG film from 61.7 ± 4.8 to 23.2 ± 3.6 MPa (Fig. S9 and S10†). While increasing the solid weight percentage to >15 wt% (w.r.t. DMSO solvent) for 1BHETA:6PEG allowed the formation of a free-standing film in the presence of LiTFSI, free standing films could not be formed from the other polyurethanes containing lesser amounts of BHETA (i.e. 1BHETA:7PEG and PUs without BHETA) using a similar approach. This demonstrated the importance of BHETA as a structural component of the polyurethane electrolytes in improving their mechanical integrity. The 1BHETA:2PEG polymer film containing LiTFSI showed improved thermal stability, with onset of thermal decomposition at ∼350 °C (from TGA), compared to that of the polymer alone (onset ∼240 °C) (Fig. S11†), likely arising from interactions between the PEG and LITFSI salt.42
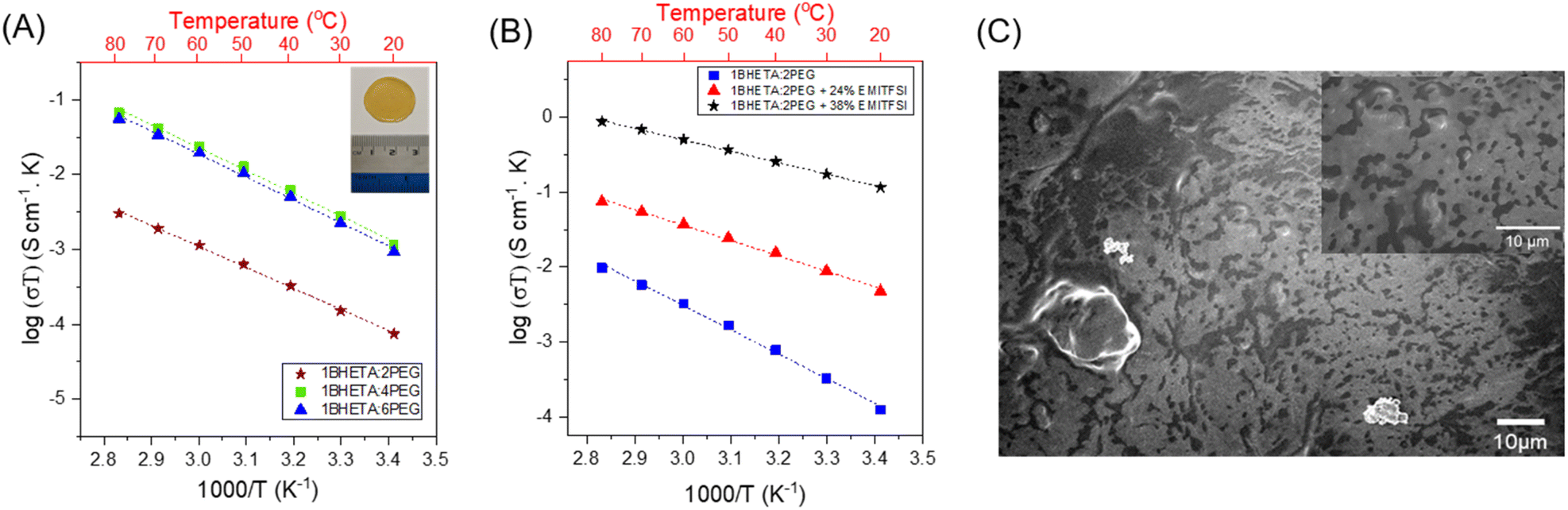 | ||
| Fig. 3 Effects of (A) modifying the BHETA:PEG ratio in the solid polymer electrolyte; inset: appearance of the 1BHETA:2PEG/LiTFSI solid polymer electrolyte film; and (B) increasing EMI-TFSI ionic liquid content in 1BHETA:2PEG gel polymer electrolytes on the ionic conductivity of the polyurethane; (C) SEM surface morphology of BHETA:PEG electrolyte after hexane rinsing to remove EMI-TFSI at 1000× magnification (inset: 3000× magnification). | ||
The ionic conductivities of the BHETA:PEG SPEs were measured above their Tg values between 20–80 °C. Linear Arrhenius-type behaviour was observed with expected augmented conductivities at higher temperatures (Fig. 3A), suggesting that Li+ ion conduction is occurring via a hopping mechanism within the polymer matrix decoupled from long-chain motion of the polyurethane electrolytes.43 When the BHETA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PEG ratio increases from 1:2 to 1:4, the ionic conductivity at both 20 °C and 60 °C were increased by more than an order of magnitude (Table 2). This is expected from the higher proportions of PEG chains present, whose lower rigidity facilitated Li+ ion transport. Using the Arrhenius equation:44
:PEG ratio increases from 1:2 to 1:4, the ionic conductivity at both 20 °C and 60 °C were increased by more than an order of magnitude (Table 2). This is expected from the higher proportions of PEG chains present, whose lower rigidity facilitated Li+ ion transport. Using the Arrhenius equation:44
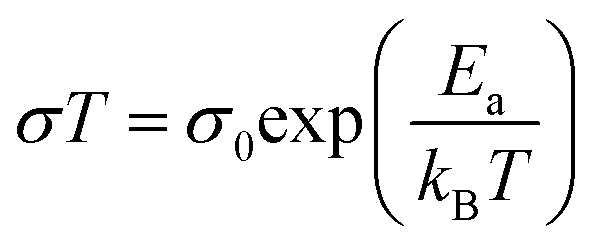 | (1) |
:PEG ratio to 1:6 did not further enhance ionic conductivity. Notably, these values are comparable to those previously reported for PEO-LiTFSI systems of 0.49 to 0.7 eV,45–47 suggesting that the dissociation of the Li+ and TFSI− was not impeded. Furthermore, the similar Ea to neat PEO-LiTFSI systems would also suggest that the presence of hydrogen bonding between the amides and the urethanes, as well as the rigid aromatic groups which enhances the SPE films' mechanical integrity, are decoupled from the ion conducting EO motifs. The decoupling of functionalities is an important strategy to achieve both facile ion transport and structural stability simultaneously without falling into the rule of mixtures.48
| Polymer matrix | EO:LITFSI molar ratioa | Ionic conductivity at 20 °C (S cm−1) | Ionic conductivity at 60 °C (S cm−1) | E a (eV) | R 2 |
|---|---|---|---|---|---|
| a Amount of LiTFSI determined using the no. of mols of PEG (Mn 17.5 kDa) based on the mass percentage of PEG from each polymer (Table 1). b Determined from eqn (1). c Goodness-of-fit when log(σT) was plotted against (1000/T). d Mass percentage of EMITFSI ionic liquid content calculated w.r.t. the total mass of polymer, LiTFSI and EMITFSI present (see ESI). | |||||
| Solid polymer electrolytes | |||||
| 1BHETA:2PEG | 10:1 |
2.5 × 10−7 | 3.4 × 10−6 | 0.56 | 0.999 |
| 1BHETA:4PEG | 10:1 |
3.9 × 10−6 | 7.2 × 10−5 | 0.61 | 0.997 |
| 1BHETA:6PEG | 10:1 |
3.2 × 10−6 | 5.9 × 10−5 | 0.61 | 0.997 |
| 1BHETA:2PEG + 29% 100 kDa PEO | 10:1 |
3.3 × 10−6 | 4.0 × 10−5 | 0.53 | 0.998 |
| 1BHETA:6PEG + 28% 100 kDa PEO | 10:1 |
8.3 × 10−6 | 1.2 × 10−4 | 0.57 | 0.998 |
|
|||||
| Gel polymer electrolytes | |||||
| 1BHETA:2PEG | 8:1 |
4.2 × 10−7 | 9.7 × 10−6 | 0.65 | 0.997 |
| 1BHETA:2PEG + 24% EMITFSId | 8:1 |
1.6 × 10−5 | 1.1 × 10−4 | 0.41 | 0.996 |
| 1BHETA:2PEG + 38% EMITFSId | 8:1 |
3.9 × 10−4 | 1.5 × 10−3 | 0.30 | 0.999 |
While PEO has been explored extensively as an electrolyte for lithium polymer batteries, replacing part of the polymer electrolyte content with a waste plastic-derived alternative can offer more opportunities for sustainable battery design, as well as to enhance the electrolyte's mechanical robustness. To explore this possibility, we formulated mixed polymer electrolytes containing the PET-derived BHETA:PEG polymers with PEO (100 kDa) and measured their ionic conductivity. Compared with the pure BHETA:PEG polymers, an increase in ionic conductivity was observed at 60 °C in the presence of PEO (Table 2, Fig. S12 and S13†). Notably, these conductivities are comparable to those of PEO/LiTFSI polymer electrolytes.49 For solid polymer electrolyte systems based solely on semicrystalline polymers such as PEO, an appreciable ionic conductivity can only be observed near the polymer's melting point (Tm ∼65 °C).50 However, this is not ideal as the polymer will have negligible structural properties in the liquid phase and will not offer any improvements in battery safety. In contrast, the combination of BHETA:PEG/PEO could give linear Arrhenius behavior for ion transport between 20–80 °C without any significant thermal phase transitions. This shows that despite the plasticizing effect of LiTFSI, the BHETA:PEG polymers were able to provide sufficient mechanical support to resist significant phase changes. Indeed, maintaining the mechanical rigidity of the electrolyte without compromising on the ionic conductivity is important to the overall safety of the battery. This is especially true for next generation of energy storage devices like Li-metal and structural batteries.
Compared to SPEs, GPEs are considered to be more practically applicable in lithium-ion batteries.51 Comprising of a solution of lithium salt in an ionic liquid embedded within a solid polymer matrix, GPEs combine desirable properties of higher conductivities and good mechanical properties.52–54 As GPEs matrices, polyurethanes can hydrogen bond to the liquid electrolytes, which enhances interaction and wettability with the liquid phase.55 Encouraged by the favourable aforementioned mechanical properties of the 1BHETA:2PEG SPE films, we investigated its use as a polymer matrix material for GPEs to further enhance the films' ionic conductivity. 1BHETA:2PEG and LiTFSI were cast into homogeneous free-standing non-leaky gel electrolyte films containing different quantities of EMI-TFSI ionic liquid, whose conductivities at different temperatures were then determined.
As shown in Fig. 3B and Table 2, the presence of the EMI-TFSI/LiTFSI liquid phase resulted in considerable enhancements to the ionic conductivity of the GPE of nearly 3 orders of magnitude compared with just 1BHETA:2PEG alone. Arrhenius-like behavior with the expected higher conductivity at elevated temperatures were observed. In addition, the lack of conspicuous phase changes (see Fig. S14† for DSC) and sharp changes in the conductivity over the range of temperatures investigated is indicative of the amorphous nature of the 1BHETA-2PEG polymer electrolyte. Increasing EMI-TFSI ionic liquid content also reduced the activation energy for ion conduction, likely due to improved liquid phase percolation through the porous polymer matrix (Fig. 3C), generating conducting channels which facilitate ion conduction. Indeed, the ionic conductivity (∼10−4 S cm−1) observed in the presence of 38% EMI-TFSI loading at ambient temperature is comparable to those of liquid electrolytes.56
The lithium transference number of the 1BHETA:2PEG GPE was estimated to be 0.48 (Fig. S15†) using the Bruce and Vincent method,57 which indicated that Li+ and TFSI− contributed almost equally to ionic conductivity. These values are comparable with similar GPE systems based on PEO-ionic liquids58 and shows that the diffusion of Li+ under polarisation was unhindered by the PU structural framework. Measurements of the oxidative stability of this GPE system revealed an onset of oxidation at ∼3.3 V (Fig. S16†). While this precluded usage with high-voltage cathodes, our GPE system was still compatible with the widely-used LiFePO4 (LFP) cathode material.
Thus, we next evaluated the feasibility of BHETA:PEG gel-polymer as a electrolyte by assembling proof-of-concept Li-ion cells with Li4Ti5O12 (LTO) as anode and LFP as cathode. The cycling performance was investigated in the voltage range of 1.5 V to 2.5 V at room temperature and at 60 °C at constant current density of 0.1C. Fig. 4A and B show the room temperature charge/discharge voltage plateaus and coulombic efficiency of LTO:BHETA:PEG gel-polymer: LFP for 150 cycles, respectively.
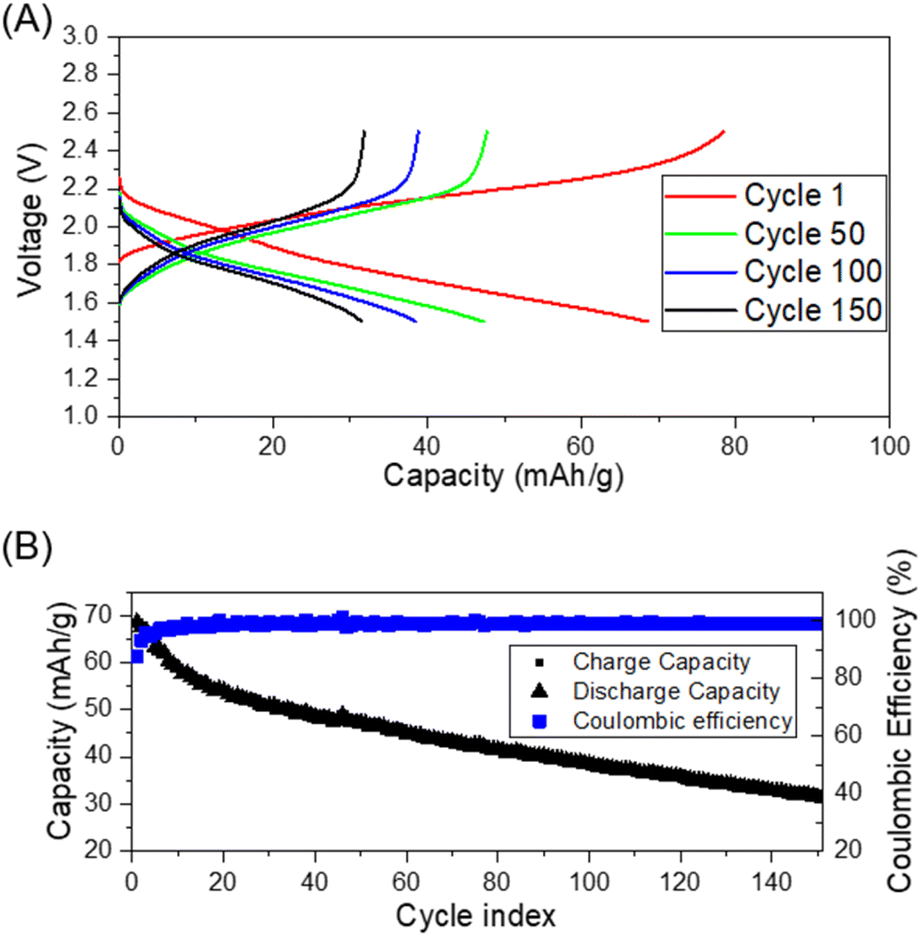 | ||
| Fig. 4 (A) Room temperature galvanostatic charge–discharge plots of Li-ion cell using the gel polymer electrolyte containing 1BHETA:2PEG + 38% EMI-TFSI, LTO anode and LFP cathode; (B) coulombic efficiency and charge/discharge capacity of the same cell for 150 cycles. | ||
The cells can be successfully charged and discharged both at room temperature and 60 °C (Fig. S17†). The charge stored and delivered at room temperature stabilised after the initial 12 cycles, with coulombic efficiency of >98% throughout the 150 cycles. For the cells running at elevated temperature of 60 °C, higher capacities (close to theoretical capacity of 170 mA h g−1) was achieved. The more dramatic capacity loss observed for these cells may be attributed to the increase of cell resistance and loss of effective contact due to the gradual extraction of ionic liquid while cycling at higher temperature. This may be due to the limited absorption capacity of the BHETA:PEG polymer.59
In conclusion, we have demonstrated successfully for the first time, the upcycling of PET from waste beverage bottles into polymer electrolytes. PET was first organocatalytically depolymerised into BHETA, which was then re-polymerised with PEG segments to form polyurethanes. The rigid aromatic BHETA components were essential for providing mechanical support for the formation of free-standing conductive films in the presence of LiTFSI, while the PEG segments provided the flexibility for ion conduction. Overall, our findings clearly confirm the potential of these waste PET-derived polyurethanes as polymer electrolytes for Li-ion batteries, achieving a room temperature conductivity of 10−4 S cm−1 as a GPE. To the best of our knowledge, this is the first report of a working Li-ion battery assembled using upcycled waste plastic-derived polymers as the electrolyte. The promising performance of our BHETA:PEG electrolyte opens new pathways and possibilities towards application of upcycling high-volume waste commodity plastics as battery materials, contributing to a sustainable energy future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the structural power for portable and electrified transportation grant (Grant number A20H3b0140, AME programmatic) for the support of this work. J. Y. C. L. acknowledges the A*STAR Central Research Fund for generous funding support. The authors acknowledge Ms Tan Sze Yu for helpful advice and discussions on thermal characterisations of the BHETA-PEG polymers.Notes and references
- C. Bremer, Global Plastics Outlook, Organisation for Economic Co-operation and Development (OECD), 2022 Search PubMed.
- I. Tiseo, Statista: Production Capacity of Polyethylene Terephthalate Worldwide from 2014 to 2024, https://www.statista.com/statistics/242764/global-polyethylene-terephthalate-production-capacity/ Search PubMed.
- Improving Plastics Management: Trends, Policy Responses, and the Role of International Co-operation and Trade, Organisation for Economic Co-operation and Development (OECD), 2018 Search PubMed.
- A. J. Ragauskas, G. W. Huber, J. Wang, A. Guss, H. M. O'Neill, C. S. K. Lin, Y. Wang, F. R. Wurm and X. Meng, ChemSusChem, 2021, 14, 3982–3984 CrossRef PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- A. B. Raheem, Z. Z. Noor, A. Hassan, M. K. Abd Hamid, S. A. Samsudin and A. H. Sabeen, J. Clean. Prod., 2019, 225, 1052–1064 CrossRef.
- European Virgin PET Rises above RPET as Virgin PET Prices Hit 10-year High, 2022, accessed 03 August 2022, https://www.spglobal.com/commodityinsights/en/market-insights/latest-news/petrochemicals/110421-european-virgin-pet-rises-above-rpet-as-virgin-pet-prices-hit-10-year-high Search PubMed.
- D. Platt, Recycled PET Buyers Switch to Virgin PET as Price Gap Widens in Europe, 2022, accessed 03 August 2022, https://www.plasticsnews.com/resin-pricing/why-recycled-pet-buyers-switching-virgin-pet-price-gap-widens-europe Search PubMed.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef.
- J. Y. Q. Teo, A. Ong, T. T. Y. Tan, X. Li, X. J. Loh and J. Y. C. Lim, Green Chem., 2022, 24, 6086–6099 RSC.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Rep Phys Sci, 2021, 2, 100514 CrossRef.
- J. B. Williamson, S. E. Lewis, R. R. Johnson III, I. M. Manning and F. A. Leibfarth, Angew. Chem., Int. Ed., 2019, 58, 8654–8668 CrossRef PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef.
- J. Y. Q. Teo, C. W. S. Yeung, T. T. Y. Tan, W. W. Loh, X. J. Loh and J. Y. C. Lim, Green Chem., 2022, 24, 6287–6294 RSC.
- M. D. de Dios Caputto, R. Navarro, J. L. Valentín and Á. Marcos-Fernández, J. Polym. Sci., 2022, 1 DOI:10.1002/pol.20220137.
- S. T. Kenny, J. N. Runic, W. Kaminsky, T. Woods, R. P. Babu, C. M. Keely, W. Blau and K. E. O'Connor, Environ. Sci. Technol, 2008, 42, 7696–7701 CrossRef PubMed.
- C. Song, B. Zhang, L. Hao, J. Min, N. Liu, R. Niu, J. Gong and T. Tang, Green Energy Environ., 2022, 7, 411–422 CrossRef.
- W. P. R. Deleu, I. Stassen, D. Jonckheere, R. Ameloot and D. E. De Vos, J. Mater. Chem. A, 2016, 4, 9519–9525 RSC.
- J. C. Barbosa, R. Gonçalves, C. M. Costa and S. Lanceros-Méndez, ACS Omega, 2022, 7, 14457–14464 CrossRef.
- A. Mirjalili, B. Dong, P. Pena, C. S. Ozkan and M. Ozkan, Energy Storage, 2020, 2, e201 CrossRef.
- J. Min, X. Wen, T. Tang, X. Chen, K. Huo, J. Gong, J. Azadmanjiri, C. He and E. Mijowska, Chem. Commun., 2020, 56, 9142–9145 RSC.
- S. Villagómez-Salas, P. Manikandan, S. F. Acuña Guzmán and V. G. Pol, ACS Omega, 2018, 3, 17520–17527 CrossRef PubMed.
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo-Martínez, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC.
- M. Y. Tan, D. Safanama, S. S. Goh, J. Y. C. Lim, C.-H. Lee, J. C. C. Yeo, W. Thitsartarn, M. Srinivasan and D. W. H. Fam, Chem.–Asian J., 2022, e202200784 Search PubMed.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef.
- J. P. Greene, in Automotive Plastics and Composites, ed. J. P. Greene, William Andrew Publishing, 2021, pp. 107–125, DOI:10.1016/B978-0-12-818008-2.00022-2.
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Nat. Rev. Mater., 2019, 4, 312–330 CrossRef.
- F. Danzi, R. M. Salgado, J. E. Oliveira, A. Arteiro, P. P. Camanho and M. H. Braga, Molecules, 2021, 26(8), 2203 CrossRef PubMed.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- J. Y. C. Lim, Q. Lin, C. K. Liu, L. Guo, K. Xue and X. J. Loh, Mater. Adv., 2020, 1, 3221–3232 RSC.
- H. Sardon, A. Pascual, D. Mecerreyes, D. Taton, H. Cramail and J. L. Hedrick, Macromolecules, 2015, 48, 3153–3165 CrossRef.
- Y. Schellekens, B. Van Trimpont, P.-J. Goelen, K. Binnemans, M. Smet, M.-A. Persoons and D. De Vos, Green Chem., 2014, 16, 4401–4407 RSC.
- Z. Lv, Y. Tang, S. Dong, Q. Zhou and G. Cui, Chem. Eng. J., 2022, 430, 132659 CrossRef.
- S. Wei Chien, J. J. M. Tay, C. P. T. Chee, X. Jun Loh, D. W. H. Fam and J. Y. C. Lim, ChemSusChem, 2021, 14, 3237–3243 CrossRef.
- H. Mu, X. Huang, W. Wang, X. Tian, Z. An and G. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 622–632 CrossRef.
- K. Fukushima, J. M. Lecuyer, D. S. Wei, H. W. Horn, G. O. Jones, H. A. Al-Megren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. McNeil, J. E. Rice and J. L. Hedrick, Polym. Chem., 2013, 4, 1610–1616 RSC.
- I. Kammakakam, K. E. O'Harra, G. P. Dennis, E. M. Jackson and J. E. Bara, Polym. Int., 2019, 68, 1123–1129 CrossRef.
- B. S. Gregorí Valdés, C. S. B. Gomes, P. T. Gomes, J. R. Ascenso, H. P. Diogo, L. M. Gonçalves, R. Galhano dos Santos, H. M. Ribeiro and J. C. Bordado, Polymers, 2018, 10(10), 1170 CrossRef.
- I. Yilgor, E. Yilgor, I. G. Guler, T. C. Ward and G. L. Wilkes, Polymer, 2006, 47, 4105–4114 CrossRef.
- S. Sylla, J. Y. Sanchez and M. Armand, Electrochim. Acta, 1992, 37, 1699–1701 CrossRef.
- N. Angulakshmi, R. B. Dhanalakshmi, M. Kathiresan, Y. Zhou and A. M. Stephan, Mater. Chem. Front., 2020, 4, 933–940 RSC.
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Devices, 2018, 3, 1–17 Search PubMed.
- R. B. Nuernberg, Ionics, 2020, 26, 2405–2412 CrossRef.
- M. Unge, H. Gudla, C. Zhang and D. Brandell, Phys. Chem. Chem. Phys., 2020, 22, 7680–7684 RSC.
- R. Bakar, S. Darvishi, T. Li, M. Han, U. Aydemir, S. Nizamoglu, K. Hong and E. Senses, ACS Appl. Polym. Mater., 2022, 4, 179–190 CrossRef.
- B. N. Choi, J. H. Yang, Y. S. Kim and C.-H. Chung, RSC Adv., 2019, 9, 21760–21770 RSC.
- D. G. Mackanic, X. Yan, Q. Zhang, N. Matsuhisa, Z. Yu, Y. Jiang, T. Manika, J. Lopez, H. Yan, K. Liu, X. Chen, Y. Cui and Z. Bao, Nat. Commun., 2019, 10, 5384 CrossRef.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- B. K. Money and J. Swenson, Macromolecules, 2013, 46, 6949–6954 CrossRef CAS.
- F. Baskoro, H. Q. Wong and H.-J. Yen, ACS Appl. Energy Mater., 2019, 2, 3937–3971 CrossRef CAS.
- M. Zhu, J. Wu, Y. Wang, M. Song, L. Long, S. H. Siyal, X. Yang and G. Sui, J. Energy Chem., 2019, 37, 126–142 CrossRef.
- C. Ma, W. Cui, X. Liu, Y. Ding and Y. Wang, InfoMat, 2022, 4, e12232 CAS.
- X. Cheng, J. Pan, Y. Zhao, M. Liao and H. Peng, Adv. Energy Mater., 2018, 8, 1702184 CrossRef.
- D. Li, L. Yuan, G. Liang and A. Gu, Ind. Eng. Chem. Res., 2020, 59, 6600–6608 CrossRef CAS.
- J. Wu, L. Yuan, W. Zhang, Z. Li, X. Xie and Y. Huang, Energy Environ. Sci., 2021, 14, 12–36 RSC.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef.
- C. Zhu, H. Cheng and Y. Yang, J. Electrochem. Soc., 2008, 155, A569 CrossRef.
- P. F. R. Ortega, J. P. C. Trigueiro, G. G. Silva and R. L. Lavall, Electrochim. Acta, 2016, 188, 809–817 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06692k |
| ‡ Due to the limited solubility of the BHETA:PEG polymers in THF and DMF, we were unable to determine the relative molecular weights of these polymers by gel permeation chromatography (GPC). |
| § The melting points of the BHETA-PEG polymers were too close to their decomposition temperatures for accurate values to be obtained. |
| This journal is © The Royal Society of Chemistry 2022 |