
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-stage electrolysis of H2O and CO2 to methanol: CO2-to-methane reduction at the cathode and subsequent methane-to-methanol oxidation at the anode†
Takashi
Hibino
 *,
Kazuyo
Kobayashi
,
Masahiro
Nagao
,
Zhou
Dongwen
and
Chen
Siyuan
*,
Kazuyo
Kobayashi
,
Masahiro
Nagao
,
Zhou
Dongwen
and
Chen
Siyuan
Graduate School of Environmental Studies, Nagoya University, Nagoya 464-8601, Japan. E-mail: hibino@urban.env.nagoya-u.ac.jp
First published on 16th October 2022
Abstract
Co-electrolysis of H2O and N/C sources to valuable chemicals has the potential to contribute to global warming mitigation. We here report the electrochemical conversion of H2O and CO2 into methanol in a gas flow system at temperatures between 100 and 200 °C. CO2 was reduced to methane with voltage-boosted H2 at a Ru/C cathode. Introduced Fe2O3 nanoparticles promoted the adsorption and activation of CO2 on the surface of Ru because of their high redox activity. Methane was oxidized to methanol through the formation of ˙CH3 and active oxygen species at a Pt anode. The addition of Au to the anode enhanced the selectivity toward methanol by reducing the activity of Pt in the complete oxidation of methanol. Combining these processes with gas penetration into the electrolyte membrane enabled the synthesis of methanol from H2O and CO2, with a maximum faradaic efficiency of 54%. This work establishes a methodology for providing a good balance between the activity and selectivity of electrodes for the CO2 reduction reaction.
Introduction
Hydrogen is an important resource that allows renewable electricity to be transported from the energy sector to end-use sectors without transmission lines.1 However, the storage and transportation of hydrogen still present challenges. Ammonia, alcohols, and hydrocarbons have been extensively investigated as potential hydrogen carriers.2–4 To maximize the sustainability of hydrogen, in terms of energy conversion efficiency and CO2 emissions, one-step production of these carriers is regarded as more suitable than multi-step production. Ideally, one-step production can be accomplished by the co-electrolysis of H2O and nitrogen/carbon sources using renewable energies. Therefore, numerous reports on the electrochemical synthesis of ammonia5,6 from H2O and N2 and the electrochemical synthesis of alcohols7–9 or hydrocarbons10–12 from H2O and CO2 have been reported. In particular, the CO2 reduction reaction (CO2RR) has recently been used for carbon capture and utilization.13–16Here, we focus on the CO2RR to yield methanol, which is a liquid fuel for the transportation industry and a raw material for the chemical industry. Most of the electrolysis processes reported to date have been carried out in aqueous solutions at room temperature (eqn 1):17–20
| CO2 + 6H+ + 6e− → CH3OH + H2O | (1) |
In the present study, we introduce a new CO2RR method in which all the reactions are conducted in the gas phase rather than in the liquid phase and proceed at temperatures greater than room temperature. The presented method enhances the CO2RR kinetics, thereby enabling the design of a more compact and efficient reactor unit than those used for liquid-phase electrolysis. However, in the proposed reaction system, as the temperature increases, the evolution of H2 becomes substantial, followed by CO2 methanation,33 which reduces the selectivity toward methanol in the products. Note that methane has been anodically oxidized to methanol at temperatures of 100 °C or higher.34 Therefore, excellent balance between the CO2 reactivity and methanol selectivity can be achieved using not only the cathode but also the anode as working electrodes (Scheme 1). In the first stage, CO2 is reduced to methane through an eight-electron reaction at the cathode (eqn (2)):
| CO2 + 8H+ + 8e− → CH4 + 2H2O | (2) |
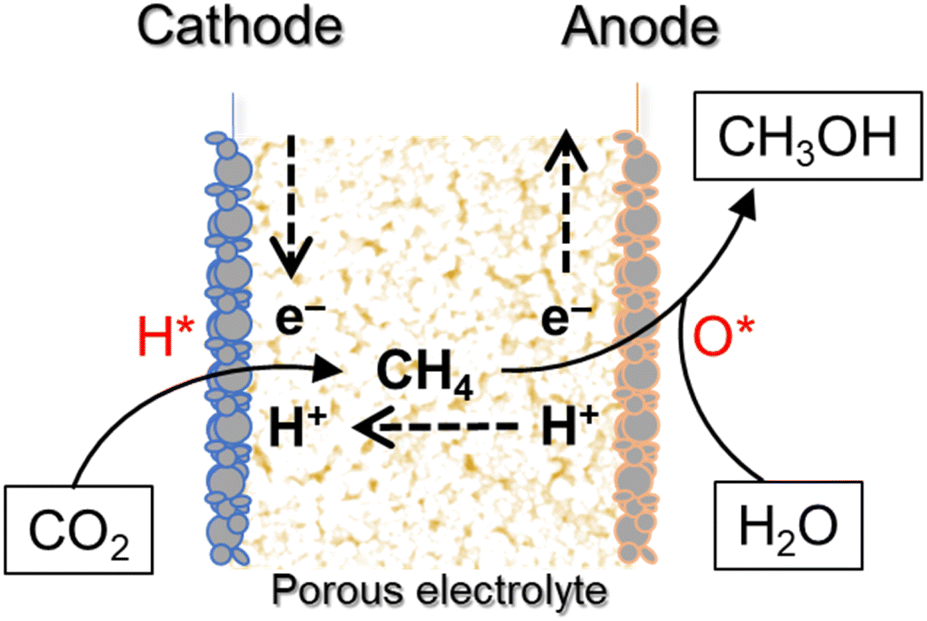 | ||
| Scheme 1 Schematic of two-stage electrolysis of H2O and CO2 to methanol using a porous electrolyte membrane. H* and O* are boosted hydrogen and active oxygen, respectively. | ||
The produced methane is then transferred from the cathode to the anode. In the second stage, methane is oxidized to methanol via a two-electron reaction at the anode (eqn (3)):
| CH4 + H2O → CH3OH + 2H+ + 2e− | (3) |
Lamoureux et al. reported a similar two-stage electrolysis for the synthesis of nitroso compounds from nitrobenzene via the formation of hydroxylamines.35
To implement the proposed protocol, we first separately optimized the cathode and anode materials for methane and methanol formation, respectively. We also improved the transport of methane from the cathode to the anode using a porous electrolyte membrane. Finally, we assessed CO2RR methanol production by combining these techniques. All experiments were carried out in the temperature range 100–200 °C. We assume that solar thermal, geothermal, and industrial waste heat would be appropriate heat sources for operating the cell.
Experimental
Materials
The metal/C cathodes (metal = Ni, Cu, Ru, Rh, Pd, Ir, Pt and Au; C = Vulcan, XC-72R; metal content = 30 wt%; metal loading = 4 mg cm−2) were prepared by reduction of metal chloride salts with NaBH4 in a carbon suspension. In the case of Ru/C, 0.56 g of the carbon source and 0.50 g of RuCl3·nH2O were ultrasonically mixed in a solution of 100 mL 2-propanol and 100 mL deionized water for 1 h. The pH of the solution was adjusted to 10 by addition of 0.1 M NaOH, and the solution was then stirred at 80 °C. The NaBH4 solution was added dropwise to the suspension to obtain a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of NaBH4:Ru. Finally, the suspension was stirred for 2 h before the solid was collected by filtration, washed with deionized water, and dried under vacuum at 100 °C. The electrode powder and an aqueous polytetrafluoroethylene (PTFE) dispersion were blended in a mixture of 2-propanol and deionized water using a Thinky AR-100 mixer. The electrode–PTFE slurry was then screen-printed onto the surface of the gas-diffusion layer (Toray TGPH-090) of a cathode. Fe-, Ce-, Co-, and In-based co-catalysts were added to the cathode by impregnation with the corresponding metal nitrate solutions, followed by thermal decomposition at 200 °C. The amount of each additive was adjusted by controlling the weight of the nitrate in the solution (40 mL). Various metal anodes (Ti, V, Fe, Cu, Mo, Pd, Ag, W, Pt, and Au) were deposited onto one side of the electrolyte by sputtering the corresponding metal targets. The metal targets (49 mm diameter, 0.05 mm thick, 99.99%) were purchased from Nilaco. Metal deposition was carried out under an Ar atmosphere using a Sanyu Electron SC-701MkII sputterer. The current was maintained at 5 mA by adjusting the Ar pressure in the chamber during sputtering. Unless otherwise stated, the loading of each deposited metal was controlled to be ∼0.1 mg cm−2 by manipulating the sputtering time. The ionic conductor was Sn0.9In0.1P2O7 with a proton conductivity of ∼0.05 S cm−1 at 100 °C or higher.36 A mixture of SnO2, In2O3, and 85% H3PO4 was calcined in a covered alumina crucible at 650 °C for 2.5 h. Sn0.9In0.1P2O7 powder (1 g) was mixed with 0.04 g of PTFE powder in a mortar and then cold-rolled to thicknesses of 150–250 μm using a laboratory rolling mill. Pt and Pt/C electrodes were used as counter electrodes for the cathodic and anodic electrolysis measurements, respectively.
:1 molar ratio of NaBH4:Ru. Finally, the suspension was stirred for 2 h before the solid was collected by filtration, washed with deionized water, and dried under vacuum at 100 °C. The electrode powder and an aqueous polytetrafluoroethylene (PTFE) dispersion were blended in a mixture of 2-propanol and deionized water using a Thinky AR-100 mixer. The electrode–PTFE slurry was then screen-printed onto the surface of the gas-diffusion layer (Toray TGPH-090) of a cathode. Fe-, Ce-, Co-, and In-based co-catalysts were added to the cathode by impregnation with the corresponding metal nitrate solutions, followed by thermal decomposition at 200 °C. The amount of each additive was adjusted by controlling the weight of the nitrate in the solution (40 mL). Various metal anodes (Ti, V, Fe, Cu, Mo, Pd, Ag, W, Pt, and Au) were deposited onto one side of the electrolyte by sputtering the corresponding metal targets. The metal targets (49 mm diameter, 0.05 mm thick, 99.99%) were purchased from Nilaco. Metal deposition was carried out under an Ar atmosphere using a Sanyu Electron SC-701MkII sputterer. The current was maintained at 5 mA by adjusting the Ar pressure in the chamber during sputtering. Unless otherwise stated, the loading of each deposited metal was controlled to be ∼0.1 mg cm−2 by manipulating the sputtering time. The ionic conductor was Sn0.9In0.1P2O7 with a proton conductivity of ∼0.05 S cm−1 at 100 °C or higher.36 A mixture of SnO2, In2O3, and 85% H3PO4 was calcined in a covered alumina crucible at 650 °C for 2.5 h. Sn0.9In0.1P2O7 powder (1 g) was mixed with 0.04 g of PTFE powder in a mortar and then cold-rolled to thicknesses of 150–250 μm using a laboratory rolling mill. Pt and Pt/C electrodes were used as counter electrodes for the cathodic and anodic electrolysis measurements, respectively.
Characterization
X-ray diffraction (XRD) patterns were collected using a Rigaku MiniFlex II diffractometer equipped with a monochromator and a Cu Kα radiation source (1.5432 Å). X-ray photoelectron spectroscopy (XPS) was performed using a VG Scientific ESCALab220i-XL spectrometer equipped with a monochromatic Al Kα radiation source (1486.6 eV). Transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDX) images were recorded using a Jeol JEM2100HK instrument operated at an accelerating voltage of 200 kV and with a beam current of 92 μA. Thermogravimetric–differential thermal analysis (TG–DTA) curves were obtained from room temperature to 500 °C using a Shimadzu DTG-60 instrument. Ultraviolet-visible (UV-vis) absorbance was measured in the wavelength range 300–700 nm using a Jasco V-630 spectrophotometer. X-ray absorption fine structure (XAFS) measurements were carried out on the BL11XU beamline at SPring-8, Japan. Extended XAFS (EXAFS) data were analyzed using WinXAS ver.3.2.37 Scanning electron microscopy (SEM) images were acquired at accelerating voltages of 2 and 10 kV using a Keyence VE-8800. The composition of the outlet gas from the electrode was monitored using two online gas chromatographs (Varian CP-4900 and Shimadzu GC-2014), in which the off-gas line was heated to ∼90 °C.Electrochemical measurements
The electrochemical cell was constructed by placing the cell assembly between two alumina tube systems, where each system was composed of double tubes with outer diameters of 6 and 13 mm (Fig. S1†). This cell was modified by attaching an additional bypass line to the chamber (Fig. S2(a)†) or removing the 6 mm tube from the chamber (Fig. S2(b)†) for the two-stage electrolysis. A single-chamber cell configuration was also used to evaluate the effect of the frequency of the applied voltage in the AC electrolysis on the formation rate of individual products (Fig. S3†). The following gas mixtures were used as the reactant gases: CO2 (20%) and He (80%) for the cathodic electrolysis; methane (20%), H2O (6%), and He (balance) for the anodic electrolysis; and CO2 (20%), H2O (6%), and He (balance) for the two-stage electrolysis. A mixture of H2O (6%) and He (94%) or pure He was supplied to the anode or cathode as the counter gas. A mixture of methane (20%), H2O (6%), and He (balance) was introduced into both the anode and cathode of the cell placed horizontally in the single chamber. The flow rate of all gases was maintained at 15 mL min−1 by a GL Sciences electronic mass flow controller. All electrochemical measurements were performed by the four-probe method using a potentiostat–galvanostat (Solartron 1287) and a frequency-response analyzer (Solartron 1260). A potential as negative as −4 V and as positive as +3 V was applied for the cathodic electrolysis and the anodic electrolysis, respectively. The faradaic efficiency (FE) for methanol formation was determined using the following equation:| FE = ((observed number of moles methanol)/(theoretical number of moles methanol)) × 100 | (4) |
The theoretical number of moles of methanol was calculated according to Faraday's law on the basis of the six-electron reaction represented by eqn (1). Impedance spectra were collected at various bias voltages over the frequency range from 0.1 to 106 Hz. The recorded curves were fitted with an equivalent circuit consisting of the serial resistance, the parallel resistance, and a constant phase element (CPE). Electrolysis of H2O was also carried out at various voltages in a phosphoric acid solution containing methyl violet for 3 h, where the bottom of the container was heated at 200 °C.
Catalytic and redox measurements
The catalytic properties of the electrodes were measured using the same electrochemical cell at open-circuit voltage (OCV). The reactant gases were a mixture of CO2 (20%), H2 (1–80%), and He (balance) for the Ru/C electrode and a mixture of methane (20%), O2 (0.3 or 1.0%), and He (balance) for the Pt electrode. The redox properties of Fe2O3 used as a co-catalyst were evaluated via TG–DTA analysis upon introducing alternately and repeatedly air and a mixture of H2 (1%) and He (99%).Results and discussion
CO2-to-methane reduction at the cathode
The electrolysis of H2O and CO2 was carried out using various metal/C cathodes with a cell voltage of −4 V (vs. Pt counter electrode) at 200 °C. Fig. 1(a) shows that Ru provided the highest methane formation rate of 3.6 × 10−5 mol m−2 s−1, with an FE of 5.9%. Fig. 1(a) also shows that Cu exhibited no electrocatalytic activity for methane formation, which contrasts with previously reported results for the liquid-phase CO2RR toward methane production.38–40 | ||
| Fig. 1 (a) Distribution of products and current density recorded using various metal/C cathodes with a cell voltage of −4 V at 200 °C. Electrolysis characteristics of the Ru/C cathode: (b) the voltage dependence at 200 °C and (c) the temperature dependence at a cell voltage of −4 V. | ||
The electrolysis characteristics of the Ru/C cathode were investigated under various conditions. Methane, along with H2, was generated beginning at −2 V, whereas no other products were observed at cell voltages as high as −4 V (Fig. 1(b)). Notably, both the rate and FE for methane formation increased with increasing cell voltage, indicating that the formation of methane accelerated as the cathode was reduced. Another important result is that the rate of methane formation increased with increasing temperature, independently of the rate of H2 evolution (Fig. 1(c)). Therefore, these reactions likely compete with each other but might not be associated with each other.
The impedance spectra obtained for the electrolysis cell under various bias voltages are shown in Fig. 2(a). The experimental conditions were the same as those used for the electrolysis of H2O and CO2. The impedance curves were large at OCV and under a bias voltage of −1 V but became much smaller as the bias voltage was made more negative than −2 V. The sum of the serial and parallel resistances determined through the equivalent-circuit model were calculated to be 2210, 1910, 150, 75, and 110 Ω cm2 at OCV, −1, −2, −3, and −4 V, respectively, where the latter four resistances roughly agree with the DC resistances estimated from the current–voltage (I–V) curves (Fig. 2(b)). However, the CPE was in the 100 μF range irrespective of the bias voltage because this value is strongly dependent upon the electrode area.
 | ||
| Fig. 2 (a) Impedance spectra recorded when various bias voltages were applied to the cell at 200 °C. (b) I–V curve of the electrolysis cell at 200 °C. XRD profiles for the Ru/C cathode after the impedance measurements: (c) the diffraction peaks assigned to Ru (101) and RuO2 (211) and (d) their peak intensities. | ||
The crystalline structure of the Ru/C cathode after the impedance measurements was analyzed and correlated with the bias voltage. As shown in Fig. 2(c), the application of a bias voltage led to a change in the RuO2 and Ru crystalline phases—specifically, the growth of the Ru (101) phase and the decay of the RuO2 (211) phase. This reduction of RuO2 to Ru might also be related to the decrease in impedance with increasing bias voltage, as previously described. Note that, when the electrolysis cell was restored to the open-circuit condition and remained under this condition for 1 h at 200 °C, the intensities of the diffraction peaks of Ru decreased and those of the diffraction peaks of RuO2 increased (Fig. 2(d)). We therefore concluded that the metallic state of Ru was maintained only at high cathode potentials. Such a metallic state might also be favored at high temperatures.
Comparing the electrochemical CO2 methanation with thermal CO2 methanation (the so-called Sabatier reaction41,42) for the Ru/C cathode used in the present work is important because these reactions likely proceed under the same reaction mechanism. Fig. 3(a) shows the rate of methane formation on the Ru/C cathode at the OCV as a function of the H2 concentration in the reactant gas. The methane formation rate increased approximately linearly with increasing H2 concentration. Fig. 3(a) also includes data for the electrolysis conducted at a current density of 50 mA cm−2, which is comparable to the current density that generates approximately 1% H2 in the reactant gas. The rate of methane formation for the electrochemical reaction at 50 mA cm−2 was markedly higher than the rate of the thermal reaction at 1% H2.
 | ||
| Fig. 3 (a) Catalytic characteristics of the Ru/C cathode for methanation of CO2 with H2 under open-circuit conditions at 200 °C. Data for the methanation of CO2 at 50 mA cm−2 in the absence of H2 in the reactant gas are included for comparison. (b) XRD patterns and (c) XPS spectra for three samples after testing: sample 1 (current density = 50 mA cm−2 and H2 concentration in the reactant gas = 0%); sample 2 (0 mA cm−2 and 1% H2); sample 3 (0 mA cm−2 and 80% H2). | ||
To understand the observed difference, we carried out XRD and XPS measurements for three Ru/C samples denoted as samples 1, 2, and 3 (refer to the caption of Fig. 3). As shown in Fig. 3(b), the crystalline phases of sample 3 contained more metallic Ru than the crystalline phases of sample 2. By contrast, the diffraction profile of sample 1 was more similar to that of sample 3 than to that of sample 2. Similarly, as evident in the XPS spectra (Fig. 3(c)), whereas Ru was not observed on the surface of sample 2, it was detected on the surface of samples 1 and 3. These results indicate that sample 1 was more extensively reduced than sample 2 when exposed to H2 at the same concentration (∼1%). Electrochemically produced H2 has been reported to be a better reductant than natural H2.43,44 In addition, the work function of electrodes is known to be easily tuned via polarization,45 which may also contribute to the reduction of RuOx at the cathode.
In an effort to improve the electrocatalytic activity of the Ru/C cathode, particularly at low voltages, we added various materials to the cathode by thermally decomposing the corresponding metal nitrates at 200 °C (Fig. S4†). Among the tested additives, the Fe additive was the most effective in enhancing methane formation at the cathode (Fig. 4(a)). No specific peaks assigned to Fe compounds were observed in the XRD pattern of the Fe-impregnated Ru/C (data not shown). However, iron (green) was detected on the surface of Ru particles (red) in the EDX elemental maps (Fig. 4(b)). A comparison of the Fe–K XANES spectra of the cathode with the spectra of α- and γ-Fe2O3 powder samples indicates that the chemical state of iron in the cathode was consistent with that of iron in Fe2O3 (Fig. S5(a)†). The formed Fe2O3 also showed a considerably lower second neighbor peak than α- and γ-Fe2O3 in the EXAFS Fourier transform magnitudes (Fig. S5(b)†). These results indicate that nanosized or amorphous Fe2O3 particles were deposited around the Ru particles on the carbon support.
 | ||
| Fig. 4 (a) Effect of additives on the methane formation rate at 200 °C. (b) Dark-field TEM image and EDX elemental maps of C (blue), O (violet), Fe (green), Ru (red), and C–Fe–Ru combined. (c) Redox characteristics of Fe2O3 powder at 200 °C in different atmospheres: a mixture of H2 (1%) and He (99%) and air: TGA (blue) and DTA (orange). (d) Rate and FE of methane formation as a function of weight of Fe2O3 in the electrolysis at a cell voltage of −3 V. | ||
The redox characteristics of Fe2O3 powder produced from iron(III) nitrate were characterized using TG–DTA. Air and a mixture containing 1% H2 were alternately and repeatedly introduced to the sample at 200 °C. As shown in Fig. 4(c), the Fe2O3 powder showed a cyclic increase and decrease in weight upon the supply of oxidizing (air) and reducing (H2 mixture) gases, respectively. This process was also accompanied by the evolution and adsorption of heat, indicating that Fe2O3 could be reduced and re-oxidized at this temperature.
CeO2 is one of the most important co-catalysts46 or support materials47 for Ru-based catalysts in the Sabatier reaction, where its oxygen vacancies play an important role as adsorption or reaction sites for CO2; the oxygen vacancies extract an O atom from the adsorbed CO2 molecule and promote the subsequent methanation. In the present study, the addition of CeO2 also positively affected the electrocatalytic activity of Ru (Fig. 4(a)). We therefore speculate that Fe2O3 acts as a co-catalyst in the same manner as CeO2 but more powerfully than CeO2.
The amount of Fe2O3 added to the Ru/C cathode was optimized for methane formation at a cell voltage of −3 V. Fig. 4(d) shows that both the rate and FE for methane formation showed maximum values at an Fe2O3 loading of 0.27 mg cm−2, which is much lower than the loading of Ru (4 mg cm−2). This difference is attributable to the effective deposition of Fe2O3 around Ru particles, as previously described. However, sustaining such deposition at loadings greater than 0.27 mg cm−2 is difficult, resulting in a decrease in methane formation at a loading of 0.39 mg cm−2 (Fig. 4(d)).
Methane-to-methanol oxidation at the anode
Anode preparation was achieved by sputtering of metal particles onto the surface of the electrolyte. This method can control the amount of metal deposited via modification of the sputtering time, without changing the crystallite size (data obtained for Pt are shown in Fig. S6† as an example). We evaluated the electrolysis characteristics of various metal anodes by applying a voltage of +3 V (vs. Pt/C counter electrode) to the cell at a temperature of 200 °C (Fig. 5(a)). Both the methanol formation rate and the current density varied depending on the metal species. However, we identified a pattern, where the use of Pt, Pd, or Ag with metal–O or metal–OH binding energies of approximately 1.5 eV48,49 resulted in relatively high electrocatalytic activities for methanol formation. In particular, Pt provided the highest methanol formation rate of 2.0 × 10−5 mol m−2 s−1. Pt is known to substantially promote the dissociation of the C–H bonds of methane, which is another factor that contributes to the increase of the rate of methanol formation.50 | ||
| Fig. 5 (a) Distribution of products and current density recorded using various metal anodes: data were obtained with a cell voltage of +3 V at 200 °C. Electrolysis characteristics of the Pt anode: (b) the voltage dependence at 200 °C and (c) the temperature dependence at a cell voltage of +3 V. | ||
Fig. 5(b) shows the dependence of the cell voltage of the electrolysis characteristics on the Pt anode at 200 °C. The formation rates of methanol, ethane, and CO2 increased in proportion with the cell voltage in the range from +1.5 to +2.5 V. However, the rate of O2 formation increased rapidly at +3 V, indicating that the main reaction substrate was shifted from methane to H2O. The temperature dependence of the electrolysis characteristics was also evaluated at a cell voltage of +3 V in the temperature range 100–200 °C. As evident in Fig. 5(c), the formation rates for all the products increased with increasing temperature. More importantly, increasing the temperature increased the FE.
The reaction of methane with O2 at the Pt anode is non-negligible. Fig. 6(a) shows that no anode products were detected when the anode was supplied with a mixture of methane, H2O, O2 (0, 0.3, or 1.0%), and He under open-circuit conditions at 200 °C. Therefore, methanol—as well as ethane and CO2—was not generated through the nonelectrochemical reaction of methane with O2 during the electrolysis.
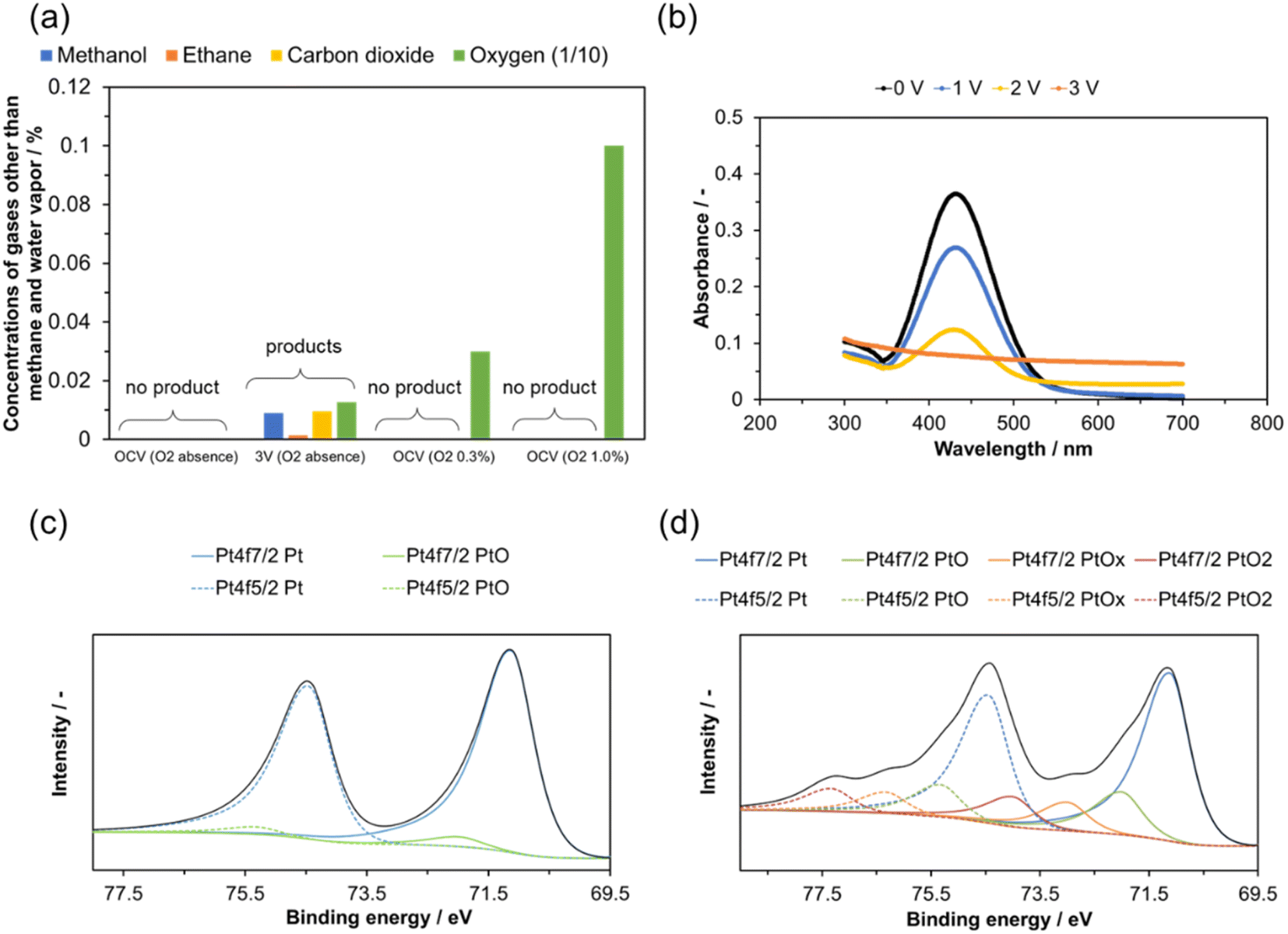 | ||
| Fig. 6 (a) Gas composition of the outlet gases from the anode at OCV or +3 V at 200 °C (excluding H2O and methane). The two reactant gases on the right contained 0.3% and 1.0% O2. (b) Change in UV-vis absorbance of methyl violet in a phosphoric acid solution as a result of H2O electrolysis. Deconvolved XPS Pt 4f spectrum for the Pt anode (c) before and (d) after electrolysis. | ||
Polarization of the anode enables the activation of O2. Methyl violet can be used to detect ˙OH radicals because it reacts with the radicals to form a colorless compound.51 Electrolysis of H2O was carried out using a Pt anode in a solution containing methyl violet. The change in the absorbance of the methyl violet as the cell voltage was varied is shown in Fig. 6(b), along with the change in the color of the solution (Fig. S7†). Whereas the peak at 432 nm remained almost unchanged at OCV during the experimental period, the intensity of this peak decreased with increasing cell voltage and was no longer detected at +3 V, indicating that ˙OH radicals were generated at the Pt anode at cell voltages of +1 V or higher.
The oxidation state of Pt in the anode before and after electrolysis was investigated by deconvolution of the Pt 4f signals in the XPS spectra. For the anode before electrolysis, the Pt 4f signal was fitted using two components indexed to Pt and PtO, where the Pt peaks were more intense than the PtO peaks (Fig. 6(c)). By contrast, for the anode after electrolysis, in addition to the aforementioned peaks, two new double peaks indexed to PtOx and PtO2 were detected by deconvolution analysis (Fig. 6(d)). These results indicate that metallic Pt was oxidized to high valence states by the anodic polarization.
On the basis of our previous observation of free radicals (˙OH) and surface oxygen species (PtO, PtOx, and PtO2), we can assume two reaction pathways: one pathway that produces ˙CH3 radicals via dehydrogenation of methane by ˙OH radicals and one that produces ˙CH3 radicals via dehydrogenation of methane by surface oxygen. This assumption led us to propose a reaction scheme (Fig. 7(a)) similar to those reported in numerous previous studies.52–55 According to this scheme, methanol is a coupling product of a ˙CH3 radical with an ˙OH radical (blue route). CO2 is synthesized by the oxidation of methanol and ethane (yellow route). Oxygen is the final oxidation product from the ˙OH radicals (green route).
 | ||
| Fig. 7 (a) Proposed reaction scheme for the electrolysis of methane and H2O. (b) Diagrams of the single-chamber cell and the AC voltage. (c) AC electrolysis characteristics of the Pt anode (amplitude voltage: 3 V). | ||
To support the proposed reaction scheme, we investigated the response of electrode reactions to the voltage change by introducing a mixture of methane, H2O, and He into a single-chamber cell with two identical Pt electrodes (Fig. 7(b)), in which one-half the mixture was supplied to the anode and one-half was supplied to the cathode. Fig. 7(c) shows the frequency dependence of the formation rates for all the products in AC electrolysis. At a low frequency of 0.1 Hz, the amounts of methanol, ethane, CO2, and O2 were close to one-half of those obtained by DC electrolysis at a voltage of +3 V (Fig.5(a)). H2 was also simultaneously observed as a product at the cathode. As the frequency increased, the formation rates of methanol, ethane, and CO2 decreased substantially compared with those of O2 and H2. In particular, the rate of CO2 formation at 10 Hz decreased to 8% of that at 0.1 Hz. The formation of CO2 involves numerous reaction steps (Fig. 7(a)). Accordingly, the concentration of intermediates near the electrode could not reach a nearly steady state at high frequencies; the mass transfer at the electrode surface tends to be rate-determining under such conditions.56 Therefore, subsequent deep oxidation could only proceed minimally. We propose that an additional effect of the AC electrolysis is alternating anodic and cathodic polarizations, which further suppresses excessive oxidation of the Pt anode compared with the DC electrolysis process.
Because the results in Fig. 5(a) indicate that no CO2 was produced when only Au was used as the anode, we sputtered Au onto the surface of the Pt anode to suppress the formation of CO2. The weight of deposited Au was proportional to the sputtering time, the slope of which was 2.3–2.8 times greater than that of Pt (Fig. 8(a)). The intensities of peaks indexed to the Au (111) and (220) planes increased with increasing Au sputtering time (Fig. 8(b)). EDX elemental maps of the Pt–Au particles showed that the distribution of Pt and Au almost completely overlapped (Fig. 8(c)), indicating that the Au particles were deposited onto the surface of the Pt particles. The effect of the Au addition on the CO2 formation was evaluated for the electrolysis of methane. The rate of CO2 formation decreased more than the rate of methanol formation as a result of increasing the Au loading (Fig. 8(d)). Consequently, the ratio between the methanol formation rate and the CO2 formation rate was maximal (4.9) when the Au sputtering time was 1.5 min, corresponding to an Au loading of 0.02 mg cm−2 (calculated from the calibration data shown in Fig. 8(a)) for a Pt loading of 0.12 mg cm−2.
 | ||
| Fig. 8 (a) Comparison of the Au and Pt sputtering profiles. (b) XRD patterns for the electrolyte before and after sputtering Pt and then Au. (c) Dark-field TEM image and EDX elemental maps of Pt (red), Au (green), and Pt–Au combined. (d) Electrolysis characteristics of the Pt–Au anodes prepared over different Au sputtering times (cell voltage: +3 V). | ||
Two-stage CO2RR toward methanol
To determine the critical thickness, we prepared electrolyte membranes with thicknesses ranging from 150 to 250 μm. The surface in the low-magnification SEM image (left) appears smooth, whereas the surface in the high-magnification image (right) appears to contain sub-micrometer pores (Fig. S8†). Whereas the ohmic resistance of the membrane decreased with decreasing thickness, the He permeation rate increased to 14 mL min−1 at a thickness of 150 μm (Fig. S9†). This permeation rate is equal to 93% of the flow rate (15 mL min−1) of the inlet gas. Note that CO2 (reactant gas), methane, and H2 (cathode products) permeated through the electrolyte membrane at sufficiently high rates. Subsequent experiments were conducted using 250 and 150 μm-thick electrolyte membranes for bypass- and penetration-mode electrolysis, respectively (hereafter denoted as cells A and B, respectively (Fig. 9(a))). | ||
| Fig. 9 (a) Schematic of cells A and B. (b) I–V curves and (c) product formation rates for cells A and B at 200 °C. Changes in (d) methanol formation rate and current density and (e) the faradaic efficiency with time during electrolysis in cells A and B at a cell voltage of 2.5 V. | ||
Fig. 9(b) shows I–V curves for cells A and B at 200 °C. A difference was observed in the current density between the two cells, particularly at voltages greater than 2 V, because of the difference in mass transfer at the electrode. All of the permeated gases contacted the anode catalyst in cell B; however, some of the bypassed gases passed through without contacting the anode catalyst in cell A. Accordingly, sufficient gas diffusion occurred at the electrode in the case of penetration mode but not in the case of bypass mode. Also, compared with the bypassed H2O, the permeated H2O might have a greater humidification effect on the electrolyte membrane, decreasing the ohmic or polarization resistance of cell B.
Notably, we synthesized methanol in both cells A and B by applying voltages greater than 1 V (Fig. 9(c)), which indicates that the electrolysis of H2O and CO2 into methanol occurred. In addition, the formation of methane as a by-product indicates that the electrolysis proceeded in two stages. The product profiles of the electrolysis were also characterized by the absence of volcano-shaped dependence of the methanol formation rate on the cell voltage, which is consistent with the observation that the rates of methane and methanol formation at the cathode (Fig. 1(b)) and anode (Fig. 5(b)), respectively, increased with increasing cell voltage.
The continuous operating characteristics of the electrolysis were monitored at a cell voltage of 2.5 V, where the highest FE for methanol formation was expected on the basis of the results obtained for methane-to-methanol oxidation at the anode (Fig. 5(b)). The current density decreased gradually with time in both cells A and B (Fig. 9(d)). Nevertheless, increases in the methanol formation rate were observed during the initial 60 and 180 min in cells A and B, respectively. As a result, the FEs for methanol formation increased to 24% in cell A and to 54% in cell B after 300 min (Fig. 9(e)). Given that the rate of methane formation was almost unchanged during the electrolysis (Fig. S10†), the change in FE with time is attributable to the transition behavior of the anode reaction rather than to that of the cathode reaction because the complete oxidation of methanol to CO2 (yellow route in Fig. 7(a)) can be suppressed by the deactivation of the Pt–Au anode during the electrolysis. More importantly, this deactivation can result in the formation of H2O2 as an additional active oxygen species at the anode where H2 and O2 coexist. H2O2 formed in situ can reportedly be used as an oxidant for nonelectrochemical57,58 or electrochemical methane-to-methanol synthesis.59,60 This effect is expected to be more pronounced for cell B than for cell A because of the difference in contact time of the gases with the anode catalyst between the two cells, as previously described. Alternatively, a pressurization effect might have occurred on the cathode chamber of cell B, causing fluctuations in the current in the cell (Fig. 9(d)); however, this effect cannot explain the high FE of cell B.
The aforementioned experimental results indicate that the product is a mixture; thus, methanol should be separated using the difference in melting points of the mixture components. For future studies aimed at improving electrolysis performance, increasing the temperature will be beneficial because of its positive effect on the formation rates of methane at the cathode and methanol at the anode. Because the melting point of PTFE contained in the electrolyte membrane is 225 °C, using a binder that is more heat resistant than PTFE would enable higher electrolysis temperatures.
Conclusions
To establish a highly efficient method for synthesizing methanol from H2O and CO2, two-stage electrolysis processes were proposed and applied to the CO2RR. In the electrolysis at the cathode, Ru/C promoted methanation more effectively when used as an electrode rather than as a catalyst. Impregnation with Fe2O3 imparted the electrode with reducing activity, thereby enhancing the production of methane. In the electrolysis at the anode, Pt showed high activity for the oxidation of methane-to-methanol, ethane, and CO2. Sputtering of Au onto the Pt surface selectively suppressed the formation of CO2 while maintaining the methanol formation. The two electrolysis processes were combined by feeding methane produced at the cathode into the anode chamber. Methanol was continuously synthesized and the faradaic efficiency increased over time. This effect was greater for the penetration mode than for the bypass mode. These results were achieved as a result of the formation of a more controllable intermediate in the electrolysis process compared with that formed in conventional electrolysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by a Kakenhi Grant-in-Aid (No. 21H03661) from the Japan Society for the Promotion of Science (JSPS), and by a grant (No. JPMJCR18R2) from the CREST Program of the Japan Science and Technology Agency (JST).Notes and references
- G. He, D. S. Mallapragada, A. Bose, C. F. Heuberger-Austin and E. Genger, Energy Environ. Sci., 2021, 14, 4635–4646 RSC.
- M. Bowker, ChemCatChem, 2019, 11, 4238–4246 CrossRef CAS PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- F. Jiao and B. Xu, Adv. Mater., 2019, 31, 1805173 CrossRef PubMed.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- D. Yang, Q. Zhu, C. Chen, H. Liu, Z. Liu, Z. Zhao, X. Zhang, S. Liu and B. Han, Nat. Commun., 2019, 10, 677 CrossRef CAS PubMed.
- S. Mou, T. Wu, J. Xie, Y. Zhang, L. Ji, H. Huang, T. Wang, Y. Luo, X. Xiong, B. Tang and X. Sun, Adv. Mater., 2019, 31, e1903499 CrossRef PubMed.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- A. Liu, M. Gao, X. Ren, F. Meng, Y. Yang, L. Gao, Q. Yang and T. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- R. Zhao, P. Ding, P. Wei, L. Zhang, Q. Liu, Y. Luo, T. Li, S. Lu, X. Shi, S. Gao, A. M. Asiri, Z. Wang and X. Sun, Adv. Funct. Mater., 2021, 31, 2009449 CrossRef CAS.
- C. Du, X. Wang, W. Chen, S. Feng, J. Wen and Y. A. Wu, Mater. Today Adv., 2020, 6, 100071 CrossRef.
- J. Wang, S. Ning, M. Luo, D. Xiang, W. Chen, X. Kang, Z. Jiang and S. Chen, Appl. Catal., B, 2021, 288, 119979 CrossRef CAS.
- S. Ning, J. Wang, D. Xiang, S. Huang, W. Chen, S. Chen and X. Kang, J. Catal., 2021, 399, 67–74 CrossRef CAS.
- W. Li, Z. Zhang, W. Liu, Q. Gan, M. Liu, S. Huo and W. Chen, J. Colloid Interf. Sci., 2022, 608, 2791–2800 CrossRef CAS PubMed.
- P. R. Yaashikaa, P. S. Kumar, S. J. Varjani and A. Saravanan, J. CO2 Util., 2019, 33, 131–147 CrossRef CAS.
- J. Feng, J. Ni and H. Pan, J. Mater. Chem. A, 2021, 9, 10546–10561 RSC.
- P. K. Jiwanti, S. Sultana, W. P. Wicaksono and Y. Einaga, J. Electroanal. Chem., 2021, 887, 161449 Search PubMed.
- Y. Liu, F. Li, X. Zhang and X. Ji, Curr. Opin. Green Sustainable Chem., 2020, 23, 10–17 CrossRef.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- J. Hazarika and M. S. Manna, Electrochim. Acta, 2019, 328, 135053 CrossRef CAS.
- C. Azenha, C. Mateos-Pedrero, M. Alvarez-Guerra, A. Irabien and A. Mendes, Electrochim. Acta, 2020, 363, 137207 CrossRef CAS.
- X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- S. Zhao, S. Guo, C. Zhu, J. Gao, H. Li, H. Huang, Y. Liu and Z. Kang, RSC Adv., 2017, 7, 1376–1381 RSC.
- J. Z. Huang, Q. Hu, X. R. Guo, Q. Zeng and L. S. Wang, Green Chem., 2018, 20, 2967–2972 RSC.
- A. Dutta, M. Rahaman, N. C. Luedi, M. Mohos and P. Broekmann, ACS Catal., 2016, 6, 3804–3814 CrossRef CAS.
- L. Lu, X. Sun, J. Ma, D. Yang, H. Wu, B. Zhang, J. Zhang and B. Han, Angew. Chem., Int. Ed., 2018, 43, 14149–14153 CrossRef PubMed.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong, B. Wu and N. Zheng, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS PubMed.
- L. Ji, L. Chang, Y. Zhang, S. Mou, T. Wang, Y. Luo, Z. Wang and X. Sun, ACS Catal., 2019, 9, 9721–9725 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castano and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS PubMed.
- Y. Cheng, X. Chu, M. Ling, N. Li, K. Wu, F. Wu, H. Li, G. Yuan and X. Wei, Catal. Sci. Technol., 2019, 9, 5668–5675 RSC.
- J. Kubota, T. Okumura and R. Hayashi, Sustainable Energy Fuels, 2022, 6, 1362–1372 RSC.
- B. Lee and T. Hibino, J. Catal., 2011, 279, 233–240 CrossRef CAS.
- C. Lamoureux, C. Moinet and A. Tallec, J. Appl. Electrochem., 1986, 16, 819–824 CrossRef CAS.
- T. Hibino, K. Kobayashi, M. Ito, Q. Ma, M. Nagao, M. Fukui and S. Teranishi, ACS Sustainable Chem. Eng., 2018, 6, 9360–9368 CrossRef CAS.
- T. Ressler, J. Synch. Rad., 1998, 5, 118–122 CrossRef CAS PubMed.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- L. M. Aeshala, R. G. Uppaluri and A. Verma, J. CO2 Util., 2013, 3, 49–55 CrossRef.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed.
- T. Abe, M. Tanizawa, K. Watanabe and A. Taguchi, Energy Environ. Sci., 2009, 2, 315–321 RSC.
- G. Garbarino, D. Bellotti, E. Finocchio, L. Magistri and G. Busca, Catal. Today, 2016, 277, 21–28 CrossRef CAS.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- Y. Zhang, X. Chen, W. Wang, L. Yin and J. C. Crittenden, Appl. Catal. B Environ., 2022, 310, 121346 CrossRef CAS.
- Y. Song, D. Johnson, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, J. Huang, F. Yang, F. Zhang, R. Qiao, A. P. Baddorf, T. J. Tschaplinski, N. L. Engle, M. C. Hatzell, Z. Wu, D. A. Cullen, H. M. Meyer, B. G. Sumpter and A. J. Rondinone, Sci. Adv., 2018, 4, e1700336 CrossRef PubMed.
- D. Méndez-Mateos, V. L. Barrio, J. M. Requies and J. F. Cambra, Catalysts, 2021, 11, 353 CrossRef.
- J. M. Rynkowski, T. Paryjczak, A. Lewicki, M. I. Szynkowska, T. P. Maniecki and W. K. Jozwiak, React. Kinet. Catal. Lett., 2000, 71, 55–64 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- N. V. Long, Y. Yang, C. M. Thi, N. V. Minh, Y. Cao and M. Nogami, Nano Energy, 2013, 2, 636–676 CrossRef.
- M. J. Boyd, A. A. Latimer, C. F. Dickens, A. C. Nielander, C. Hahn, J. K. Nørskov, D. C. Higgins and T. F. Jaramillo, ACS Catal., 2019, 9, 7578–7587 CrossRef CAS.
- Y. Xue, Q. Luan, D. Yang, X. Yao and K. Zhou, J. Phys. Chem. C, 2011, 115, 4433–4438 CrossRef CAS.
- L. Arnarson, P. S. Schmidt, M. Pandey, A. Bagger, K. S. Thygesen, I. E. L. Stephens and J. Rossmeisl, Phys. Chem. Chem. Phys., 2018, 20, 11152–11159 RSC.
- J. C. Fornaciari, D. Primc, K. Kawashima, B. R. Wygant, S. Verma, L. Spanu, C. B. Mullins, A. T. Bell and A. Z. Weber, ACS Energy Lett., 2020, 5, 2954–2963 CrossRef CAS.
- A. H. Bagherzadeh Mostaghimi, T. A. Al-Attas, M. G. Kibria and S. Siahrostami, J. Mater. Chem. A, 2020, 8, 15575–15590 RSC.
- Y. Kang, Z. Li, X. Lv, W. Song, Y. Wei, X. Zhang, J. Liu and Z. Zhao, J. Catal., 2021, 393, 20–29 CrossRef CAS.
- B. Lee, H. Naito, M. Nagao and T. Hibino, Angew. Chem., Int. Ed., 2012, 51, 6961–6965 CrossRef CAS PubMed.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F.-S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- A. Tomita, J. Nakajima and T. Hibino, Angew. Chem., Int. Ed., 2008, 47, 1462–1464 CrossRef CAS PubMed.
- R. F. B. de Souza, D. Z. Florio, E. Antolini and A. O. Neto, Catalysts, 2022, 12, 217 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See: https://doi.org/10.1039/d2ta04011e |
| This journal is © The Royal Society of Chemistry 2022 |