
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface stability of ionic-liquid-passivated mixed-cation perovskite probed with in situ photoelectron spectroscopy†
Suresh
Maniyarasu
 a,
Ben F.
Spencer
bc,
Hongbo
Mo
b,
Alex S.
Walton
d,
Andrew G.
Thomas
*bc and
Wendy R.
Flavell
*ac
a,
Ben F.
Spencer
bc,
Hongbo
Mo
b,
Alex S.
Walton
d,
Andrew G.
Thomas
*bc and
Wendy R.
Flavell
*ac
aPhoton Science Institute, Department of Physics and Astronomy, School of Natural Sciences, The University of Manchester, M13 9PL, UK. E-mail: wendy.flavell@manchester.ac.uk; andrew.g.thomas@manchester.ac.uk
bPhoton Science Institute, Department of Materials, School of Natural Sciences, The University of Manchester, M13 9PL, UK
cHenry Royce Institute, The University of Manchester, M13 9PL, UK
dPhoton Science Institute, Department of Chemistry, School of Natural Sciences, The University of Manchester, M13 9PL, UK
First published on 23rd August 2022
Abstract
In recent times, mixed-cation metal halide perovskites have shown promising photovoltaic performance, and the long-term stability of these metal halide perovskites has also been considerably improved by incorporating additives into the perovskite precursor. Here, the role of ionic liquid additives in improving the stability of perovskite is investigated by in situ surface sensitive studies. A small amount (0.3 mol%) of 1-octyl-3-methylimidazolium chloride ionic liquid (IL) is incorporated into FA0.9Cs0.1PbI3 (FACs) (where FA represents the formamidinium cation, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) (NH2)2+). The thermal- and moisture-induced decomposition of FACs and IL-FACs is investigated using near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS). A comparative study of the pristine and IL-incorporated FACs compositions shows that the IL additive prevents the out-diffusion of organic ions (FA+) from the lattice for temperatures up to 100 °C under 9 mbar water vapour and up to 150 °C under UHV conditions. Both compositions exhibit better stability under 9 mbar water vapour (equivalent to ∼30% relative humidity) compared with conventional methylammonium lead iodide (MAPbI3). The champion device fabricated with IL additive exhibits an improved power conversion efficiency (PCE) of 16% compared with the 13% PCE of the pristine FACs sample. Overall, the results suggest that the IL additive acts to improve the device performance as well as the stability of perovskites under thermal annealing in dry environments, but that a careful choice of IL will be necessary for full passivation in wet environments.
(NH2)2+). The thermal- and moisture-induced decomposition of FACs and IL-FACs is investigated using near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS). A comparative study of the pristine and IL-incorporated FACs compositions shows that the IL additive prevents the out-diffusion of organic ions (FA+) from the lattice for temperatures up to 100 °C under 9 mbar water vapour and up to 150 °C under UHV conditions. Both compositions exhibit better stability under 9 mbar water vapour (equivalent to ∼30% relative humidity) compared with conventional methylammonium lead iodide (MAPbI3). The champion device fabricated with IL additive exhibits an improved power conversion efficiency (PCE) of 16% compared with the 13% PCE of the pristine FACs sample. Overall, the results suggest that the IL additive acts to improve the device performance as well as the stability of perovskites under thermal annealing in dry environments, but that a careful choice of IL will be necessary for full passivation in wet environments.
1. Introduction
In recent times, mixed-cation hybrid perovskites have emerged as the leading energy-harvesting material among photovoltaic researchers due to their superior optoelectronic properties. The power conversion efficiency (PCE) of perovskite solar cells (PSCs) is now 25.7% (certified 25.2%), which has almost reached the PCE of commercialized silicon photovoltaic technology.1,2 However, both extrinsic (temperature, moisture, and oxygen) and intrinsic (defects) factors limit the long-term stability of perovskite material and hinder the large-scale commercialization of PSCs.3,4 Most of the high-efficiency reported PSCs are processed by a low-temperature and low-cost solution method, which often results in polycrystalline thin films with numerous grain boundaries and defects on the surface.5 The photovoltaic performance and stability of PSCs are highly dependent on the morphology of the perovskite film, and defects facilitate the decomposition of perovskite upon contact with environmental conditions or through ion migration under device operational conditions.6–8 Above a certain humidity level, reactions with moisture/oxygen cause the perovskite phase to turn into a hydrated intermediate phase. In addition, thermal stress also leads to the organic components in the lattice decomposing into volatile by-products.9,10 Hence, the development of improved quality of perovskite thin films is paramount in order to achieve better operational stability and device performance.Several approaches have been developed to control the perovskite crystal growth and morphology, in which additives incorporated into the precursor solution play an important role as dopants, crystallization agents, or in passivating defect sites.11 Alkali cations introduced from caesium iodide, rubidium iodide or potassium iodide have been used as additives to the perovskite precursor, induce uniform crystal growth and grain boundary passivation, resulting in improved device performance and retarded thermal decomposition.12,13 In recent years, ionic liquids (ILs) have been intensively investigated as promising additives in PSCs due to their attractive and unique features such as low vapour pressure, high ionic conductivity, and high thermal stability.14,15 Introduction of these liquid-crystalline compounds into the perovskite precursor can effectively tune the crystallization kinetics of the perovskite layer, and passivate the defects at grain boundaries and surfaces, which results in improved device performance and stability against device operational conditions.16,17 Shahiduzzaman et al. incorporated 1 wt% of 1-hexyl-3-methylimidazolium chloride in the methylammonium lead iodide (MAPbI3) precursor in N,N-dimethylformamide (DMF) to form a uniform MAPbI3 thin film by a spin-coating method. The presence of IL in the perovskite precursor slowed down the evaporation of the solvent (DMF) during annealing and facilitated the formation of homogeneous nucleation sites, which then grew together to form uniform spherical MAPbI3 nanoparticles.18 Seo et al. added 5 mol% of methylammonium formate (MaF) to the MAPbI3 precursor and improved the PCE to 19.5%.19 The improvement in efficiency was ascribed to the larger grain size, which resulted in efficient charge collection and thus better photovoltaic performance. The authors proposed a mechanism for crystal growth. HCOO− in MaF initially coordinates with Pb2+ in the precursor, then iodide (I−) gradually replaces HCOO− during sample annealing and produces a homogeneous and highly oriented crystal with no impurity phase.19 Bai et al., reported the use of 1 butyl 3 methylimidazolium tetrafluoroborate IL in the triple mixed cation perovskite composition (FA0.83MA0.17)0.95Cs0.05Pb(I0.9Br0.1)3. PSCs fabricated with 0.3 mol% of IL in this triple cation composition exhibited a PCE of 19.8%, which remained stable under full-spectrum sunlight at 70–75 °C for more than 1800 hours.20
Although there have been several investigations of the use of ILs to tune the perovskite crystal growth, there have been very few investigations of the surface stability. R. Xia et al. used ex situ X-ray photoelectron spectroscopy (XPS) to investigate the thermal decomposition of 1-(4-ethenylbenzyl)-3-(3,3,4,4,5,5,6,6,7,7,8,8,8) tridecafluorooctylimidazolium iodide- (ETI-) incorporated MAPbI3 thin films after heating at 60 °C in a N2 atmosphere. They demonstrated that the MAPbI3 sample without ETI content decomposed forming PbI2 and metallic Pb0, whereas, in samples containing 1% and 4% of ETI the formation of metallic Pb was suppressed. The authors proposed that the ETI additive was segregated to the interfacial-grain boundaries, inhibiting the out-diffusion of MA+ cation from the lattice.21 However, to the best of our knowledge there have been no in situ investigations of the separate effects of thermal annealing and exposure to atmospheric gases on the surfaces, and so a detailed understanding of the thermal decomposition of these IL-assisted perovskite materials requires further investigation.
In this work, we prepared a methylammonium- (MA-) free mixed-cation composition FA0.9Cs0.1PbI3 (denoted as FACs hereafter) and incorporated 0.3 mol% of 1-octyl-3-methylimidazolium chloride (OMIM Cl) into the FACs (denoted as IL-FACs) to fabricate high-quality perovskite thin films with large crystal domains. These samples were entirely processed under environmental conditions at room temperature with relative humidity (RH) of around 30–50%. The thermal and moisture-induced thermal decomposition of these samples was studied using XPS and near-ambient pressure XPS (NAP-XPS) at temperatures up to 150 °C. NAP-XPS was used to investigate moisture-induced degradation at room temperature (RT) and thermal decomposition under humid conditions, recording spectra in situ in the presence of 9 mbar of water vapour pressure (which is approximately equivalent to 30% RH). Our XPS results suggest that the incorporation of IL hinders the decomposition of organic cation from the lattice, even when not every FACs unit at the surface is protected by an IL cation. The incorporation of OMIM Cl into FACs was also found to improve the crystallinity, which resulted in a significant improvement of PCE from 13% to 16%. However, the IL-FACs bonding is disrupted on annealing in water and the organic cation in the lattice is decomposed into gaseous by-products. The combination of XPS and NAP-XPS investigations provides insights into designing a stable perovskite composition.
2. Experimental section
2.1 Materials
Indium tin oxide (ITO) substrates (Ossila), pre-patterned ITO substrates for OPV testing (Ossila S211), titanium diisopropoxide bis(acetylacetonate) (Sigma-Aldrich), 1-butanol (Sigma-Aldrich), formamidine acetate salt (Sigma-Aldrich), hydroiodic acid (57 wt% in H2O from Sigma-Aldrich), lead iodide (Alfa Aesar, 99.9985% metal basis), caesium iodide (Sigma-Aldrich), 1-octyl-3-methylimidazolium chloride (Sigma-Aldrich, ≥97%), N,N-dimethylformamide (DMF, Sigma-Aldrich), dimethyl sulfoxide (DMSO, Sigma-Aldrich), ethyl acetate (Sigma-Aldrich), Spiro-OMeTAD (Sigma-Aldrich, 99%), chlorobenzene (Sigma-Aldrich, 99.8%), 4-tert-butylpyridine (Sigma-Aldrich, 98%), bis(trifluoromethane) sulfonimide lithium salt (Li-TFSI, Sigma-Aldrich).2.2 Preparation of perovskite precursor solution
The FA0.9Cs0.1PbI3 precursor was prepared by dissolving 1.2 M of PbI2 in a mixed solvent of DMF and DMSO (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio). The solution was stirred on a hot plate at 70 °C. Once obtained, the saturated PbI2 solution, FAI (1.08 M) and CsI (0.12 M) were added together at room temperature and stirred until the FAI and CsI dissolved completely. The ionic-liquid-incorporated perovskite precursor was prepared by adding 0.05 M of ILs (1-octyl-3-methylimidazolium chloride) into the FA0.9Cs0.1PbI3 precursor solution and stirring at room temperature for 12 hours. This nominal concentration of IL passivates the FACs with ca. 0.05 OMIM Cl molecules per FACs unit. All precursors were filtered using a 0.25 μm PTFE filter prior to deposition.
:1 volume ratio). The solution was stirred on a hot plate at 70 °C. Once obtained, the saturated PbI2 solution, FAI (1.08 M) and CsI (0.12 M) were added together at room temperature and stirred until the FAI and CsI dissolved completely. The ionic-liquid-incorporated perovskite precursor was prepared by adding 0.05 M of ILs (1-octyl-3-methylimidazolium chloride) into the FA0.9Cs0.1PbI3 precursor solution and stirring at room temperature for 12 hours. This nominal concentration of IL passivates the FACs with ca. 0.05 OMIM Cl molecules per FACs unit. All precursors were filtered using a 0.25 μm PTFE filter prior to deposition.
2.3 Deposition of perovskite samples
The preparation of formamidinium iodide (FAI) powder and TiO2 electrodes followed the same procedure as reported in our previous work.13 The perovskite films were processed using a one-step solution processing method (Fig. 1) under ambient conditions at 35–40% relative humidity. 100 μl of perovskite solution was dropped onto a pre-heated TiO2 substrate held at 70 °C and spin-coated using a two-step program of 1000 rpm for 10 s followed by 4000 rpm for 30 s. 200 μl of antisolvent (ethyl acetate) was dripped onto the substrate during the second program at 10 s prior to completion. The substrates were then immediately transferred to a pre-heated hot plate and annealed at 100 °C for 10 minutes. The final thickness of the prepared sample was estimated to be ∼350 nm, as measured using a Veeco Dektak 8 Surface profilometer. All samples were placed in an air-tight container and stored in a vacuum desiccator prior to and between measurements.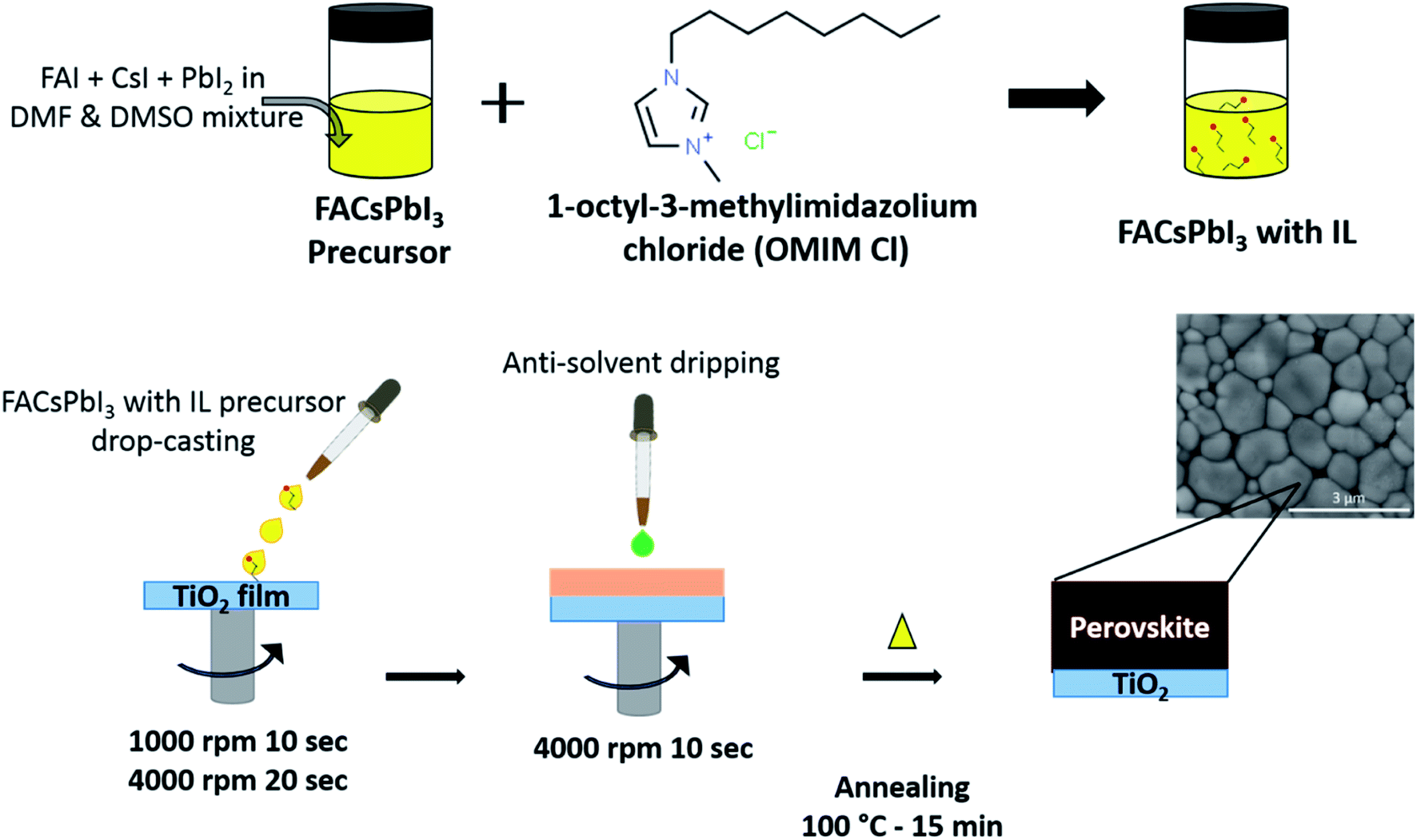 | ||
| Fig. 1 Schematic representation of perovskite precursor preparation and the thin film fabrication procedure. The inset image shows SEM images of an IL-incorporated FACs thin film. | ||
2.4 Device preparation (planar architecture)
ITO-patterned substrates were ultra-sonicated with Hellmanex detergent solution, DI water, acetone, and ethanol for 10 minutes each. The cleaned substrates were blown dry with air and treated with a UV ozone cleaner for 15 minutes. The compact-layer TiO2 and perovskite layers were deposited as detailed above. The hole transport layer (Spiro-OMeTAD) solution was prepared by dissolving 70 mg of Spiro-OMeTAD in 1 ml of chlorobenzene. To further improve the conductivity, 35 μl of 4-tert-butylpyridine and 20 μl of bis(trifluoromethane)sulfonimide lithium salt (520 mg dissolved in acetonitrile) were added to the prepared Spiro-OMeTAD solution. 80 μl of this solution was spin coated at 4000 rpm for 20 s on top of the perovskite thin films. Finally, 80 nm of gold electrode was deposited using a thermal evaporator.2.5 Characterization
The crystalline properties of both thin films was measured using Cu Kα X-ray diffractometer (D8 Advance, Bruker). The films were measured in the 2θ range of 7–42° at a scan rate of 4° per minute. The surface morphology of both films was measured using a scanning electron microscope (Quanta 250). Light absorption and photoluminescence measurements were carried out using an ultraviolet-visible-near-infrared spectrometer (Lambda 1050, PerkinElmer) and a FLS980 spectrometer (Edinburgh Instruments), respectively. The measured wavelength range in the light absorption and PL measurements was 550–850 nm and the excitation wavelength in the PL measurements was 450 nm. The UV and PL spectra were recorded from samples deposited on cleaned soda-lime glass substrates to avoid the IR absorbance of ITO. The photovoltaic properties (J–V characteristics) were measured at RT (25 °C) under illumination from a class AAA solar simulator (Oriel) equipped with AM 1.5G spectral distribution (450 W xenon lamp). The PSCs were measured using a solar cell I–V test system (Ossila, Manual-S211) to define the active area of 0.24 cm2. The J–V parameters of the devices were measured using Keithley source meter.2.6 XPS
Thermal degradation of both compositions in the absence of water vapour was measured using an ESCA 2SR X-ray photoelectron spectrometer (ScientaOmicron GmBH) under ultra-high vacuum (UHV, <10−8 mbar) conditions. The instrument consists of a monochromatic Al Kα (1.49 keV, 20 mA emission at 300 W) source and Argus CU hemispherical electron energy analyzer. To minimise air exposure, samples were loaded immediately from the vacuum desiccator into the load lock chamber and transferred to the analysis chamber for core level measurements. The sample plate was heated using a resistive heating filament attached to the sample base plate. The spectral acquisition was started once the set temperature was stabilized and this was maintained throughout the experiment. The survey spectra and high-resolution core level spectra were measured at pass energies of 80 eV and 20 eV, respectively. Spectra were aligned on the binding energy scale relative to the C 1s peak (285.3 eV).22 In addition, a check was made by aligning to the 4f peak of metallic Pb (137 eV);23 both methods generated consistent binding energies. All the spectra were processed by subtracting the background using Shirley (I 3d, Pb 4f and N 1s) or linear (C 1s and Cs 3d) background models and the spectra were deconvoluted using Gaussian/Lorentzian [GL(30), 70% Gaussian and 30% Lorentzian] line shapes in CasaXPS software. The peak intensities were corrected using their corresponding relative sensitivity factors (RSF) from the Scofield library in order to calculate the surface stoichiometries.242.7 NAP-XPS
The NAP-XPS instrument specifications (Specs-group) and the measurement details have been reported in our previous publications.13 The moisture-induced degradation was carried out in the NAP gas cell using a pressure of 9 mbar water vapour. The samples were measured before exposure to water, during exposure, and after pumping out the water vapour to attain UHV conditions (after exposure). A similar procedure was followed for the thermal stress measurements. Once the pressure reached 9 mbar, the temperature was raised to 100 °C and this temperature was maintained throughout spectral acquisition. The temperature was then raised to 150 °C and a further data set was acquired. After-exposure measurements were conducted at room temperature under UHV conditions.2.8 Control of degradation by X-ray irradiation
For each sample condition (in XPS and in NAP-XPS), the samples were exposed to X-ray irradiation for around 5 hours. Sample degradation leading to the formation of metallic lead may occur through X-ray irradiation, for example using intense synchrotron radiation.23 In previous work, we have assessed the extent of X-ray-induced degradation of MAPI-based perovskites.13,25 This indicates that, using lab-based sources such as those used here, beam-induced degradation is not significant over the timescale of our experiments, compared (for example) with the degradation induced by annealing. The sample position was moved by 0.5 mm to a fresh position (as the X-ray spot size is 300 μm) for each new condition.3. Results
3.1 Crystallography, optical, morphological properties and device performance
The preparation of FACsPbI3 and OMIM Cl-incorporated FACsPbI3 thin films is illustrated schematically in Fig. 1 and described in detail in the experimental section (Section 2). The influence of the OMIM Cl on FACsPbI3 crystal growth was investigated by X-ray diffraction (XRD). Fig. 2(a) shows the XRD patterns of the FACs and IL-FACs films. All observed reflections are attributable to the perovskite, with strong reflections at 13.9° and 28.1° corresponding to the (101) and (201) plane of α-FAPbI3 phase.26 Upon incorporating the IL into the perovskite precursor, the diffraction peaks become somewhat narrower, suggesting an enhanced crystallinity. There is an increase in the relative intensities of the {110} and (111) reflections, indicating changes in the texture of the film. Neither sample shows any reflection at 2θ = 12.7°, attributable to PbI2,26 indicating the complete conversion of perovskite from its precursor phase and an absence of degradation. The top-view surface morphology of both films was measured by scanning electron microscopy (SEM) and is shown in Fig. 2(b) and (c). The FACs perovskite thin film in Fig. 2(b) shows a smaller grain size with lots of pinholes, whereas the IL-incorporated FACs sample exhibits larger domains with a smooth and flat morphology as shown in Fig. 2(c). The improvement in the grain size is ascribed to the higher boiling point of IL than DMF and DMSO, which retards the crystal growth resulting in larger grain size with reduced grain boundaries compared with the pristine FACs film.27,28 The stability of perovskite thin films is highly dependent on a smooth morphology and large crystal size, because the degradation is believed to be initiated from the surface and grain boundaries.6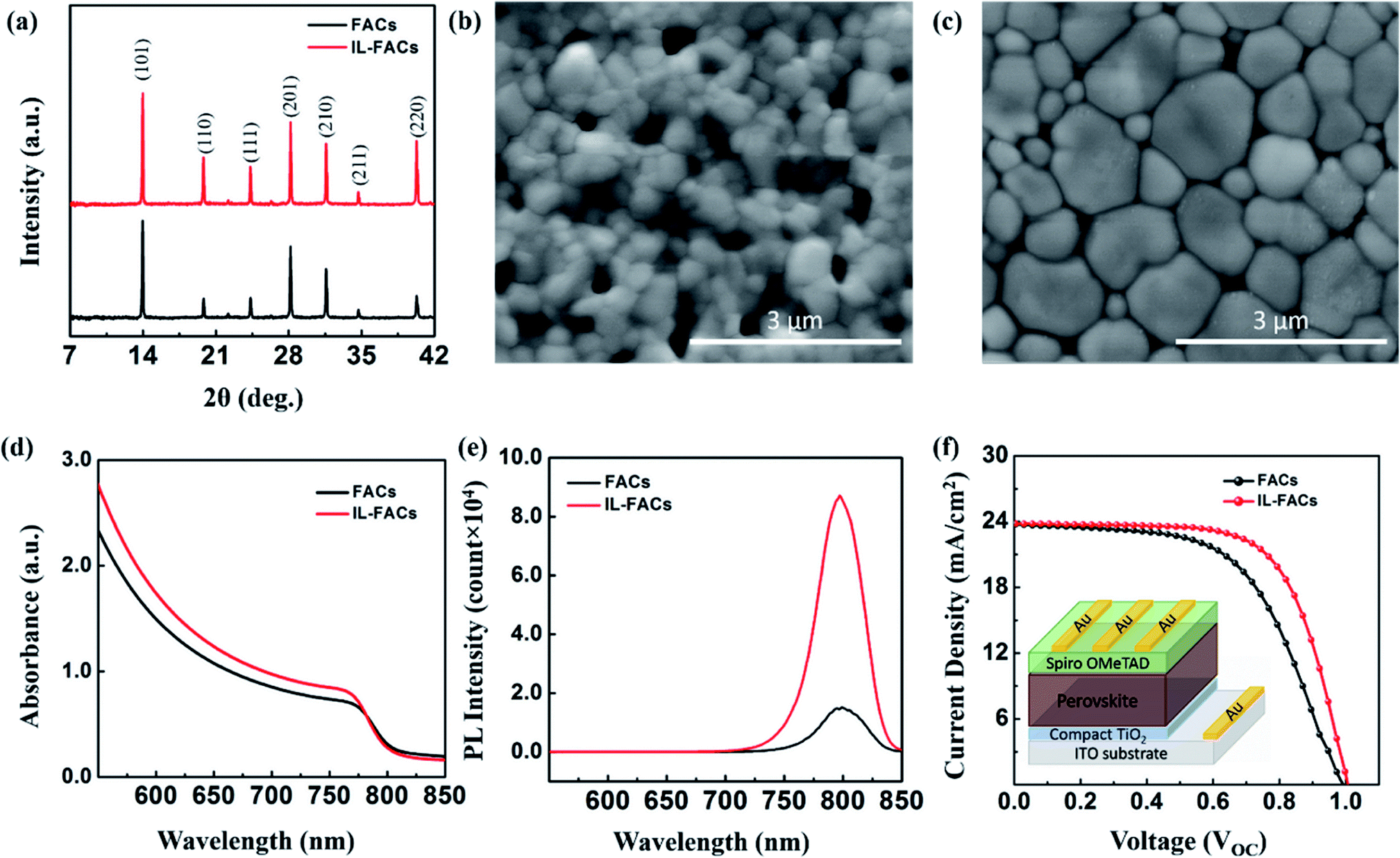 | ||
| Fig. 2 (a) XRD diffraction patterns of the FACs (black) and OMIM Cl-incorporated FACs (red, denoted as IL-FACs) thin films. (b, c) SEM images of FACs and IL-FACs thin films. (d, e) UV absorbance and photoluminescence spectra of FACs (black) and IL-FACs thin films (red). (f). Reverse scan J–V curves of the device prepared with FACs (black) and IL-FACs (red) thin films. The inset image in (f) shows a schematic figure of the planar device architecture. | ||
In order to investigate the effect of OMIM Cl on the optoelectronic properties, we acquired absorbance and steady-state photoluminescence (PL) spectra of perovskite thin film in the presence/absence of OMIM Cl. Fig. 2(d) shows absorption characteristics of FACs and IL-FACs thin films directly grown on a bare glass substrate. Both samples exhibit a narrow absorption peak at ∼803 nm, however, the IL-incorporated sample shows a higher absorbance across the entire wavelength region which can be related to the uniform crystal growth. Both compositions give a bandgap value of 1.54 ± 0.01 eV, which is consistent with the reported literature.29Fig. 2(e) shows the corresponding PL spectra showing that the IL-FACs sample shows a threefold increase in the PL intensity compared with the pristine FACs sample. The improved PL intensity in the IL-FACs sample suggests the reduction of trap/defect density sites in the perovskite thin film, which might otherwise lead to non-radiative recombination losses in the device.30 The influence of OMIM Cl on the photovoltaic performance was studied by fabricating planar devices. The current density–voltage (J–V) characteristics of FACsPbI3 and FACsPbI3/OMIM Cl devices are shown in Fig. 2(f). The champion device prepared with a FACsPbI3/OMIM Cl film exhibits a short-circuit current density (JSC) of 23.8 ± 1.0 mA cm−2, open-circuit voltage (VOC) of 1.01 ± 0.05 V, fill factor (FF) of 66.7 ± 1.5%, yielding a PCE (ƞ) of 16.0 ± 1.0%, whereas the FACsPbI3 device configuration demonstrates a JSC of 23.8 ± 1.0 mA cm−2, VOC of 0.99 ± 0.05 V, FF of 57.2 ± 1.5%, yielding a PCE (ƞ) of 13.5 ± 1.0%. The enhancement of overall performance in the IL-based device is consistent with the suppression of non-radiative recombination losses (as evident from the PL results) and uniform crystal growth and film formation.28,31 The effect of hysteresis was studied and the graphs are shown in Fig. S1 in the ESI.† Both devices exhibit hysteresis, however, a significant improvement was observed in FACsPbI3/OMIM Cl-compared to FACsPbI3-based devices. The lower hysteresis in IL-based devices is attributed to the larger crystal domains having fewer grain boundaries, which reduces the charge carrier recombination losses during device operation.20 The IL-additive-based devices retained 80% of their original PCE after 1000 hours of storage under dark conditions at room temperature, compared to 50% for cells based on FACs only. We associate the drop with the intercalation of some water with the IL, a point we return to below.
3.2 Thermal decomposition of FACs and IL-FACs under UHV conditions
The thermal decomposition of the FACs and IL-FACs perovskite thin films was studied using XPS at two different temperatures (100 °C and 150 °C) under UHV conditions. Fig. 3(a)–(d) illustrates the high-resolution (Pb 4f, I 3d, N 1s and Cs 3d) core level spectra of the FACs and IL-FACs films obtained at RT, 100 °C, and 150 °C respectively. The XPS overview spectra of FACs and IL-FACs are shown in Fig. S2 in the ESI.† As shown in Fig. 3(a)(i), at room temperature, the Pb 4f core-level spectra of pristine FACs showed two characteristic peaks at 138.7 ± 0.1 eV (Pb 4f7/2) and 143.6 ± 0.1 eV (Pb 4f5/2) binding energy (BE), representing the chemical state of Pb2+ in the perovskite phase.13,23 In addition, there is a small additional doublet at lower binding energy (137 ± 0.1 eV and 141.9 ± 0.1 eV BE), which can be assigned to metallic Pb (herein denoted as Pb0).13 This is an indicator either of incomplete conversion to perovskite from the precursor solution32 or of sample degradation.33 In contrast the spectrum of the IL-incorporated sample (Fig. 3(a)(iv)) does not show any peak at 137 ± 0.1 eV BE, suggesting that the IL in the perovskite precursor induced complete conversion of perovskite and/or suppressed initial degradation, consistent with the XRD results. Relative to FACs, the position of the Pb 4f core doublet in pristine IL-FACs is shifted −0.1 eV towards lower binding energies, to 138.6 ± 0.1 eV and 143.5 ± 0.1 eV BE, an effect which has been noted previously,28,31 and which we discuss further below. As depicted in Fig. 3(a)(ii) and (iii), the Pb 4f core level of FACs shows a BE shift of +0.1 eV on heating in UHV to 100 °C and 150 °C, previously assigned to the decomposition of perovskite into the PbI2 phase (and supported by the chemical quantification discussed below).23 As reported in our previous work, PbI2 readily decomposes into Pb0 and I2 on thermal annealing (and under intense X-ray irradiation), which leads to a sharp increase in the amount of Pb0 detected at the surface (Fig. 3(a)(iii)).13,25 In contrast, the sample containing OMIM Cl showed no shift in the Pb 4f BE position on heating, and no peak due to Pb0, except for a very small feature in the sample annealed at 150 °C. This indicates the initiation of decomposition of the perovskite. | ||
| Fig. 3 High-resolution core level spectra of FACs13 and IL-FACs thin films measured at RT, and at two different temperatures (100 °C and 150 °C) under UHV conditions (a) Pb 4f, (b) I 3d, (c) N 1s, and (d) Cs 3d. Spectra labelled (i, iv), (ii, v), and (iii, vi) correspond to the FACs and IL-FACs samples measured at RT, 100 °C, and 150 °C, respectively. For each condition, all spectra are normalized to the intensity of the Pb 4f7/2 core level from Pb2+. | ||
The high-resolution I 3d core level spectra are used to investigate the X-site (halogen) component of the ABX3 lattice. Fig. 3(b) shows the I 3d doublet, which, in FACs, exhibits two characteristic peaks at 619.5 ± 0.1 eV and 631.0 ± 0.1 eV, corresponding to the I 3d5/2 and I 3d3/2 peaks respectively.13 Relative to FACs, the I 3d core level in IL-FACs also showed a shift to low binding energy (−0.2 eV), similar to the Pb 4f core level. Shifts to low binding energy on IL incorporation have been attributed to bonding interaction between the perovskite halide lattice and imidazolium in the IL structure; we return to this topic below.27,28,31 With increasing temperature, the I 3d core level in the FACs sample showed a BE shift of +0.1 eV (Fig. 3(b)(ii) and (iii)), consistent with the decomposition of perovskite into PbI2 during sample heating under UHV conditions.13,23 There was no significant change in the I 3d BE of IL-FACs on heating as shown in Fig. 3(b)(iv)–(vi).
The surface chemical composition of the samples was calculated by correcting the peak intensities using the RSFs; the evolution with heating is shown in Fig. 4 and Table S1 in the ESI.†
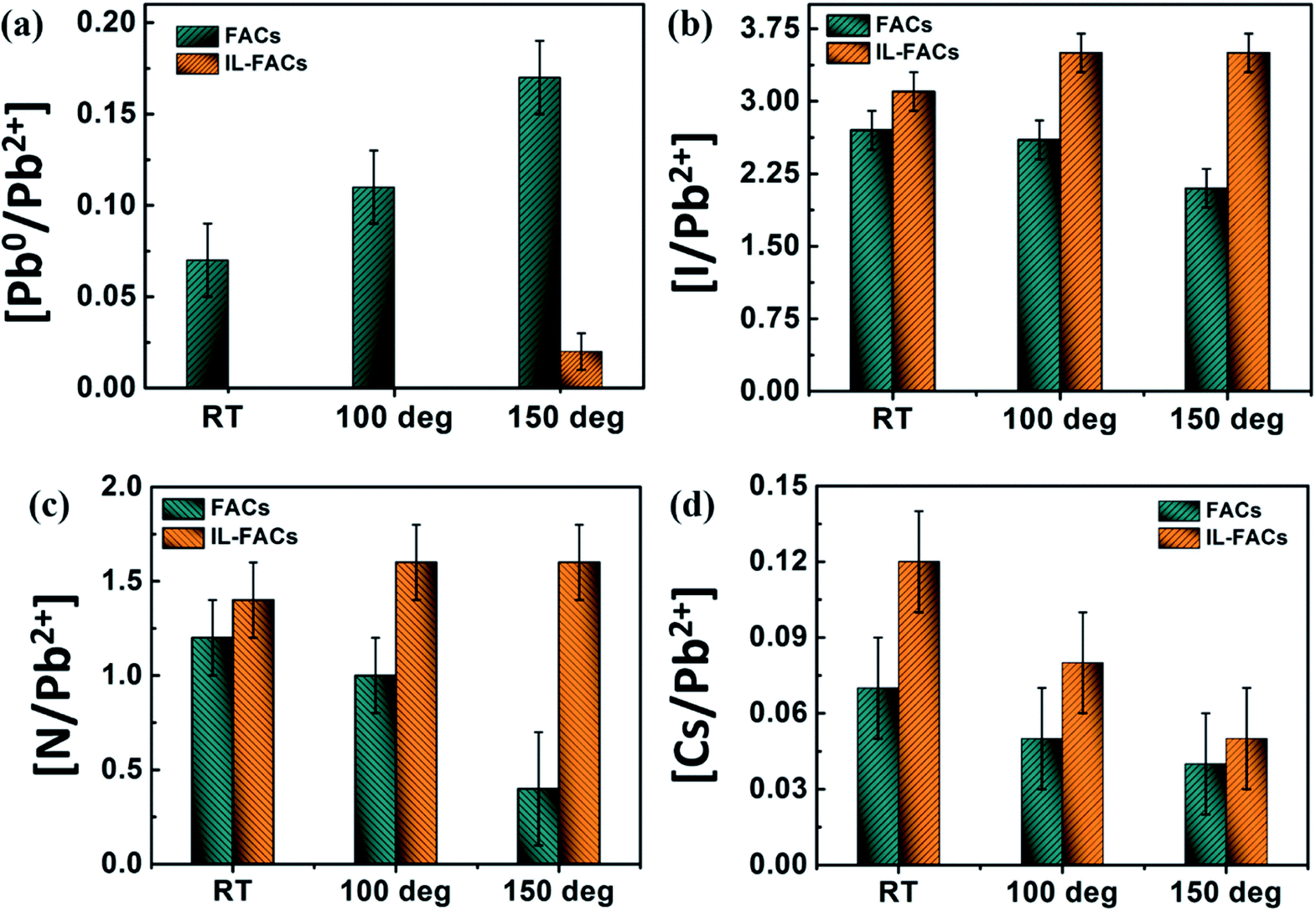 | ||
| Fig. 4 (a–d) Elemental composition changes of the FACs and IL-FACs samples determined from XPS as a function of sample heating (a) Pb0/Pb2+, (b) I/Pb2+, (c) N*/Pb2+, and (d) Cs/Pb2+ (* the N/Pb2+ ratio presented in (c) includes only the N signal from the FA cation of the perovskite). | ||
The decomposition of the organic components under thermal stress is analysed using the C 1s and N 1s core-level spectra. The C 1s spectra of FACs and IL-FACs are shown in Fig. S3 in the ESI.† The C 1s spectra of the FACs sample show two peaks at 285.3 ± 0.1 eV and 288.6 ± 0.1 eV BE, which correspond to the adventitious carbon at the surface of the perovskite and to the C atom in the formamidinium cation, respectively.34 Following heat treatment in UHV conditions, the intensity of both peaks gradually decreases, which implies the decomposition of organic cation into gaseous products as reported in our previous study.13 The C 1s spectra of the IL-FACs composition are also shown in Fig. S3.† As can be seen in the figure, additional broad structure is present at 286.0–288.0 eV BE, which is fitted using the established fit for OMIM Cl, with three components at slightly different BEs, corresponding to the three different environments of carbon atoms bound to N in the imidazolium ring.35 The signal from the hydrocarbon chain of the IL enhances the intensity at ca. 285 eV BE.35 Upon increasing the temperature to 100 °C and 150 °C, no significant changes are observed in the peak intensities or BE positions. The N 1s core-level spectra are shown in Fig. 3(c). The peak located at 400.9 ± 0.1 eV BE represents the N atom in the FA cation and in FACs, the intensity of the N signal is reduced on heating, indicating the release of nitrogen from the surface.13 The changes in C and N stoichiometry (Table S1†) show that within error, the percentage rates of reduction of the FA C 1s and N 1s signals are similar, reinforcing the conclusion that the organic cation is lost via decomposition into gaseous products that are not adsorbed on the surface; species including NH3, HCN and C3N3H3 have been proposed.13,36 The N 1s spectra of IL-FACs showed a −0.2 eV BE shift as compared to FACs, and an additional peak at higher binding energy (402.2 ± 0.1 eV) which can be assigned to the N atoms in the OMIM Cl ionic liquid.35 In this case, there is no change (within error) observed in the chemical ratios of C/Pb2+ and N/Pb2+ on annealing (Table S1† and Fig. 4(c)), implying that the incorporation of IL into the perovskite suppresses decomposition under thermal annealing.
The C 1s and N 1s photoelectrons have similar kinetic energies and thus probing depths, and so the data in Table S1† allow us to make a rough estimate of the proportions of perovskite (FA cation) and IL at the surface of the sample. We note that the OMIM cation, if incorporated intact, has two N atoms, five C atoms attached to N (labelled C–N(IL) in Table S1†) and a 7-atom hydrocarbon chain. The C–H stoichiometry in Table S1† includes both the atoms in this chain, and the adventitious carbon normally observed at the perovskite surface. The FA cation contains two N atoms. Using these facts, we can see (from comparison of the measured N(IL) and C–N(IL) signals) that the imidazolium moiety appears to be incorporated intact. Comparison of the N(FA), N(IL) and C–N(IL) stoichiometries suggests a FA:IL ratio at the surface of roughly 3:1. We also observe a large increase in the C–H signal when the IL is incorporated, from the hydrocarbon chain of the IL. Because this signal also includes adventitious carbon, any conclusions drawn must be tentative, but if we ascribe the ‘excess’ C–H signal over that observed in pristine FACs to the IL, then the measured C–H and C–N(IL) stoichiometries are roughly consistent with the nominal stoichiometry of the IL, again suggesting that the OMIM cation is incorporated intact.
The effect of temperature on the alkali-metal cation (Cs+) is investigated using the Cs 3d core-level. The Cs 3d core level spectra of FACs and IL-FACs are shown in Fig. 3(d). The doublet peaks at BEs of 725.1 ± 0.1 eV and 739.0 ± 0.1 eV can be assigned to the Cs 3d5/2 and Cs 3d3/2 peaks respectively.13 In both samples, the intensity of the Cs 3d5/2 peak and the Cs/Pb2+ stoichiometric ratio are gradually reduced with increasing temperature as seen in Fig. 3(d) and in Table S1 in the ESI.†
In general, the signal observed from Cl 2p was either small or absent. An absence of the IL anion in XPS of perovskite-IL composites has also been noted by other authors,20 and it has been suggested that the Cl− ion exchanges readily with the lattice I− at the surface.28 As our XPS shows that the OMIM cation is incorporated intact, we infer that the IL bonds to the surface via the interaction between the OMIM cation and the lattice I−, displacing Cl−. In general, we observe a small shift of the core levels to low binding energy when IL is incorporated into FACs. However, we do not necessarily regard this as a confirmation of bonding between the two components. Binding energy calibration in XPS of ILs is notoriously difficult, due to charging under X-ray irradiation.37 Shifts to both high and low BE are observed, dependent on anion, chain length and time under X-rays.37 Here, we choose to calibrate our BE scale for FACs so that the aliphatic hydrocarbon peak of FACs appears at 285.3 eV, consistent with previous literature.13,22 As we noted above, this generates BEs that are consistent with choosing the BE of Pb0 to be 137 eV BE.23 However, the accepted calibration of the aliphatic C 1s of OMIM Cl is C 1s = 285.0 eV BE,37 and indeed, it was necessary to use this calibration to satisfactorily fit the OMIM C 1s spectrum (for example in Fig. S3†). Relative to C 1s at 285.3 eV BE, I 3d, N 1s, Pb 4f and Cs 3d levels all show a small shift (0.1–0.2 eV) to low BE on IL incorporation, which we here attribute to BE calibration issues in the IL-incorporated samples rather than to a bonding shift, as we would not expect all core levels to shift in the latter scenario.
3.3 FACs and IL-FACs under 9 mbar water vapour pressure
The moisture stability of both compositions was studied by employing NAP-XPS, in which the environment of the analysis chamber may be adjusted to a specific pressure (up to a maximum of ca. 9 mbar water vapour pressure, corresponding to ∼30% RH). The NAP-XPS measurements of the FACs composition exposed to 9 mbar of water vapour have been reported in our previous study.13 The IL-FACs film was exposed to 9 mbar of water vapour during spectral acquisition for approximately 5 hours in the NAP-XPS measurement. A peak assigned to O 1s was observed in the XPS spectrum of pristine IL-FACs (Fig. S2†), which has been observed by other authors,31,38 and is likely to be associated with water incorporation into the IL.39,40 During NAP-XPS measurements, a peak at ∼536 eV BE associated with gas phase water vapour was observed.41 The O 1s core level signal showed an increase in intensity after the NAP-XPS measurements (Fig. S4 and Table S2†), suggesting further incorporation of water after exposure.The high-resolution core-level spectra (Pb 4f, I 3d, C 1s, N 1s and Cs 3d) of the IL-FACs measured at UHV conditions before exposure, during exposure, and after exposure to water vapour are shown in Fig. 5(a)–(e). The Pb 4f, I 3d, N 1s and Cs 3d core levels are located at 138.6 eV, 619.3 eV, 400.7 eV, and 725 ± 0.1 eV, respectively. These core levels remain in the same BE position during and after water exposure, indicating that the sample did not undergo significant degradation upon exposure to water vapour at room temperature. A similar result was found previously for FACs.13 The lack of change in these spectra also acts as a useful confirmation that there is insignificant beam damage under the conditions of our experiment; for example, in the Pb 4f spectrum, no additional doublet due to Pb0 is observed at lower binding energy after exposure.
 | ||
| Fig. 5 High-resolution core level spectra of IL-FACs measured under UHV conditions, during exposure and after exposure to 9 mbar water vapour at room temperature (a) Pb 4f, (b) I 3d, (c) C 1s, (d) N 1s, and (e) Cs 3d. Spectra labelled (i), (ii), and (iii) correspond to the sample measured before, during, and after exposure, respectively. For each condition, all spectra are normalized to the intensity of Pb 4f7/2 core level from Pb2+. | ||
As expected, the core level intensities are reduced during exposure (Fig. 5), which is attributed to the attenuation of the signals in the presence of moisture. The elemental ratios I/Pb2+, N/Pb2+, Cs/Pb2+, and Pb0/Pb2+ before and after exposure are calculated for both compositions and reported in Table S2 in the ESI.†13 Since the RSFs are not known for the analyser under NAP conditions, the chemical composition during water exposure is not calculated. The ratio of I/Pb2+ and N/Pb2+ for the pristine FACs sample was found to be 2.6 ± 0.2 and 1.2 ± 0.2, respectively, showing (within error) an insignificant decrease to 2.4 ± 0.2 and 1.0 ± 0.2, respectively after exposure.13 Similar values, which also show no significant change after exposure, are seen for the IL-incorporated sample (Table S2†).
3.4 Thermal decomposition of FACs and IL-FACs under 9 mbar water vapour pressure
In order to further understand the decomposition, we performed the moisture and thermal stress measurements simultaneously for both pristine and IL-FACs samples. Fig. 6 depicts the high-resolution core level Pb 4f, I 3d and N 1s spectra of FACs and IL-FACs samples measured under 9 mbar of water vapour pressure with increasing temperature. Both samples were measured initially at UHV conditions, then during heating with simultaneous water exposure and again at room temperature, after pumping to regain UHV conditions after the experiment. As seen in Fig. 6, the positions of the 4f Pb2+ and I 3d signals of both samples are shifted slightly to higher BE when the temperature is increased to 150 °C, suggesting the decomposition of perovskite into lead iodide (PbI2).13 This shift appears to be larger in the case of IL-FACs. The N 1s spectra of both samples show some loss of nitrogen at 100 °C in water vapour, and a further increase of temperature to 150 °C completely removed the FA cation from the lattice as no N signal from FA (at 400.9 eV BE) was observed. In the case of IL-FACs, the N 1s signal due to the N atoms in the IL (at 402.2 eV BE) remains. The release of nitrogen is also confirmed by the corresponding C 1s spectra which are shown in Fig. S5 in the ESI.† As can be seen from Fig. S5,† for both samples, the peak at 288.6 ± 0.1 eV due to C bound to N in the FA cation was attenuated with increasing temperature and completely removed at 150 °C. Fig. 6 shows that (as in Fig. 3) the pristine FACs sample showed the presence of some metallic Pb, and this increased somewhat on annealing in water. The IL-FACs sample showed no significant signal due to Pb(0) at room temperature and on annealing to 100 °C in water vapour, but it showed a strong metallic Pb feature (Fig. 6(b)(iii)) and complete quenching of the FA N 1s signal (Fig. 6(f)(iii)) on increasing the temperature to 150 °C. These results contrast with those obtained on UHV annealing, and suggest that the passivation effect provided by the IL is largely removed on annealing in the presence of water vapour. Indeed, the degradation via PbI2 to metallic Pb appears to be rather more severe in the case of IL-FACs. | ||
| Fig. 6 High-resolution core level NAP-XPS spectra of (a, b) Pb 4f, (c, d) I 3d, (e, f) N 1s obtained from FACs (a, c, e) and IL-FACs (b, d, f) measured at different conditions as noted. Spectra labelled (i), (ii), (iii) and (iv) correspond to the sample measured under UHV conditions, at 100 °C in 9 mbar water vapour, 150 °C in 9 mbar water vapour and at room temperature in UHV conditions (after exposure), respectively. For each condition, all spectra are normalized to the intensity of the Pb 4f7/2 core level from Pb2+. | ||
To provide a clear picture of degradation, the changes in the elemental ratios between I/Pb2+, N/Pb2+, Cs/Pb2+, and Pb0/Pb2+ are calculated for both compositions and reported in Table S3 in the ESI.† The I/Pb2+ ratio for both pristine samples was found to be 2.8 ± 0.2 which is close to nominal stoichiometry. After the thermal treatment, the I/Pb2+ ratios were reduced to 1.7 ± 0.2 and 2.0 ± 0.2 for FACs and IL-FACs samples, respectively. The corresponding N/Pb2+ ratios drop to 0.1 ± 0.2 and 0.2 ± 0.2 from 1.8 ± 0.2 and 1.4 ± 0.2, respectively. The alkali metal content is analysed using Cs 3d spectra and reported in Fig. S5 in the ESI.† The Cs/Pb2+ ratio is calculated and reported in Table S3 in the ESI.† The ratios of Cs/Pb2+ were calculated to be 0.08 ± 0.02 and 0.09 ± 0.02 for the FACs and IL-FACs samples respectively, and after the thermal treatment in the presence of water vapour, the corresponding ratios were 0.03 ± 0.02 and 0.12 ± 0.02, respectively. As we have previously noted,13 quantification of the low alkali-ion concentrations by XPS is subject to large errors, and a cautious interpretation is necessary. Nevertheless, it appears that the loss of Cs+ from the perovskite on heating in water is suppressed in IL-FACs compared to FACs.
The changes in surface O content of the samples during annealing in water are presented in Table S4 and Fig. S6 of the ESI.† At room temperature before exposure, very little O is detected at the surface of FACs (Table S4†). However, a significant peak is observed at 532.8 ± 0.1 eV BE for IL-FACs, consistent with surface-adsorbed water and hydroxide (Fig. S4 and S6†).42 After annealing to 150 °C in 9 mbar water vapour, in IL-FACs, this peak is reduced somewhat in intensity, and (for both samples) a second feature appears at 530.8 ± 0.1 eV BE, consistent with incorporation of oxygen into the lattice at the surface of the sample,42 in other words, a chemical oxidation of the sample surface. This contrasts with the results obtained after room temperature exposure to water (Fig. S4 and Table S2†), where an increase in the amount surface-adsorbed water is seen for IL-FACs.
4. Discussion
The long-term stability of metal halide perovskites has been recently improved by altering the perovskite composition or adding additives into the perovskite precursor which modulates the perovskite growth kinetics and improves the crystal size.43 The comparative in situ study of FACs and IL-FACs thin films under thermal stress emphasizes the importance of including additives into the lattice in order to improve the device performance and stability. In general, the degradation of perovskite is initiated by the migration of ions (predominantly the halide anion, here I−) through vacancies in the perovskite lattice.13,44 Consistent with our previous results, ambient-processed FACs shows a small amount of Pb0 and a lower than nominal value of I/Pb2+ (2.7 ± 0.2) (as shown in Fig. 3a(i) and 4(b)), which indicates some initial degradation or incomplete conversion of perovskite during sample preparation at ambient conditions.13 Under thermal annealing in vacuum at temperatures up to 150 °C, the FACs sample undergoes significant decomposition which is evidenced by the drop in the I/Pb2+ and N/Pb2+ ratios, leaving PbI2 on the surface (as shown in Fig. 4). This is further decomposed into Pb0 and I2 under thermal annealing and X-ray irradiation consistent with previously reported literature.13,45 The incorporation of a small amount of IL into the precursor facilitates the complete conversion to perovskite with an improvement in crystal size (Fig. 1(a) and (c)). Pristine IL-FACs shows an I/Pb2+ ratio that is close to nominal and an absence of metallic Pb0 as seen in Fig. 3(a) and 4(b). The IL-FACs composition appears to be stable on annealing; there is no significant drop in the I/Pb2+ and N/Pb2+ ratios and no significant rise in metallic Pb0 on thermal annealing up to 150 °C. On annealing, the organic cation in the FACs sample is lost from the surface and decomposes into gaseous by-products,36 and the proportion of adventitious carbon content also decreases as reported in Table S1 and Fig. S3 in the ESI.† In contrast, the IL sample shows no significant change in surface carbon content from perovskite or IL sources on annealing, and a slight rise in aliphatic carbon signal as seen in Fig. S3 and Table S1.†Overall, these results clearly demonstrate that a small amount of IL additive in the precursor (corresponding to 5 at% of IL per FACs unit) improves the stability to thermal annealing in the absence of water. The improvement in stability can be related to the presence of IL molecules at the surface or grain boundaries (where the experimentally-determined IL:FACs ratio is ca. 1:3), which reduces the out-diffusion of the organic component at elevated working temperatures.21 As we have previously noted, the quantification of the dilute alkali-metal cation by XPS is subject to large errors.13 However, it appears that alkali cations are lost from both FACs and IL-FACs on UHV annealing (but only from FACs on annealing in water). As Cs+ occupies the same site as the organic cation,46 it is lost concomitantly with it as the perovskite lattice degrades.13 Its loss from the surface of the IL-FACs composition on UHV annealing is therefore perhaps surprising, but we note that only about one third of FACs units are passivated by IL, and Cs is quite volatile on heating in UHV.47–49
In general, humidity is known to influence perovskite degradation, but under ambient conditions it is difficult to control precisely in order to deconvolute the separate effects of moisture and temperature. This led us to investigate the samples using NAP-XPS, in which the humidity can be controlled. The degradation of FACs and IL-FACs was first investigated under 9 mbar of water vapour (equivalent to 30% of RH) at room temperature. The results in Section 3.3 demonstrate that neither of the compositions studied here show a change in the metallic Pb content and no significant change in the I/Pb2+ and N/Pb2+ ratios after exposure to water. The core level signals of all the elements remain at the same binding energy positions after exposure, which demonstrates that there is no significant change in the chemical environments at the surface of the samples. However, the O 1s signal from IL-FACs is significantly larger than from FACs and increases after water exposure, suggesting incorporation of water into the IL. The results demonstrate that both the ambient processed mixed-cation and the IL-incorporated sample are more stable to water than the conventional MAPbI3 perovskite.25 The comparison between the results from thermal annealing and water exposure suggests that the decomposition of the perovskite is largely triggered by the migration of ions.
To thoroughly understand the stability of the IL-FACs composition, we further carried out the thermal annealing and moisture treatments simultaneously for both compositions. Section 3.4 shows that, in contrast to the results from annealing in UHV, on annealing in the presence of water, both FACs and IL-FACs undergo decomposition. The metallic Pb feature emerges at 137 eV and becomes significant with increasing temperature as shown in Fig. 6, which indicates that the sample is decomposed via the production of PbI2, consistent with the BE shift in the I 3d and Pb 4f spectra.13,23 and the changes in surface quantification. The organic cation is completely lost after annealing in water at 150 °C, evidenced by the loss of the N 1s and C 1s signals associated with FA. In contrast to the FACs sample, there is no metallic Pb observed up to 100 °C for IL-FACs, but overall, it appears that the presence of water vapour has largely removed the passivating effect of the IL that was evident on thermal annealing in UHV. A number of authors have studied the miscibility of OMIM-based ILs with water,50,51 and it is clear that even the most hydrophobic ILs show some miscibility.52 The presence of halide anions (as at the surface of the perovskite) increases this miscibility, and conformational changes in the IL cation are induced in the presence of water.53 We recall that XPS showed that the ratio of FA:IL at the surface is ca. 3:1, in other words the I:IL ratio is expected to be around 9:1 (assuming nominal stoichiometry for the perovskite). Thus there a significant amount of perovskite surface I− which is not coordinated to OMIM cations. In this scenario, we suggest that upon exposure to water, water molecules can readily become intercalated with the surface-adsorbed OMIM (as suggested by the O 1s signal in water exposure experiments, Table S3†). On annealing to 150 °C, oxygen becomes incorporated into the lattice at the surface of the sample, a process that we anticipate is facilitated when the water is trapped close to the surface as in the IL-FACs sample. The OMIM passivating layer is disrupted, allowing lattice decomposition (aided by ion migration) to proceed when the sample is annealed.13 The drop in the measured PCE of 20% over 1000 hours may therefore be attributed to this. Thus, although the IL provides good passivation of the perovskite surface to thermal annealing in dry environments, the choice of IL for full passivation in environments needs some care.
5. Conclusions
We have investigated the decomposition behaviour of ionic-liquid-incorporated mixed-cation perovskites under thermal annealing in vacuum and in the presence of water. It is shown that the addition of a small amount of IL to FACs improves the perovskite nucleation and promotes larger crystal domains, enhancing light-harvesting, device performance and stability. A solar cell based on IL-FACs exhibited a power conversion efficiency of 16%, which is higher than the equivalent device prepared with FACs (13%). A significant improvement of fill factor is found in the IL-FACs devices due to the larger crystal domains, which helps to suppress the charge carrier recombination.28,54 XPS results clearly demonstrate that a small amount of IL additive in the precursor (producing an FA:IL ratio at the surface of ca. 3:1) improves the stability to thermal annealing in the absence of water. The improvement in stability can be related to the binding of OMIM cations to the perovskite surface, which reduces the out-diffusion of the organic component on annealing. Both FACs and IL-FACs exhibit superior moisture stability under 9 mbar of water vapour at room temperature compared with the conventional MAPbI3 perovskite.25 However, the passivating effect of the IL is disrupted by the addition of water, due to the miscibility of the IL with water in the presence of the surface halide anions of the perovskite lattice, and, on annealing in water, oxidation of the surface occurs. Thus, although the IL provides good passivation of the perovskite surface to thermal annealing in dry environments, a careful choice of IL for full passivation in wet environments will be needed.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author S. M. acknowledges the University of Manchester for the award of an Overseas Research Scholar Awards and a Research Impact Scholarship. XPS and Near-Ambient Pressure XPS access was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1 and EP/P025021/1.References
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y. K. Kim, C. S. Moon, N. J. Jeon, J. P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- R. Wang, M. Mujahid, Y. Duan, Z. K. Wang, J. Xue and Y. Yang, Adv. Funct. Mater., 2019, 29, 1–25 Search PubMed.
- J. M. Ball and A. Petrozza, Nat. Energy, 2016, 1(11), 1–13 Search PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Q. Wang, B. Chen, Y. Liu, Y. Deng, Y. Bai, Q. Dong and J. Huang, Energy Environ. Sci., 2017, 10, 516–522 RSC.
- W. Zhou, J. Gu, Z. Yang, M. Wang and Q. Zhao, J. Phys. D: Appl. Phys., 2020, 54(6), 063001 CrossRef.
- S. Kim, S. Bae, S. W. Lee, K. Cho, K. D. Lee, H. Kim, S. Park, G. Kwon, S. W. Ahn, H. M. Lee, Y. Kang, H. S. Lee and D. Kim, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- F. Zhang and K. Zhu, Adv. Energy Mater., 2020, 10, 1–26 Search PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 14173–14180 CrossRef CAS PubMed.
- S. Maniyarasu, J. C. R. Ke, B. F. Spencer, A. S. Walton, A. G. Thomas and W. R. Flavell, ACS Appl. Mater. Interfaces, 2021, 13(36), 43573–43586 CrossRef CAS PubMed.
- W. Yu, L. Huang, D. Yang, P. Fu, L. Zhou, J. Zhang and C. Li, J. Mater. Chem. A, 2015, 3, 10660–10665 RSC.
- D. Yang, R. Yang, X. Ren, X. Zhu, Z. Yang, C. Li and S. Liu, Adv. Mater., 2016, 28(26), 5206–5213 CrossRef CAS PubMed.
- S. Akin, E. Akman and S. Sonmezoglu, Adv. Funct. Mater., 2020, 30(28), 2002964 CrossRef CAS.
- C. Luo, G. Li, L. Chen, J. Dong, M. Yu, C. Xu, Y. Yao, M. Wang, Q. Song and S. Zhang, Sustain. Energy Fuels, 2020, 4, 3971–3978 RSC.
- M. Shahiduzzaman, K. Yamamoto, Y. Furumoto, T. Kuwabara, K. Takahashi and T. Taima, RSC Adv., 2015, 5, 77495–77500 RSC.
- J. Y. Seo, T. Matsui, J. Luo, J. P. Correa-Baena, F. Giordano, M. Saliba, K. Schenk, A. Ummadisingu, K. Domanski, M. Hadadian, A. Hagfeldt, S. M. Zakeeruddin, U. Steiner, M. Grätzel and A. Abate, Adv. Energy Mater., 2016, 6, 1–6 Search PubMed.
- S. Bai, P. Da, C. Li, Z. Wang, Z. Yuan, F. Fu, M. Kawecki, X. Liu, N. Sakai, J. T. W. Wang, S. Huettner, S. Buecheler, M. Fahlman, F. Gao and H. J. Snaith, Nature, 2019, 571, 245–250 CrossRef CAS PubMed.
- R. Xia, Z. Fei, N. Drigo, F. D. Bobbink, Z. Huang, R. Jasiūnas, M. Franckevičius, V. Gulbinas, M. Mensi, X. Fang, C. Roldán-Carmona, M. K. Nazeeruddin and P. J. Dyson, Adv. Funct. Mater., 2019, 29(22), 1902021 CrossRef.
- L. Liu, J. A. McLeod, R. Wang, P. Shen and S. Duhm, Appl. Phys. Lett., 2015, 107(6), 061904 CrossRef.
- B. Philippe, B. W. Park, R. Lindblad, J. Oscarsson, S. Ahmadi, E. M. J. Johansson and H. Rensmo, Chem. Mater., 2015, 27, 1720–1731 CrossRef CAS.
- A. G. Shard, J. Vac. Sci. Technol., A, 2020, 38, 041201 CrossRef CAS.
- J. Chun-Ren Ke, A. S. Walton, D. J. Lewis, A. Tedstone, P. O'Brien, A. G. Thomas and W. R. Flavell, Chem. Commun., 2017, 53, 5231–5234 RSC.
- T. Singh and T. Miyasaka, Adv. Energy Mater., 2018, 8(3), 1700677 CrossRef.
- Y. Wang, Y. Yang, D. W. Han, Q. F. Yang, Q. Yuan, H. Y. Li, Y. Yang, D. Y. Zhou and L. Feng, Sol. Energy Mater. Sol. Cells, 2020, 212, 110553 CrossRef CAS.
- M. Shahiduzzaman, L. Wang, S. Fukaya, E. Y. Muslih, A. Kogo, M. Nakano, M. Karakawa, K. Takahashi, K. Tomita, J. M. Nunzi, T. Miyasaka and T. Taima, ACS Appl. Mater. Interfaces, 2021, 13, 21194–21206 CrossRef CAS PubMed.
- S. H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS PubMed.
- D. Liu, Z. Shao, J. Gui, M. Chen, M. Liu, G. Cui, S. Pang and Y. Zhou, Chem. Commun., 2019, 55, 11059–11062 RSC.
- W. Zhang, X. Liu, B. He, Z. Gong, J. Zhu, Y. Ding, H. Chen and Q. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 4540–4548 CrossRef CAS PubMed.
- A. Bahtiar, S. Rahmanita and Y. D. Inayatie, IOP Conf. Ser. Mater. Sci. Eng., 2017, 196, 012037 CrossRef.
- W. C. Lin, W. C. Lo, J. X. Li, Y. K. Wang, J. F. Tang and Z. Y. Fong, npj Mater. Degrad., 2021, 5, 1–6 CrossRef.
- J. Yang, X. Liu, Y. Zhang, X. Zheng, X. He, H. Wang, F. Yue, S. Braun, J. Chen, J. Xu, Y. Li, Y. Jin, J. Tang, C. Duan, M. Fahlman and Q. Bao, Nano Energy, 2018, 54, 218–226 CrossRef CAS.
- D. A. Beattie, A. Arcifa, I. Delcheva, B. A. Le Cerf, S. V. MacWilliams, A. Rossi and M. Krasowska, Colloids Surf. A Physicochem. Eng. Asp., 2018, 544, 78–85 CrossRef CAS.
- E. J. Juarez-Perez, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 16912–16919 RSC.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- X. Xia, J. Peng, Q. Wan, X. Wang, Z. Fan, J. Zhao and F. Li, ACS Appl. Mater. Interfaces, 2021, 13, 17677–17689 CrossRef CAS PubMed.
- A. Broderick, Y. Khalifa, M. B. Shiflett and J. T. Newberg, J. Phys. Chem. C, 2017, 121, 7337–7343 CrossRef CAS.
- Y. Khalifa, A. Broderick and J. T. Newberg, J. Phys. Condens. Matter, 2018, 30(32), 325001 CrossRef PubMed.
- M. Kot, L. Kegelmann, H. Köbler, M. Vorokhta, C. Escudero, P. Kúš, B. Šmíd, M. Tallarida, S. Albrecht, A. Abate, I. Matolínová, D. Schmeißer and J. I. Flege, ChemSusChem, 2020, 13, 5722–5730 CrossRef CAS PubMed.
- M. J. Jackman, A. G. Thomas and C. Muryn, J. Phys. Chem. C, 2015, 119, 13682–13690 CrossRef CAS.
- S. Wang, Z. Li, Y. Zhang, X. Liu, J. Han, X. Li, Z. Liu, S. (Frank) Liu and W. C. H. Choy, Adv. Funct. Mater., 2019, 29(15), 1900417 CrossRef.
- S. Van Reenen, M. Kemerink and H. J. Snaith, J. Phys. Chem. Lett., 2015, 6, 3808–3814 CrossRef CAS PubMed.
- R. I. Dawood, A. J. Forty and M. R. Tubbs, Proc. R. Soc. London, Ser. A, 1965, 284(1397), 272–288 CAS.
- J. W. Lee, D. H. Kim, H. S. Kim, S. W. Seo, S. M. Cho and N. G. Park, Adv. Energy Mater., 2015, 5(20), 1501310 CrossRef.
- M. Madadi, J. Heiska, J. Multia and M. Karppinen, ACS Appl. Mater. Interfaces, 2021, 13, 56793–56811 CrossRef CAS PubMed.
- B. Bendler, R. Barrahma, P. Philipp and T. Wirtz, Surf. Interface Anal., 2011, 43, 514–517 CrossRef CAS.
- H. H. Sønsteby, J. E. Bratvold, V. A.-L. K. Killi, D. Choudhury, J. W. Elam, H. Fjellvåg and O. Nilsen, J. Vac. Sci. Technol., A, 2020, 38, 060804 CrossRef.
- F. Di Francesco, N. Calisi, M. Creatini, B. Melai, P. Salvo and C. Chiappe, Green Chem., 2011, 13, 1712–1717 RSC.
- M. Klahn, C. Stüber, A. Seduraman and P. Wu, J. Phys. Chem. B, 2010, 114, 2856–2868 CrossRef CAS PubMed.
- M. G. Freire, L. M. N. B. F. Santos, A. M. Fernandes, J. A. P. Coutinho and I. M. Marrucho, Fluid Phase Equilib., 2007, 261, 449–454 CrossRef CAS.
- S. Rivera-Rubero and S. Baldelli, J. Am. Chem. Soc., 2004, 126, 11788–11789 CrossRef CAS PubMed.
- Y. Zhao, H. Tan, H. Yuan, Z. Yang, J. Z. Fan, J. Kim, O. Voznyy, X. Gong, L. N. Quan, C. S. Tan, J. Hofkens, D. Yu, Q. Zhao and E. H. Sargent, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, Fig. S1–S6, Tables S1–S4. See https://doi.org/10.1039/d2ta03748c |
| This journal is © The Royal Society of Chemistry 2022 |