
Iridium nanohollows with porous walls for acidic water splitting†
Xiaobing
Bao
a,
Sijia
Li
a,
Chentao
Hao
a,
Yifan
Qin
a,
Yutong
Gong
 a,
Yong
Yang
*a,
Aoni
Xu
*b and
Mingchuan
Luo
*c
a,
Yong
Yang
*a,
Aoni
Xu
*b and
Mingchuan
Luo
*c
aState Key Laboratory of Solidification Processing, Center of Advanced Lubrication and Seal Materials, Northwestern Polytechnical University, Xi'an, Shaanxi 710072, PR China. E-mail: yongyangfj@nwpu.edu.cn
bCatalysis Theory Center, Department of Physics, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark. E-mail: aonixu@dtu.dk
cLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. E-mail: m.luo@lic.leidenuniv.nl
First published on 11th July 2022
Abstract
Engineering iridium (Ir)-based electrocatalysts towards high activity and satisfactory durability for the oxygen evolution reaction (OER) in acidic media has been long pursued to commercialize proton exchange membrane-based electrolyzers. Here we report a novel class of Ir porous nanohollows (p-NHs) with tunable wall thickness, which electrocatalyze acidic OER with much enhanced performance relative to conventional Ir nanoparticles. The p-NH structure is deliberately-tailored via a facile hydrothermal approach, in which the initially-formed solid Ir spheres were in situ etched via the Kirkendall effect. At an overpotential of 300 mV, the Ir p-NH catalyst delivers a mass activity of 1.75 A mgIr−1, which is 6.25 and 3.20 times higher than those of commercial Ir/C and control Ir solid nanosphere catalysts, respectively. Ir p-NHs as an anode enable voltages of 1.50 V and 1.59 V at 10 and 100 mA cm−2, respectively, for acidic water splitting. We explore how porosity energetically promotes OER activity of Ir-based catalysts using density functional theory (DFT) calculations, which reveal that the adsorption of *OOH and thus OER activity can be described by the generalized coordination number of surface Ir sites. Our findings offer new insights into the rational design of highly-open Ir-based nanostructures for efficient OER electrocatalysis.
 Mingchuan Luo | Dr Mingchuan Luo is a Marie Skłodowska-Curie Fellow at Leiden University. His research focuses on understanding and engineering the mysterious electrified interface for renewable and sustainable innovation. Dr Luo obtained his PhD in chemistry from Beijing University of Chemical Technology in 2016. Since then, Dr Luo has conducted postdoctoral research at Peking University for advancing electrocatalytic nanomaterials, at University of Toronto for upgrading carbon dioxide electrolysis systems, and now at Leiden University for studying fundamental electrochemistry at electrified interfaces. |
Introduction
Water electrolysis enables massive hydrogen production powered by renewable electricity and, in doing so, promises less dependence on fossil fuels in the industry and transportation sectors.1–5 In particular, proton exchange membrane water electrolyzers (PEMWEs), featuring high voltage efficiency, high current densities, rapid response and low gas crossover, are of wide research interest.6–10 A crucial challenge related to PEMWEs arises from the sluggish kinetics of the anodic oxygen evolution reaction (OER) – a complex reaction that requires the transfer of four electrons and protons. Iridium is the only element that catalyzes the OER in strong acid with satisfactory activity and stability.11–16 However, the scarcity of Ir has posed severe limitations to the widespread application of PEMWEs in terms of both cost and supply constraints, which has motivated considerable research efforts to upgrade Ir-based OER electrocatalysts.17–22A widely-adopted upgrading strategy is to alloy Ir with other elements.23–29 The selection of alloying elements has been mainly guided by an energetic-descriptor-based approach to pursue an optimal adsorption of OER intermediates, with transition metals being the most popular candidates.30,31 However, most of these transition metals are thermodynamically unstable during acidic OER electrolysis and, as a consequence, tend to leach out.31–34 As a result, the actual catalytic structure of an alloying catalyst is a porous Ir skeleton derived from the continuous dissolution of transition metals – a process known as dealloying.35–38 Notably, such porous Ir architectures were recently shown to possess superior OER performance over their nanoparticulate counterparts. Markovic and co-workers reported an Ir nanoporous architecture, prepared from the dealloying of an Ir25Os75 precursor, which exhibited an exceptional balance between activity and stability for the acidic OER.38
Another upgrading strategy is to engineer geometries of catalytic components, with the purpose of maximizing the atomic utilization efficiency of Ir by increasing the accessibility of all Ir atoms to reactive molecules, electrons and ions.39–42 A hollow morphology enables the liberation of buried atoms that cannot be reached by reactants or electrolytes and thus has a higher utilization efficiency for catalysis than a solid one.43–46 Furthermore, hollow structures also facilitate high mass transport during the reaction, which is crucial for industrial-scale high-rate electrolysis. Therefore, it is highly appealing to study acidic OER behavior on an Ir-based electrocatalyst that integrates porous architecture with hollow morphology.
Here we report the design and synthesis of 3-dimensional (3D) Ir nanohollows enclosed with porous walls (denoted as Ir p-NHs) for efficient OER electrocatalysis in acidic media. Ir p-NHs were deliberately constructed in a facile hydrothermal system, in which the Kirkendall process was in situ triggered to excavate the initially-formed solid Ir nanospheres. The wall thickness could be facilely manipulated by controlling the concentration of the etching agent, i.e. Co3+ ions. The Ir p-NH catalyst showed a mass activity of 1.75 A mgIr−1 at an overpotential of 300 mV, which is 6.25 and 3.20 times higher than those of commercial Ir/C and control Ir solid nanosphere catalysts, respectively. When applied as an anode for water splitting in acidic media, Ir p-NHs enabled an overall voltage of 1.50 V at 10 mA cm−2. DFT calculations were carried out to explore the effect of porosity on OER performance of Ir-based catalysts and establish a relationship between the generalized coordination number, intermediates’ adsorption strength and OER activities on Ir surfaces.
Results and discussion
Synthesis and characterization
Ir p-NHs were synthesized using a one-pot hydrothermal approach, in which iridium chloride (IrCl3) was used as a precursor, glyoxal solution and citric acid as reducing agents, and cobalt acetylacetonate (Co(acac)3) as an etching agent (Fig. S1, ESI†). The hollowing process was driven by the Kirkendall effect, through the galvanic reaction between Co3+ ions and metallic Ir domains (Fig. S2, ESI†).47,48 We tracked the synthesis by collecting products at different reaction times and examined the morphologies using transmission electron microscopy (TEM, Fig. 1). Solid spheres with a diameter of ∼1 μm were observed at the initial stage (Fig. 1b), followed by the gradual hollowing with a prolonged reaction (Fig. 1c and d). Increasing the Co(acac)3 concentration decreases the wall thickness of nanohollows, and the collapse of the hollow structure was observed when fed with an excess amount of Co(acac)3 (Fig. S3, ESI†). Only solid nanospheres (NSs, used as a control catalyst) were obtained in the absence of Co(acac)3, verifying the key role of the Kirkendall effect in driving the hollowing process (Fig. S4, ESI†). Besides, both glyoxal and citric acid are essential to obtain uniform Ir p-NHs. In the absence of glyoxal, mixture products of nanoparticles and carbon spheres were obtained (Fig. S5a, ESI†), while dendritic nanostructures were produced in the absence of citric acid (Fig. S5b, ESI†).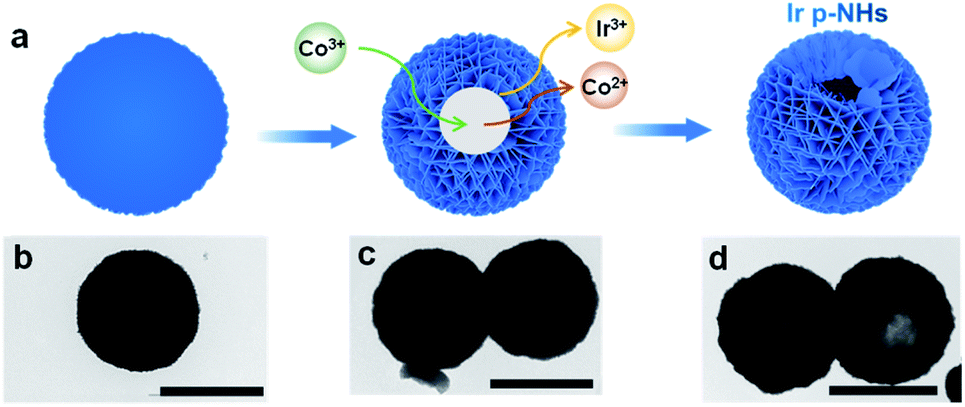 | ||
| Fig. 1 (a) Schematic illustration of the structural evolution of Ir p-NHs, with corresponding TEM images of products collected at a reaction time of (b) 1 h, (c) 3 h and (d) 5 h. Scale bar, 1 μm. | ||
High-angle annular dark-field scanning TEM (HAADF-STEM) was used to inspect Ir p-NHs for more structural details (Fig. 2a). The ratio of wall thickness to the overall hollow diameter was measured to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (Fig. 2b), indicative of high surface exposure of Ir atoms. Focusing on the wall region (Fig. 2c), we observed an assembly of two-dimensional nanosheets, as confirmed by the HRTEM image in Fig. 2d, the HAADF-STEM images in Fig. S6† (in the top view of p-NHs), and the SEM images in Fig. S7.† Abundant porosities and channels at the nanoscale were readily observed from these images, which were likely generated during the Kirkendall process. A lattice distance of 0.24 nm can be assigned to the (111) plane of a face-centered cubic (fcc) Ir crystallite (Fig. 2e and S8, ESI†), in line with the corresponding selected area electron diffraction (SAED) pattern. The STEM-EDS images suggest the uniform distribution of Ir (Fig. S9, ESI†). As confirmed by energy-dispersive X-ray spectroscopy (EDX) (Fig. S10, ESI†), Ir is the only detected metal element, excluding the residual of Co in p-NHs.
:10 (Fig. 2b), indicative of high surface exposure of Ir atoms. Focusing on the wall region (Fig. 2c), we observed an assembly of two-dimensional nanosheets, as confirmed by the HRTEM image in Fig. 2d, the HAADF-STEM images in Fig. S6† (in the top view of p-NHs), and the SEM images in Fig. S7.† Abundant porosities and channels at the nanoscale were readily observed from these images, which were likely generated during the Kirkendall process. A lattice distance of 0.24 nm can be assigned to the (111) plane of a face-centered cubic (fcc) Ir crystallite (Fig. 2e and S8, ESI†), in line with the corresponding selected area electron diffraction (SAED) pattern. The STEM-EDS images suggest the uniform distribution of Ir (Fig. S9, ESI†). As confirmed by energy-dispersive X-ray spectroscopy (EDX) (Fig. S10, ESI†), Ir is the only detected metal element, excluding the residual of Co in p-NHs.
 | ||
| Fig. 2 Structural characterization of Ir p-HNs. (a–c) HAADF-STEM images, (d and e) HRTEM images and the inserted SAED pattern, and (f) high-resolution XPS Ir 4f spectra of Ir p-HNs and Ir NSs. | ||
X-ray photoelectron spectroscopy (XPS) was carried out to compare chemical states of Ir in p-NHs and NSs. The XPS survey spectra of Ir p-NHs show the presence of C, O and Ir (Fig. S11a, ESI†) and the absence of Co (Fig. S11b, ESI†), in line with the results of STEM-EDS and EDX. The Ir 4f peaks of Ir p-NHs shifted more negatively compared to those of Ir NSs (Fig. 2f), indicative of a more electron-rich state of surface Ir on the former. Given the absence of the ligand effect on Ir (we have excluded the residence of Co above), such difference in the electronic structure is deduced to have originated from the distinction of surface geometries between p-NHs and NSs. We will provide more explanation regarding this aspect in the DFT section. In the case of Ir p-NHs, the fitted peaks at 61.04 and 64.04 eV are attributed to Ir 4f5/2 and Ir 4f7/2 of metallic iridium (Ir0), respectively; while the peaks at 61.7 and 64.7 eV can be assigned to Ir4+. The third set located at 62.5 and 65.5 eV is attributed to the satellite of Ir4+. By fitting the high-resolution XPS spectra, we found a higher ratio of the metallic Ir state on p-HNs than that on NSs (Table S1, ESI†). Intriguingly, a similar trend, i.e. an increased metallic state ratio, was also observed in the study of dealloying Ir-based materials.38
Electrocatalytic performance
We evaluated the electrocatalytic performance of Ir p-NHs for the OER using a typical three-electrode configuration (see the ESI† for details). The OER activity of Ir p-NHs was benchmarked against that of a commercial Ir/C catalyst and also compared to that of solid Ir NSs to reveal the structural effect of porosity and hollow structures on OER performance. Fig. 3a shows the OER polarization curves of various catalysts in 0.5 M H2SO4 at 1 mV s−1. The benchmark Ir/C catalyst showed a comparable OER activity, in terms of both the onset and the overpotential to deliver a current density of 10 mA cm−2 (η10), to previously-reported values.19,45 Following the customarily-adopted performance metric of η10, Ir p-NHs deliver an η10 of 243 mV, which is 54 mV and 31 mV lower than those of commercial Ir/C (η10 of 297 mV) and Ir NSs (η10 of 274 mV), respectively. Overall, Ir NHs-x samples (x represents the feeding amount of Co(acac)3 in the synthesis) showed higher activity than Ir NSs, suggesting the structural benefits of porosity and hollow structures (Fig. S12, ESI†).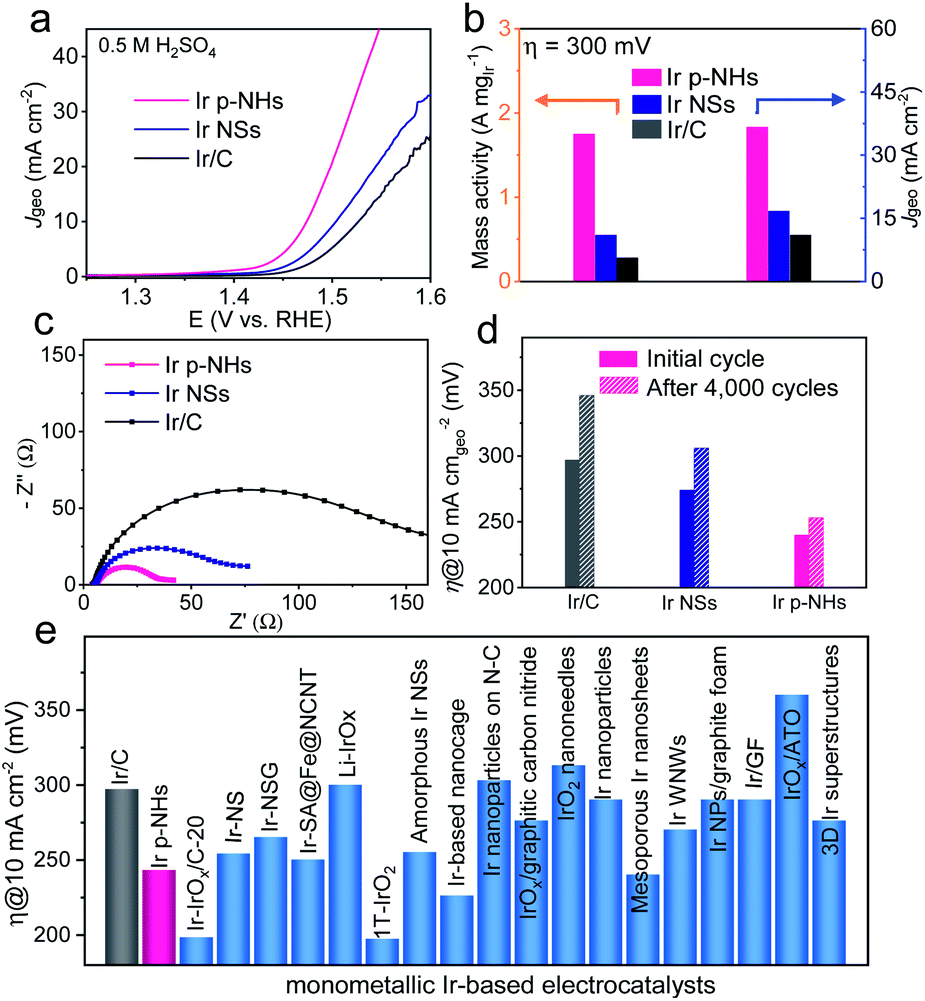 | ||
| Fig. 3 (a) Polarization curves of the OER, with currents being normalized to geometric surface areas, (b) mass activities and current densities at an overpotential of 300 mV, (c) electrochemical impedance spectroscopy, and (d) overpotentials at 10 mA cmgeo−2 before and after 4000 cycles on Ir p-NHs, Ir NSs and commercial Ir/C catalysts. (e) Activity comparison with recently reported monometallic Ir-based electrocatalysts in acidic electrolyte. | ||
Notably, the mass activity of Ir p-NHs reached 1.75 A mgIr−1, 6.25 and 3.2 times higher those that of commercial Ir/C (0.28 A mgIr−1) and Ir NSs (0.55 A mgIr−1), respectively (Fig. 3b and S13, ESI†). The electrochemical impedance spectrascopy (EIS) results reveal a smaller charge-transfer resistance of Ir p-NHs compared to that of the Ir NS counterpart (Fig. 3c), suggesting that the Kirkendall process not only generates porosities and channels but also promotes the integration of conductive Ir frameworks. This observation agrees with the previous finding that an electron-conduction ‘highway’ was constructed within the catalytic nanostructures via a dealloying process.38 All three catalysts show a similar Tafel slope of ∼60 mV dec−1 (Fig. S14, ESI†), indicative of a similar OER mechanism and identical rate-determining step. Regarding the electrocatalytic durability, the η10 of Ir p-NHs increased by only 10 mV after 4000 OER cycles, outperforming Ir/C (η10 increased by 49 mV) and Ir NSs (η10 increased by 31 mV) catalysts (Fig. 3d and S15, ESI†). The amount of dissolved Ir ions of each catalyst in the after-cycling electrolyte was further measured using ICP-AES (Fig. S16, ESI†), which verified a significantly improved stability of p-NHs (22.0 ng mL−1) compared to Ir NSs (77.3 ng mL−1) and Ir/C (51.0 ng mL−1) controls. Performance comparison shows Ir p-NHs as one promising monometallic OER catalyst in acidic electrolytes (Fig. 3e and Table S2, ESI†), though we need to be cautious about the different conditions and protocols for OER measurements across the literature, especially for the catalyst loading.
We further applied Ir p-NHs as the anode and 20% Pt/C as the cathode to evaluate water splitting performance in 0.5 M H2SO4 (Fig. 4a). As displayed in Fig. 4b, the Ir p-NH electrode delivers current densities of 10 and 100 mA cm−2 at cell voltages of 1.50 and 1.59 V, respectively, both of which are lower those of than Ir/C and Ir NSs. The catalytic durability is further compared in Fig. 4c. 81% of the initial activity was retained on the Ir p-NH catalyst after a 10 hour continuous reaction at a constant voltage of 1.49 V; while only 49% and 12% were retained on Ir NSs and Ir/C. The post-OER Ir p-NH catalyst was further characterized by TEM. As shown in Fig. S17,† the p-NH structure was well preserved, indicative of its high stability under acidic OER electrocatalysis.
 | ||
| Fig. 4 Initial electrocatalytic overall water splitting performance in 0.5 M H2SO4. (a) Scheme of the two-electrode cell electrolyzer using the Ir-based catalyst as the anode and 20% Pt/C as the cathode. (b) Polarization curves of Ir p-NHs, Ir NSs and Ir/C for overall water splitting. (c) Durability test for the three catalysts in a two-electrode configuration. | ||
DFT calculations and discussion
Despite a considerable number of OER studies reporting the beneficial effect of porosities/channels on Ir-based catalysts, the underlying mechanism remains poorly understood, especially from an energetic point of view. As revealed by the XPS results (Fig. 2f), a porosity-induced electronic modification was observed for surface Ir sites on p-NHs. Given the absence of the ligand effect (i.e., alloying with transition metals) in both structures, it is reasonable to deduce that such electronic difference is associated with the local geometries of surface Ir atoms. As is well established in surface science, the work functions of metals are highly dependent on the coordination environment of surface atoms. The methodology of using coordination numbers to bridge the electronic structure and electrocatalytic activity has proven valid in the oxygen reduction reaction on Pt surfaces but is yet to be explored for the OER.49,50We then leveraged this methodology to understand how surface local geometries impact OER electrocatalysis from an energetic point of view, by performing density functional theory (DFT) calculations on both ideal and defective IrO2(110) surfaces with tunable generalized coordination numbers (GCNs) (Fig. S18, ESI†).51 Along the OER pathway of H2O → *OH → *O → *OOH → O2, the ideal (110) facet shows a theoretical overpotential of 0.77 V with a rate-limiting step of *O → *OOH (Fig. S19 and S20a, ESI†), in agreement with previous literature reports.52 Before introducing point defects for GCN tuning, we first identified the most stable surface structure of IrO2 under OER relevant conditions (i.e., pH = 0; U > 1.4 V vs. SHE) based on a surface phase diagram.53 As shown in Fig. 5a, the most stable catalytic surface is O-terminated, suggesting that the existence of an oxygen vacancy is unlikely during OER electrocatalysis. Therefore, we only considered Ir vacancies for GCN tuning in our model.54
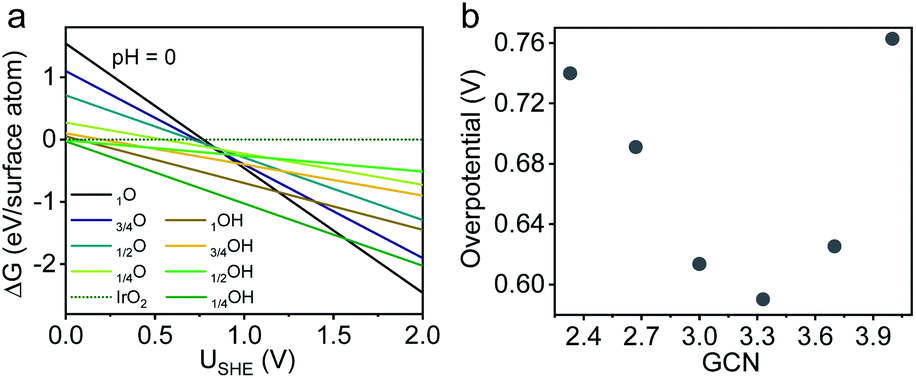 | ||
| Fig. 5 DFT calculations on IrO2(110). (a) Surface phase diagram of IrO2 at pH = 0. (b) Theoretical overpotential of the defective IrO2 surface with different GCNs. | ||
We gradually reduced the GCN by removing surface Ir atoms from our model one-by-one (Table S3, ESI†). Theoretical OER activities were then calculated on these defective surfaces (Fig. 5b). The optimal GCN region lies between 3.0 and 3.7, in which the defective IrO2 surfaces outperform the ideal counterpart by 0.15–0.2 eV due to a lower energetic barrier for *OOH formation (Fig. S20b, ESI†). Intriguingly, we found that a GCN less than 3 led to an unstable IrO3 structure, which had a lower OER activity due to an increased energetic barrier for the dehydrogenation to *O (Fig. S20c and d, ESI†). Overall, our DFT results suggest that an intermediate GCN is crucial for upgrading an Ir-based OER electrocatalyst for PEMWEs. We note here that the purpose of our DFT calculations is not to specifically explain the catalytic behavior of the Ir p-NS structure, but rather is the first attempt to testify the explanative viability of GCNs for the acidic OER on a broad family of defective Ir-based catalytic surfaces. From the above results, the GCN well bridges local geometries (related to shape, crystallinity, porosity, etc.) and adsorption energetics of the OER and could guide future structural engineering of more efficient Ir-based OER catalysts.
Conclusions
We have reported a class of efficient OER electrocatalysts, structurally featuring Ir porous nanohollows (Ir p-NHs), for acidic water splitting. The Kirkendall effect thermodynamically drives the structural evolution. Ir p-NHs show enhanced OER catalytic activities over benchmark Ir/C and Ir nanosphere control catalysts in both half-cell and full-cell measurements, revealing the beneficial role of porosity towards OER electrocatalysis. Leveraging density functional theory (DFT) calculations, we further demonstrate that the generalized coordination number (GCN) could serve as a structural descriptor to predict OER activities on Ir-based surfaces, and an intermediate GCN region was desired. Our findings offer new insights for the rational design of defective Ir-based nanostructures for efficient OER electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the National Natural Science Foundation of China (Grant No. 22171227 and No. 51902266), Key Research and Development Projects of Shaanxi Province (No. 2020GXLH-Z-032), Natural Science Basic Research Program of Shaanxi Province (No. 2022JQ-118), the State Key Laboratory of Solidification Processing (NPU), China (Grant No. 2022-QZ-03), the Opening Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (Grant No. sklpme2022-4-05), and the European Commission Horizon 2020—Research and Innovation Framework Programme (Marie Skłodowska-Curie actions Individual Fellowship awarded to M. L., No. 897818).References
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, R. Mom, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. R. Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408–413 CrossRef CAS PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- G. S. Ogumerem and E. N. Pistikopoulos, J. Process Control, 2020, 91, 37–49 CrossRef CAS.
- Y. Li, Y. Sun, Y. Qin, W. Zhang, L. Wang, M. Luo, H. Yang and S. Guo, Adv. Energy Mater., 2020, 10, 1903120 CrossRef CAS.
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- P. Lettenmeier, L. Wang, U. Golla-Schindler, P. Gazdzicki, N. A. Cañas, M. Handl, R. Hiesgen, S. S. Hosseiny, A. S. Gago and K. A. Friedrich, Angew. Chem., Int. Ed., 2016, 55, 742–746 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- Z. Chen, L. Guo, L. Pan, T. Yan, Z. He, Y. Li, C. Shi, Z.-F. Huang, X. Zhang and J.-J. Zou, Adv. Energy Mater., 2022, 2103670 CrossRef CAS.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- L. Zu, X. Qian, S. Zhao, Q. Liang, Y. E. Chen, M. Liu, B. J. Su, K. H. Wu, L. Qu, L. Duan, H. Zhan, J. Y. Zhang, C. Li, W. Li, J. Y. Juang, J. Zhu, D. Li, A. Yu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 2208–2217 CrossRef CAS PubMed.
- L. Zhuang, F. Xu, K. Wang, J. Li, C. Liang, W. Zhou, Z. Xu, Z. Shao and Z. Zhu, Small, 2021, 17, 2100121 CrossRef CAS PubMed.
- Y. Xie, X. Long, X. Li, C. Chang, K. Qu and Z. Yang, Chem. Commun., 2021, 57, 8620–8623 RSC.
- W. H. Lee, Y. J. Ko, J. H. Kim, C. H. Choi, K. H. Chae, H. Kim, Y. J. Hwang, B. K. Min, P. Strasser and H. S. Oh, Nat. Commun., 2021, 12, 4271 CrossRef CAS PubMed.
- H. Liu, H. Jang, Y. Wang, M. G. Kim, H. Li, Q. Qin, X. Liu and J. Cho, J. Mater. Chem. A, 2022, 10, 3393–3399 RSC.
- N. Li, L. Cai, C. Wang, Y. Lin, J. Huang, H. Sheng, H. Pan, W. Zhang, Q. Ji, H. Duan, W. Hu, W. Zhang, F. Hu, H. Tan, Z. Sun, B. Song, S. Jin and W. Yan, J. Am. Chem. Soc., 2021, 143, 18001–18009 CrossRef CAS PubMed.
- Q. Dang, H. Lin, Z. Fan, L. Ma, Q. Shao, Y. Ji, F. Zheng, S. Geng, S.-Z. Yang, N. Kong, W. Zhu, Y. Li, F. Liao, X. Huang and M. Shao, Nat. Commun., 2021, 12, 6007 CrossRef CAS PubMed.
- F. Bizzotto, J. Quinson, A. Zana, J. J. K. Kirkensgaard, A. Dworzak, M. Oezaslan and M. Arenz, Catal. Sci. Technol., 2019, 9, 6345–6356 RSC.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- X. Zheng, M. Qin, S. Ma, Y. Chen, H. Ning, R. Yang, S. Mao and Y. Wang, Adv. Sci., 2022, 2104636 CrossRef CAS PubMed.
- S. Chatterjee, S. Intikhab, L. Profitt, Y. Li, V. Natu, R. Gawas and J. Snyder, J. Catal., 2021, 393, 303–312 CrossRef CAS.
- J. Zhu, M. Xie, Z. Chen, Z. Lyu, M. Chi, W. Jin and Y. Xia, Adv. Energy Mater., 2020, 10, 1904114 CrossRef CAS.
- R. Li, H. Wang, F. Hu, K. C. Chan, X. Liu, Z. Lu, J. Wang, Z. Li, L. Zeng, Y. Li, X. Wu and Y. Xiong, Nat. Commun., 2021, 12, 3540 CrossRef CAS PubMed.
- J. Shan, T. Ling, K. Davey, Y. Zheng and S. Z. Qiao, Adv. Mater., 2019, 31, e1900510 CrossRef PubMed.
- H. Zhu, Z. Zhu, J. Hao, S. Sun, S. Lu, C. Wang, P. Ma, W. Dong and M. Du, Chem. Eng. J., 2022, 431, 133251 CrossRef CAS.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov, R. Schlögl, T. Jones and P. Strasser, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. Ren, X. Guo and S. Huang, Appl. Surf. Sci., 2021, 556, 149801 CrossRef CAS.
- D. Liu, Q. Lv, S. Lu, J. Fang, Y. Zhang, X. Wang, Y. Xue, W. Zhu and Z. Zhuang, Nano Lett., 2021, 21, 2809–2816 CrossRef CAS PubMed.
- J. Park, Y. J. Sa, H. Baik, T. Kwon, S. H. Joo and K. Lee, ACS Nano, 2017, 11, 5500–5509 CrossRef CAS PubMed.
- L. Fu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 24836–24841 RSC.
- M. Liu, S. Liu, Q. Mao, S. Yin, Z. Wang, Y. Xu, X. Li, L. Wang and H. Wang, J. Mater. Chem. A, 2022, 10, 2021–2026 RSC.
- Y. Pi, J. Guo, Q. Shao and X. Huang, Chem. Mater., 2018, 30, 8571–8578 CrossRef CAS.
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef.
- Y.-T. Kim, P. P. Lopes, S.-A. Park, A. Y. Lee, J. Lim, H. Lee, S. Back, Y. Jung, N. Danilovic, V. Stamenkovic, J. Erlebacher, J. Snyder and N. M. Markovic, Nat. Commun., 2017, 8, 1449 CrossRef PubMed.
- Z. Shi, Y. Wang, J. Li, X. Wang, Y. Wang, Y. Li, W. Xu, Z. Jiang, C. Liu, W. Xing and J. Ge, Joule, 2021, 5, 2164–2176 CrossRef CAS.
- Z. Li, Y. Chen, S. Ji, Y. Tang, W. Chen, A. Li, J. Zhao, Y. Xiong, Y. Wu, Y. Gong, T. Yao, W. Liu, L. Zheng, J. Dong, Y. Wang, Z. Zhuang, W. Xing, C.-T. He, C. Peng, W.-C. Cheong, Q. Li, M. Zhang, Z. Chen, N. Fu, X. Gao, W. Zhu, J. Wan, J. Zhang, L. Gu, S. Wei, P. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. Duan, Y. Huang, X.-M. Chen, D. Wang and Y. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS PubMed.
- D. Lebedev, R. Ezhov, J. Heras-Domingo, A. Comas-Vives, N. Kaeffer, M. Willinger, X. Solans-Monfort, X. Huang, Y. Pushkar and C. Copéret, ACS Cent. Sci., 2020, 6, 1189–1198 CrossRef CAS PubMed.
- T. Li, O. Kasian, S. Cherevko, S. Zhang, S. Geiger, C. Scheu, P. Felfer, D. Raabe, B. Gault and K. J. J. Mayrhofer, Nat. Catal., 2018, 1, 300–305 CrossRef.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed.
- L. Zhou, Z. Zhuang, H. Zhao, M. Lin, D. Zhao and L. Mai, Adv. Mater., 2017, 29, 1602914 CrossRef PubMed.
- J. Zhu, Z. Chen, M. Xie, Z. Lyu, M. Chi, M. Mavrikakis, W. Jin and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 7244–7248 CrossRef CAS PubMed.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G.-C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- A. Cabot, M. Ibáñez, P. Guardia and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 11326–11328 CrossRef CAS PubMed.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- W. Sun, Y. Song, X.-Q. Gong, L.-m. Cao and J. Yang, Chem. Sci., 2015, 6, 4993–4999 RSC.
- O. Vinogradova, D. Krishnamurthy, V. Pande and V. Viswanathan, Langmuir, 2018, 34, 12259–12269 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, P. Sautet and D. Loffreda, Angew. Chem., Int. Ed., 2014, 53, 8316–8319 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02193e |
| This journal is © The Royal Society of Chemistry 2022 |