
Heterostructuring cobalt sulfide with highly oxophilic 1T-tungsten sulfide for durable and efficient oxygen electrocatalysis†
Shengxiong
Yang
a,
Bin
Li
a,
Minqiu
Lan
a,
Liangsheng
Liu
a,
Yimin
Sun
c,
Fei
Xiao
ab and
Junwu
Xiao
 *a
*a
aKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, Department of Chemistry and Chemical Engineering, Huazhong University of Science & Technology, Wuhan 430074, China
bShenzhen Huazhong University of Science and Technology Research Institute, Shenzhen 518000, China
cHubei Key Laboratory of Plasma Chemistry and Advanced Materials, School of Materials Science and Engineering, Wuhan Institute of Technology, 693 Xiongchu Avenue, Wuhan, 430073, China. E-mail: chjwxiao@hust.edu.cn
First published on 9th May 2022
Abstract
First-row transition metal sulfides are one of the most promising platinum-group metal-free catalysts toward oxygen electrocatalysis, but their structural and compositional changes during catalysis result in the instability of the electrochemical performance. Herein, a heterointerface strategy is proposed for constructing 1T-phase tungsten sulfide decorated on cobalt sulfide for modulating the electronic structure and retarding the phase change of the underlying cobalt sulfide due to the high oxophilicity and strong electronic interaction, as well as the metallic feature. Consequently, an impressive electrocatalytic performance toward oxygen reduction is achieved with a comparable mass activity to platinum-group metals and no noticeable degradation regardless of what is performed in the rotating-disk electrode and gas diffusion electrode setups. Moreover, the turnover frequency is almost one order of magnitude higher than that of cobalt sulfide for catalyzing oxygen evolution. When employed as the cathode of a zinc–air battery, it delivers an improved cycling performance over 480 h of operation and a peak power density of 183.5 mW cm−2, outperforming most transition metal sulfides reported to date. This work provides an effective strategy to improve the activity and durability of first-row transition metal sulfides by interface engineering for oxygen electrocatalysis.
 Junwu Xiao | Dr Junwu Xiao is currently an associate professor in Department of Chemistry and Chemical Engineering at Huazhong University of Science & Technology (HUST). He received his PhD degree in 2012 from The Hong Kong University of Science & Technology (HKUST) under the supervision of Prof. Shihe Yang. From 2018 to 2020, he worked in Department of Chemical & Biomolecular Engineering at The University of Delaware as a visiting scholar under the supervision of Prof. Yushan Yan. His research focuses on anion exchange membrane water electrolyzers/hydrogen fuel cells. |
Introductions
One of the greatest obstacles in developing polymer exchange membrane fuel cells and rechargeable metal–air batteries is associated with designing low-cost but efficient catalysts towards the oxygen reduction/evolution reactions (ORR and OER),1 since they possess very sluggish kinetics and four proton-coupled electron transfer mechanisms. Transition-metal sulfides (TMS) categorized as layered sulfides (WS2, MoS2, etc.) and nonlayered sulfides (MxSy, M = Co, Fe, Ni, Cu, etc.), have been intensively exploited as a promising class of ORR/OER catalysts to replace expensive and scarce platinum-group metals (Pt, Ru, and Ir).2,3 In spite of incredibly boosting the activities by means of structural, compositional, and defect engineering,4,5 they still undergo self-oxidation under ORR/OER conditions to form (oxy)hydroxides,6–10 accompanied by morphological changes and even structural collapse, leading to the instability of the catalytic performance.It is widely documented that encapsulating the carbon layer and impregnating transition-metal sulfides into carbon supports effectively hinder the oxidation and impedes the catalyst aggregation/detachment,11–17 thereby showing reasonable catalytic performance when being probed in the configuration of a micrometer thin layer without the consideration of the mass transport effect. However, a low ion-accessible surface area and poor mass transport properties seriously retard the catalyst utilization and deteriorate the energy conversion efficiency, especially when the catalysts are explored for assembling a catalyst layer of several tens of micrometers in thickness. Interface engineering of sulfide-based heterostructures has been recently developed as a promising strategy for notably enhancing the stability performance without the compromise of catalytic kinetics because of the synergistic effects and strongly coupled interfaces.18 For example, Guo et al.19 reported oxygen vacancy-rich NiS2/CoS2 heterostructure nanowires as a stable and efficient cathode catalyst of Zn–air batteries. Lee et al.20 proposed that the strong interfacial interaction in the core–shell CoSx@Cu2MoS4 nanostructure ensured the intact structure and unnoticeable compositional change during catalysis, rendering rapid catalytic kinetics and much superior durability. ZnS decorated at NiCo2S4 modulated its electronic configuration and constructed more defects,21 synergistically promoting ORR and OER kinetics. The local lattice strain in the Cu–CoFS heterostructure resulting from a lattice-mismatch can modulate the electronic structure configuration and, thus, optimize the adsorption strength of oxygen-intermediates during ORR and OER catalysis.4 However, the inherent sulfur leaching phenomenon is inevitable. Tungsten and molybdenum are suggested to have a stronger oxophilicity than first-row transition metals,22,23 and, therefore, the corresponding layered sulfides, which are preferable to host oxygen-containing species, have been usually utilized for accelerating water dissociation, in conjunction with non-layered sulfides, such as NiCoS/MoS2,24 FeNiS/MoS2,25 Co9S8/MoS2,22,26 NiS/MoS2,27,28 MoS2/Co9S8/Ni3S2/Ni,29 CoNi2S4/WS2/Co9S8,30etc.
Considering the aforementioned analysis, integrating non-layered sulfides with layered sulfides may synergistically strengthen the adsorption of oxygen intermediates and hinder the oxidation of underlying first-row transition metal sulfides, and it is a promising approach for promoting oxygen electrocatalysis,31,32 especially for 1T-phase layered sulfides that with metallic features are capable of enhancing the capability of electron/charge transfer.18,33 Herein, we have designed a CoS/1T-WS2 heterostructure supported by carbon nanocubes for oxygen electrocatalysis. Highly oxophilic 1T-WS2 anchored at CoS nanocrystals modulated the electronic structure and enhanced the attachment of oxygen molecules. It also retarded the compositional change of CoS underneath and preserved its structural integrity during ORR catalysis, coupled with carbon nanocube supports. As a result, the best-performing CoS/WS2 catalyst delivered exceptional activity and durability for the ORR not only in a rotating-disk electrode (RDE) setup but also in a gas diffusion electrode (GDE) setup, and was successfully explored as a robust and stable cathode for aqueous Zn–air batteries.
Experimental
Synthesis of carbon nanocube-supported cobalt sulfide/tungsten sulfide (CoS/WS2/CNC) and cobalt sulfide (CoS/CNC)
Zeolitic imidazolate framework (ZIF)-67 nanocubes were synthesized according to previous methods.34 Cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 0.1 M) and cetyltrimethylammonium bromide (CTAB, 1.37 mM) were dissolved in 10 mL of deionized water. 2-Methylimidazole (2-Melm, 4.54 g) was dissolved in 70 mL of deionized water, and was then injected into the Co(NO3)2 and CTAB solution under vigorous stirring for 20 min. After being washed with anhydrous ethanol, the as-collected ZIF-67 nanocubes were dried overnight at room temperature. The ZIF-67 powder (0.2 g) and m-aminophenol (0.2 g) were dispersed into 80 mL of anhydrous ethanol, followed by the addition of 10 mL of sodium tungstate dihydrate (Na2WO4·2H2O) and sodium bicarbonate (NaHCO3) aqueous solution. Formaldehyde solution (1.5%; 10 mL) was then added dropwise and was vigorously stirred at 35 °C for 9 h to get ZIF-67/poly(m-aminophenol) (PmAP)-HCO3−/WO42− as the concentrations of NaHCO3 and Na2WO4 were 4 mM and 2 mM, respectively. Similarity, ZIF-67/PmAP-HCO3− and ZIF-67/PmAP-WO42− were prepared with only the addition of 8 mM NaHCO3 or 4 mM Na2WO4. After being collected by centrifugation and being washed with deionized water, the products were thermally decomposed at 700 °C for 1 h and an Ar flow of 100 sccm as ∼4 g of thiourea was located at the front end of the tube furnace, forming cobalt sulfide/tungsten sulfide supported by carbon nanocubes. The samples were named as CoS/WS2/CNC-1 and CoS/WS2/CNC-2 using ZIF-67/PmAP-HCO3−/WO42− and ZIF-67/PmAP-WO42− precursors, respectively. Likewise, carbon nanocube-supported cobalt sulfide (CoS/CNC) was synthesized using ZIF-67/PmAP-HCO3− powder. WS2 powders were prepared using commercial WO3 powder through a similar sulfurization process.Results and discussion
Fig. 1a schematically depicts the synthetic process of carbon nanocube-supported cobalt sulfide/tungsten sulfide (CoS/WS2/CNC). Using solid ZIF-67 nanocubes as raw materials (Fig. S1†),34,35 poly(m-aminophenol) (PmAP) and cobalt carbonate/tungstate co-deposit densely at the surface via the in situ polymerization of m-aminophenol and the co-precipitation of cobalt cations with bicarbonate (HCO3−) and/or tungstate (WO42−) (Fig. S2†),36 along with the preservation of the ZIF-67 phase (Fig. S3†). They are named as ZIF-67/PmAP-HCO3−, ZIF-67/PmAP-HCO3−/WO42−, and ZIF-67/PmAP-WO42−, respectively, when the reactants contain sodium bicarbonate and sodium tungstate with molar ratios of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 4:2, and 0:4. However, the nanocube surface evolves from a short nanorod to nanoplatelet aggregates with increasing the tungstate concentration in the reactants (Fig. S4†). By comparison, the precipitates are distributed randomly at the nanocube surface with only adding sodium bicarbonate or m-aminophenol and formaldehyde (Fig. S5 and S6†), named as ZIF-67/HCO3− and ZIF-67/PmAP, implying the stabilizing effect of PmAP. Fourier-transform infrared spectroscopy (FT-IR) in Fig. S7† reveals that the peaks at 1622 cm−1, 1514 cm−1, and 1240 cm−1 assigned to the stretching vibrations of C
:0, 4:2, and 0:4. However, the nanocube surface evolves from a short nanorod to nanoplatelet aggregates with increasing the tungstate concentration in the reactants (Fig. S4†). By comparison, the precipitates are distributed randomly at the nanocube surface with only adding sodium bicarbonate or m-aminophenol and formaldehyde (Fig. S5 and S6†), named as ZIF-67/HCO3− and ZIF-67/PmAP, implying the stabilizing effect of PmAP. Fourier-transform infrared spectroscopy (FT-IR) in Fig. S7† reveals that the peaks at 1622 cm−1, 1514 cm−1, and 1240 cm−1 assigned to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, CC, and C–O–C groups, respectively, are noticeably visualized in ZIF-67/PmAP-HCO3−/WO42− and ZIF-67/PmAP,37 suggesting the successful polymerization of mAP into PmAP.
N, CC, and C–O–C groups, respectively, are noticeably visualized in ZIF-67/PmAP-HCO3−/WO42− and ZIF-67/PmAP,37 suggesting the successful polymerization of mAP into PmAP.
 | ||
| Fig. 1 (a) Schematic illustration of the fabrication pathway of CoS/WS2/CNC. (b) SEM, (c and d) TEM, and (e) high-resolution TEM images of CoS/WS2/CNC-1. | ||
During the subsequent sulfurization process, ZIF-67/PmAP-HCO3−, ZIF-67/PmAP-HCO3−/WO42−, and ZIF-67/PmAP-WO42− are transformed into carbon nanocube-supported cobalt sulfide and/or tungsten sulfide, named as CoS/CNC, CoS/WS2/CNC-1, and CoS/WS2/CNC-2, respectively. The CoS phase (JCPDS: 01-070-2864) is predominant in the X-ray diffraction (XRD) pattern of CoS/CNC, with a minor Co9S8 phase at 2θ = 29.8° and 52.1° (Fig. S8†). The additional diffraction peaks at 2θ = 14.3°, 32.8°, 33.6°, 39.5°, 58.4°, and 60.5° corresponding to the WS2 phase appeared in CoS/WS2/CNC-1 and become more obvious in CoS/WS2/CNC-2. These are consolidated by microwave plasma-atomic emission spectroscopy (MP-AES) and X-ray fluorescence (XRF) analysis (Table S1†). The cobalt percentage is 20.2 wt% for CoS/CNC, 19.6 wt% for CoS/WS2/CNC-1, and 18.7 wt% for CoS/WS2/CNC-2, alongside a decrease in the Co/W molar ratio from 9.8 for CoS/WS2/CNC-1 to 6.2 for CoS/WS2/CNC-2. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images in Fig. 1b and c reveal that CoS/WS2/CNC-1 exhibits the hollow nanocube structure with a concave surface, in accordance with CoS/CNC and CoS/WS2/CNC-2 (Fig. S9 and S10†). The high-magnification TEM image in Fig. 1d reveals that the layered WS2 nanoplatelets located at the surface of CoS nanocrystals form the heterostructures, consistent with that of CoS/WS2/CNC-2 (Fig. S10c†). This is further illustrated by high-resolution TEM images in Fig. 1e and S10d,† where the lattice fringes at distances of 0.29 nm and 0.25 nm correspond to the (100) and (101) crystal faces of CoS, and 0.62 nm is assigned to the (002) planes of WS2. By comparison, a solid nanocube is formed via the direct sulfurization of ZIF-67 precursors (Fig. S11†). This is because the robust cobalt carbonate/tungstate nanostructures at the surface of ZIF-67/PmAP-HCO3−/WO42− show a much smaller shrinkage than interior ZIF-67 during the pyrolysis process and, meanwhile, the released gas promotes the out-diffusion of cobalt ions in interior ZIF-67,38 leading to the formation of hollow structures in CoS/CNC and CoS/WS2/CNC.
The electrocatalytic performance towards the ORR was first analyzed on a RRDE-3A rotating ring-disk electrode and IVIUM Verter potentiostat. Fig. 2a shows the cyclic voltammetry (CV) profiles of CoS/WS2/CNC-1 in a N2 or O2-saturated electrolyte. An oxidation peak clearly appears at ∼0.94 V versus the reversible hydrogen electrode (RHE), consistent with that of CoS/CNC and CoS/WS2/CNC-2 (Fig. S12†), while it disappears in the CV curve of physically mixed tungsten sulfide and carbon catalyst (WS2 + C), implying that it originates from the oxidization of cobalt sulfide. Moreover, a reduction peak assigned to the reduction of oxygen molecules is observed in the CV curve of CoS/WS2/CNC-1, of which a peak potential (Ep) to 0.874 V versus RHE is achieved, which is 12 mV and 24 mV more positive than that for CoS/WS2/CNC-2 and CoS/CNC. Linear sweep voltammetry (LSV) curves in Fig. 2b are in good agreement with the CV analysis, indicating a better ORR activity of CoS/WS2/CNC-1 with a half-wave potential (E1/2) of 0.868 V versus RHE as compared to CoS/CNC (0.844 V versus RHE for E1/2) and CoS/WS2/CNC-2 (0.852 V versus RHE for E1/2). They all are superior to the WS2 + C catalyst, indicating that cobalt sulfide majorly contributes to the ORR activity. With respect to the benchmark Pt/C catalyst, in spite of a lower onset potential, the most optimal CoS/WS2/CNC-1 shows a close E1/2 and even has a slightly higher diffusion-limited current. Moreover, rotating ring-disk electrode (RRDE) experiments are explored for quantitatively determining the production rates of OH− and HO2− during ORR catalysis, and can, thus, give an insight into the reaction pathway, i.e., a 2e− or 4e− process. The ring current of CoS/WS2/CNC-1 is negligible at >0.75 V versus RHE and increases to 0.017 mA at 0.4 V versus RHE (Fig. 2c), which is still close to 0.014 mA for the Pt/C catalyst. Consequently, a 4e− oxygen reduction pathway is predominant under the catalysis of CoS/WS2/CNC-1 (Fig. S13†), as illustrated by linear Koutecky–Levich (K–L) plots at potentials of 0.80–0.60 V versus RHE that have similar slopes to the Pt/C catalyst (Fig. S14†). The exceptional ORR performance of CoS/WS2/CNC-1 is further demonstrated by the high kinetic current density (Jk), mass activity (Jmass), and turnover frequency (TOF), as depicted in Fig. 2d. Jk, Jmass, and TOF values of 16.2 mA cm−2, 413.3 A gCo−1, and 0.063 s−1 at 0.80 V versus RHE are achieved, respectively, which are far superior to those of CoS/CNC and CoS/WS2/CNC-2, and even approach those of the Pt/C catalyst. Moreover, the smaller Tafel slope relative to that of the Pt/C catalyst gives a clue to the good catalytic kinetics (Fig. S15†).
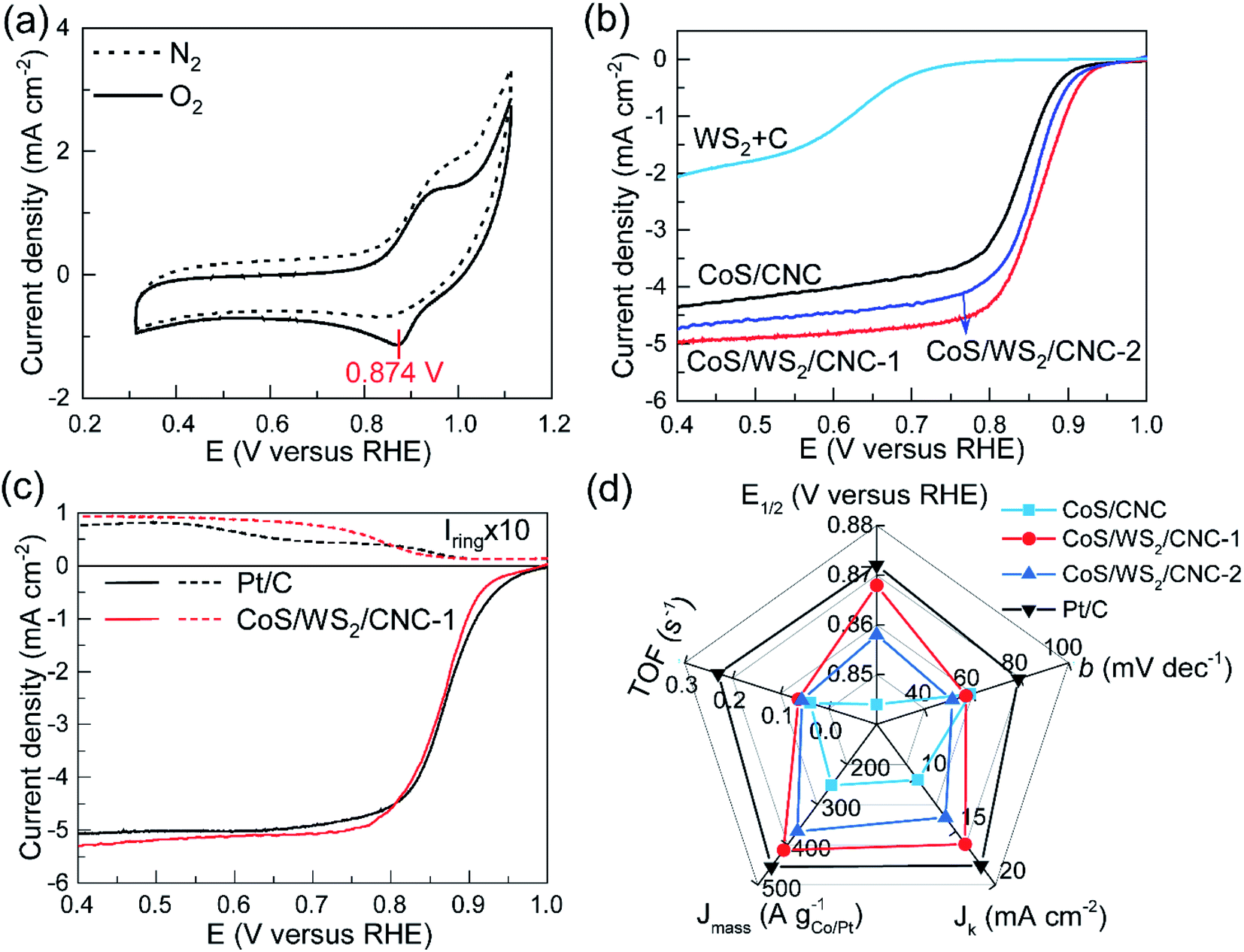 | ||
| Fig. 2 Electrocatalytic performance towards the ORR performed on a rotating disk electrode. (a) CV curves of CoS/WS2/CNC-1 catalyst in a N2 or O2-saturated 0.1 M KOH electrolyte. (b) LSV curves at a rotating speed of 1600 rpm and a scan rate of 5 mV s−1. (c) RRDE profiles of CoS/WS2/CNC-1 and Pt/C catalysts at a Pt ring potential of 1.2 V versus RHE. (d) Comparison of half-wave potential (E1/2), Tafel slope (b), kinetic current density (Jk) at 0.80 V versus RHE, mass activity (Jmass) at 0.80 V versus RHE, as well as turnover frequency (TOF) at 0.80 V versus RHE in CoS/CNC, CoS/WS2/CNC-1, CoS/WS2/CNC-2, and Pt/C catalysts. | ||
The composition and chemical states of the catalysts were probed through the X-ray photoelectron spectroscopy (XPS) technique. As displayed in Fig. 3a, the Co 2p XPS peaks at 778.5 eV and 793.8 eV are indexed to the Co–S bond in CoS/CNC,39,40 and the peaks at 781.0 eV and 796.8 eV are ascribed to a Co2+ species,41 as evidenced by the typical satellite peaks at 784.2 eV and 803.0 eV. Compared to those in CoS/CNC, the 2p3/2 and 2p1/2 peaks of Co–S bond shift to 778.3 eV and 793.4 eV in CoS/WS2/CNC-1 and CoS/WS WS2/CNC-2, which is assumed to be due to a strong interaction between CoS and WS2.20,39,41 The W 4f core level peaks at 31.9 eV and 34.0 eV suggest that the 1T-phase WS2 with a metallic feature is predominant in CoS/WS2/CNC-1 (Fig. 3b),18,42 as consolidated by the S 2p peaks at 161.8 eV and 163.0 eV in Fig. S15.†33,43 The weak W 4f signals at 35–39 eV indicate the existence of the oxidized W species, such as W5+ (35.4 eV for 4f7/2 and 37.5 eV for 4f5/2) and W6+ (36.8 eV for 4f7/2 and 38.3 eV for 4f5/2).18,44 The main W 4f7/2 and 4f5/2 peaks are upshifted by 0.2 eV as more WS2 nanoplatelets are anchored at the CoS surface, further disclosing the electron transfer from WS2 to CoS. This is validated by the S 2p core level peak upshifting from CoS/CNC, CoS/WS2/CNC-1, to CoS/WS2/CNC-2 (Fig. S16†). The electronic interaction between CoS and WS2 is beneficial for effectively manipulating the electronic structure and thereby influencing the adsorption strength of oxygen intermediates during catalysis.30,39 Moreover, other S 2p signals centered at 163.6 eV, 165.0 eV, 168.2 eV, and 169.6 eV are assigned to S22− and oxidized sulfate groups (–SOx, x = 2–4).45,46 Modulating the absorption behavior of oxygen by the CoS/WS2 interface is confirmed by O2-temperature programmed desorption (O2-TPD) plots (Fig. 3c). Note that the desorption peaks originating from the chemically absorbed oxygen occur at a higher temperature (372 °C) for CoS/WS2/CNC-1 as compared to 345 °C for CoS, and further increases to 425 °C for CoS/WS2/CNC-2.
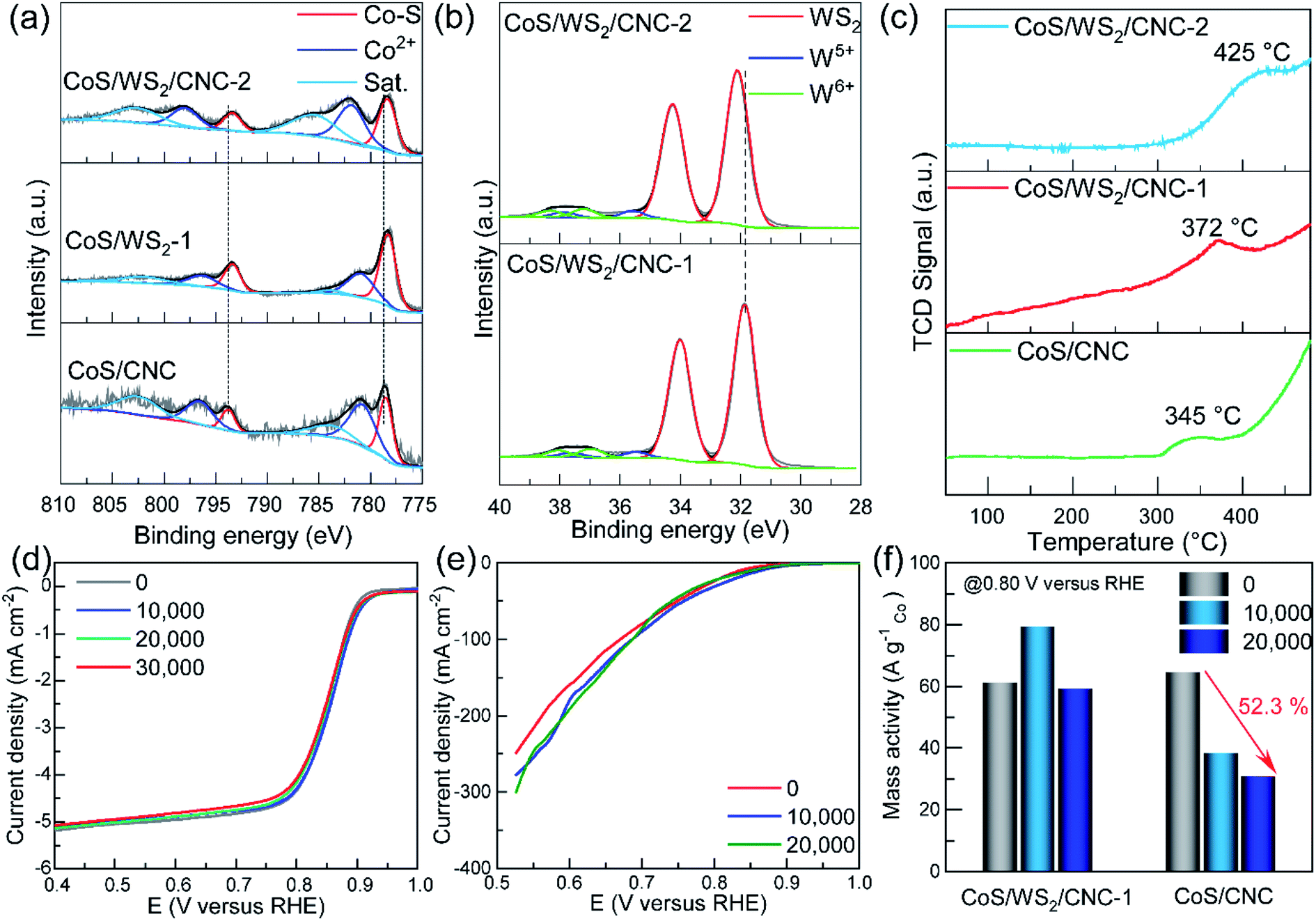 | ||
| Fig. 3 High-resolution (a) Co 2p and (b) W 4f XPS spectra, and (c) O2-TPD profiles of CoS/CNC, CoS/WS2/CNC-1, and CoS/WS2/CNC-2 catalysts. The polarization curves of CoS/WS2/CNC-1 catalyst measured in the (d) RDE and (e) GDE setups during the ADT process, and (f) mass activity (Jmass) at 0.80 V versus RHE tested in the GDE configuration. | ||
It is evidenced by the RDE results that CoS is suggested as the main ORR active component in the CoS/WS2/CNC catalyst rather than WS2. The ORR proceeds through four proton-coupled electron transfer steps, and, thus, the affinity of oxygenated intermediates on catalytically active sites (e.g. *OOH, *O, *OH, where * represents catalytically active centers) that are closely dependent on the orbital filling of transition-metal centers is considered as an indicator of ORR performance.47–49 As reported previously, cobalt centers in the Co–S configuration show a weaker affinity towards oxygenated intermediates than a promising value,50–52 agreeing well with those of spinel cobalt oxide and Co–N–C catalysts.53–56 Growing 1T-WS2 nanoplatelets with a metallic feature at the CoS surface can induce the electron transfer into CoS and strengthen the affinity of oxygenated intermediates, as manifested by a higher desorption temperature in O2-TPD plots of CoS/WS2/CNC-1 than that of CoS/CNC, thereby enhancing the ORR performance. However, more WS2 makes the desorption of intermediates difficult, owing to the overly strong binding strength, and deteriorates ORR activity. Hence, the results discussed above disclose that the electronic coupling effect between CoS and WS2 modulates the electronic structure and enhances the adsorption of oxygen-containing species at a reasonable level, leading to the improved ORR performance.
The durability performance of CoS/WS2/CNC-1 was examined according to the accelerated durability test (ADT) protocol. Fig. 3d shows the ORR polarization curves when CoS/WS2/CNC-1 catalyst is cycled from 1.0 V to 0.6 V versus RHE in the RDE setup. The ORR activity exhibits no noticeable degradation after 10000 ADT cycles and is still well retained even with prolonging to 30000 ADT cycles, but the CoS/CNC catalyst experiences a 12 mV negative shift for the E1/2 and a ∼13.1% loss for the diffusion-limited current at 0.5 V versus RHE after 10000 ADT cycles (Fig. S17†). The extraordinary long-term stability performance is further substantiated by the high retention ratio of the current measured via the chronoamperometric (CA) method, as displayed in Fig. S18,† whereby 94.8% of the initial current is retained for the CoS/WS2/CNC-1 catalyst when it was continuously operated at 0.50 V versus RHE for 50000 s, associated with a low to 8 mV negative shift for the E1/2, in contrast to 85.3% for the CoS/CNC catalyst and 77.4% for the Pt/C catalyst. Compared with the RDE setup, a GDE can more realistically reflect the catalytic performance in a membrane electrode assembly (MEA) due to the improved mass transport and high current,57–59 and is schematically shown in Fig. S19.† The CoS/WS2/CNC-1 catalyst is sprayed with a Sigracet SGL 29BC gas diffusion layer as the working electrode and is separated from the Hg/HgO reference electrode and the Pt mesh counter electrode using an anion exchange membrane. Fig. 3e shows the polarization curve of the CoS/WS2/CNC-1 catalyst recorded at an oxygen flow rate of 100 sccm. The onset potential that is determined at −1 mA cm−2 raises from 0.911 V in the initial cycle to 0.945 V versus RHE in the 10000th cycle, and subtly shifts to 0.925 V versus RHE in the 20000th cycle, along with the current at 0.60 V versus RHE increasing by 19.8%. By comparison, there is a 29 mV negative shift for the onset potential and a 31.8% current loss for the CoS/CNC catalyst under similar conditions (Fig. S20†). The good durability is further reflected by the higher retention ratio of the mass activity (Jmass), where Jmass is stable at ∼60 A gCo−1 and 0.80 V versus RHE for CoS/WS2/CNC-1 during cycling, but up to 52.3% of Jmass is lost for the CoS/CNC catalyst (Fig. 3f).
To better elucidate the compositional and structural changes during the ADT process, CoS/WS2/CNC-1 and CoS/CNC that have been cycled from 1.0 V to 0.6 V versus RHE for 20000 ADT cycles in the GDE setup, named as CoS/WS2/CNC-1-ADT and CoS/CNC-ADT, are characterized using electron microscopy, XRD, and XPS techniques. Apart from the crystal phases (CoS, Co9S8, and WS2, Fig. S21†), the nanocube morphology is well retained during the ADT process (Fig. S22a and S22b†). The TEM image in Fig. S22c† shows that WS2 nanoplatelets, labeled by red arrows, are still attached to the CoS surface, as manifested by the high-resolution TEM image in Fig. S22d.† It is also consolidated by preserving the Co–S bond in CoS/WS2/CNC-1-ADT (Fig. S23a†). The W 4f XPS spectrum discloses the existence of WS2 species after the ADT process (Fig. S23b†), consistent with XRD and TEM analysis. The Co 2p peaks indexed to Co3+ (780.1 eV and 795.2 eV) and Co2+ (782.0 eV and 797.0 eV) species and W 4f signals correlated to the oxidized W species are significantly enhanced, as compared to those in CoS/WS2/CNC-1, accompanied with weakened S2− and S22− signals in the S 2p XPS spectrum and an emergence of the metal–oxygen (M–O) bond in the O 1s XPS spectra (Fig. S23c and S23d†). This demonstrates that, in spite of conserving the crystal phases, a tiny minority of CoS and WS2 still evolved into oxides and/or (oxy)hydroxides during catalysis. By comparison, the phase transformation degree is substantially boosted for CoS/CNC catalyst under similar conditions, as seen from the disappeared Co–S bond, weaker S2− and S22− signals, as well as the stronger O 1s peak in the XPS spectra of CoS/CNC-ADT (Fig. S24†). Furthermore, the S2−/M–O ratio determined from the integrated peak area is 2.65 for CoS/WS2/CNC-1-ADT, which is ∼240 times higher than that for CoS/CNC-ADT. Therefore, because of the stronger oxophilicity of tungsten compared to cobalt,22 tungsten sulfide would be more easily converted into the oxides with respect to cobalt sulfide, thus greatly prohibiting a further phase change of cobalt sulfide underneath, coupled with a thin carbon protection layer with good crystallinity.
The electrocatalytic performance towards the OER was examined on an L-type glassy carbon electrode in a typical three-electrode configuration. Fig. 4a shows the polarization curves recorded at a scan rate of 5 mV s−1 with 85%-iR correction. The CoS/CNC catalyst shows a very poor OER activity with the onset potential of up to 1.60 V versus RHE and the overpotential potential at 10 mA cm−2 (η10) of 0.47 V. When decorating with WS2 nanoplatelets at CoS nanocrystals, the activity is dramatically improved in terms of decreasing the onset potential and η10 to 1.55 V versus RHE and 0.43 V for CoS/WS2/CNC-2, and further dropping to 1.53 V versus RHE and 0.38 V for CoS/WS2/CNC-1. The onset potential and η10 of the best-performing CoS/WS2/CNC-1 catalyst is merely 6 mV and 9 mV more positive than that of the benchmark RuO2 catalyst. Even at a current of 100 mA cm−2, the OER overpotential potential (η100) is only 7 mV higher than that of RuO2 catalyst. The improved OER performance is also verified by a smaller Tafel slope (66.9 mV dec−1) compared to 106.7 mV dec−1 for CoS/WS2/CNC-2 and 110.6 mV dec−1 for CoS/CNC (Fig. S25†).
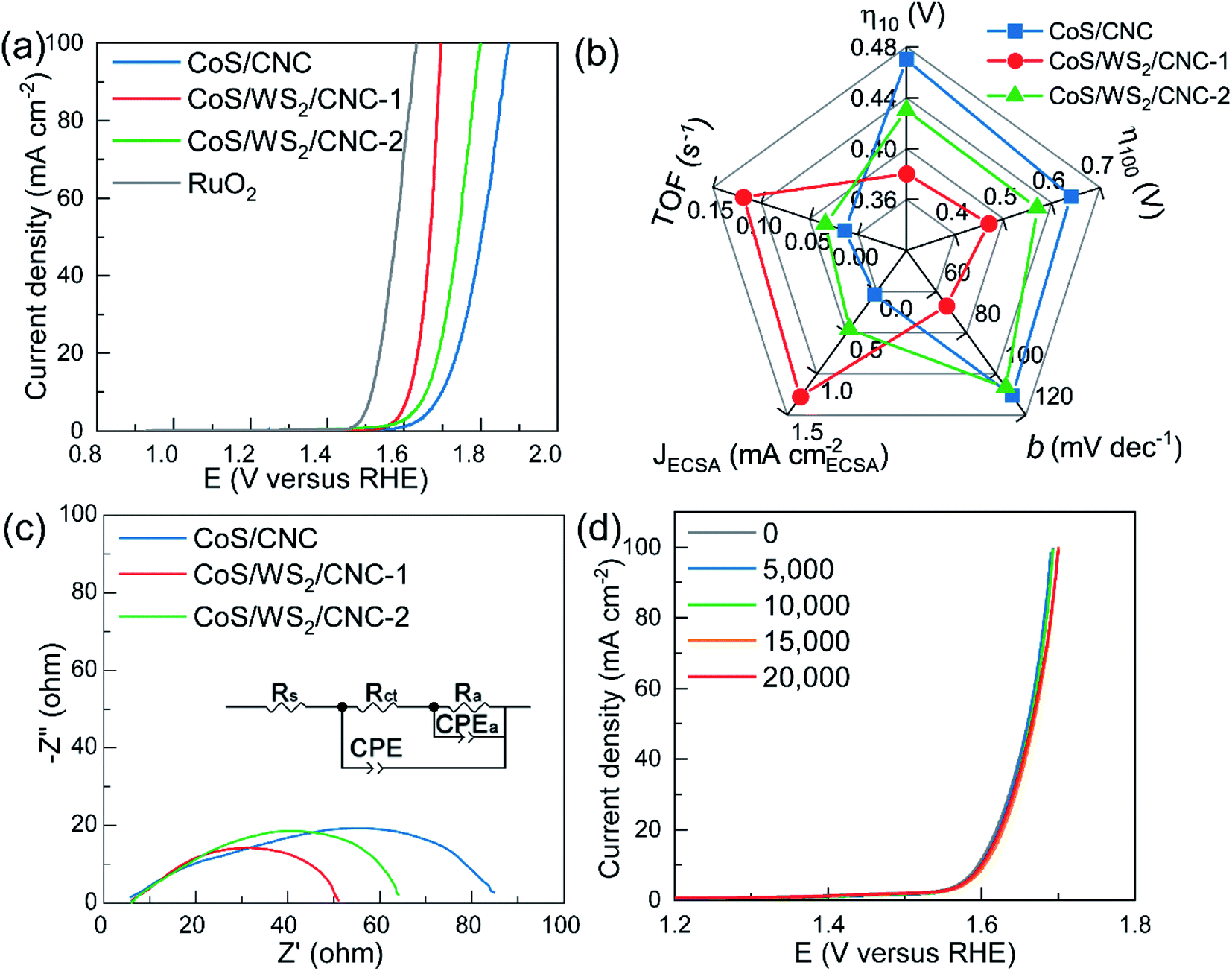 | ||
| Fig. 4 Electrocatalytic performance towards the OER performed on an L-type glassy carbon electrode. (a) OER polarization curves at 5 mV s−1 and with 85%-iR correction. (b) Summaries of an OER overpotential at 10 mA cm−2 (η10) and 100 mA cm−2 (η10), Tafel slope (b), an OER current at 1.65 V versus RHE normalized by the ECSA (JECSA), as well as the turnover frequency (TOF) at 1.65 V versus RHE. (c) EIS curves at 1.65 V versus RHE (inset: corresponding equivalent circuit diagram). (d) OER polarization curves of CoS/WS2/CNC-1 catalyst that has been scanned from 1.4 to 1.7 V versus RHE for 0–20000 cycles. | ||
The electrochemically active surface area (ECSA) is considered as a key factor to determine the OER activity and is linearly proportional to the double-layered capacitance in the non-faradic region. As depicted in Fig. S26,† CoS/CNC shows a ∼3.9 and 5.2 times higher ECSA than CoS/WS2/CNC-1 and CoS/WS2/CNC-2, while the CoS/CNS catalyst delivers a much poorer OER performance, indicating that the ECSA is not the key factor in influencing the OER performance. The OER current normalized by the ECSA (JECSA) and turnover frequency (TOF) are given for representing the intrinsic OER activity of Co sites. As displayed in Fig. 4b, the JECSA and TOF of CoS/CNC are 0.038 mA cmECSA−2 and 0.013 s−1 at 1.65 V versus RHE, while they are as high as 1.28 mA cmECSA−2 and 0.12 s−1 for CoS/WS2/CNC-1 and 0.46 mA cmECSA−2 and 0.033 s−1 for CoS/WS2/CNC-2, suggesting an impressive OER performance of the CoS/WS2 heterostructure. The capability of charge transfer is another important parameter for evaluating catalytic activity and is evaluated by using electrochemical impedance spectroscopy (EIS) at 1.65 V versus RHE. It is simulated by the equivalent circuit model, as depicted in the inset of Fig. 4c, where Rs is uncompensated ohmic resistance (Rs), Rct is the charge transfer resistance, Ra and Ca represent the resistance and capacitance contributed by adsorbed OER intermediates during catalysis, respectively, and a constant phase element (CPE) is the capacitance at the electrode/solution interface.60,61 The CoS/WS2/CNC-1 possess a much lower Rct (10.5 ohm) as compared to 16.1 ohm for CoS/WS2/CNC-2 and 61.7 ohm for CoS/CNC (Fig. 4c), indicative of a faster charge transfer during catalysis. The stable durability performance of CoS/WS2/CNC-1 was demonstrated by using the ADT protocol and the chronopotentiometric method, where the OER polarization curves were well overlapped when it was scanned from 1.4 V to 1.7 V versus RHE for 20000 ADT cycles (Fig. 4d) and the OER potential was merely raised by ∼10 mV after 100 h of continuous operation at 20 mA cm−2 (Fig. S27†).
To verify its good performance in powering electronic devices, CoS/WS2/CNC-1 was sprayed on a gas diffusion layer as an air cathode of an aqueous Zn–air battery. Fig. 5a shows the open circuit voltage (OCV) of a Zn–air battery. A stable OCV of 1.41 V is exported when the CoS/WS2/CNC-1 cathode is used, in good agreement with 1.414 V measured by the voltmeter, which is 20 mV higher than that assembled by the CoS/CNC cathode and is only 10 mV lower compared to that using the Pt/C and RuO2 cathode (Pt/C + RuO2). The discharging curve and corresponding power density in Fig. 5b reveal that the peak power density of the Zn–air battery is 151.9 mW cm−2 using CoS/CNC cathode and raises to 183.5 mW cm−2 when the CoS/WS2/CNC-1 cathode is used, which outperform those of the Zn–air batteries driven by the Pt/C + RuO2 cathode and most of the state-of-art transition-metal sulfide cathodes (Table S2†).11–15,19,21,62–68 The specific capacity normalized by consumed Zn mass reaches 683.4 mA h gZn−1 at 5 mA cm−2 for the Zn–air battery enabled by the Pt/C + RuO2 cathode, which is close to the values reported recently,11,15,64,69 and increases to 727.1 mA h gZn−1 and 819.2 mA h gZn−1 for CoS/CNC and CoS/WS2/CNC-1-based Zn–air batteries (Fig. 5c). The feasibility of powering the electronic devices is demonstrated by successfully lightening the “HUST” display composed of 61 parallel-connected blue light-emitting diodes (LEDs, rated voltage: 2.5 V) by two series-connected Zn–air batteries enabled by CoS/WS2/CNC-1 and launching a 5 V electric fan by four series-connected ones (Fig. 5d). The durability performance of the Zn–air battery is examined via repetitively charging/discharging at 5 mA cm−2 with 20 min per cycle. As displayed in Fig. 5e, the discharging end and charging end potentials are 1.16 V and 2.14 V for the Pt/C + RuO2 cathode in the initial cycle, and drastically drop to 1.12 V and 2.21 V after 24 h of operation (72 cycles of operation). By comparison, the durability performance is remarkably enhanced for the CoS/WS2/CNC-1-based Zn–air battery, where the discharging end and charging end potentials are 1.15 V and 2.13 in the initial cycles and are stable at 1.16 V and 2.08 V after 72 cycles, alongside increasing the voltaic efficiency from 54.2% to 55.8%. Even with prolonging to 1440 cycles (480 h of operation), the voltage difference between the discharging end potential (1.16 V) and the charging end potential (2.03 V) is lowered to 0.87 V, and the corresponding voltaic efficiency is increased to 57.1%, indicating an impressive long-term stability performance.
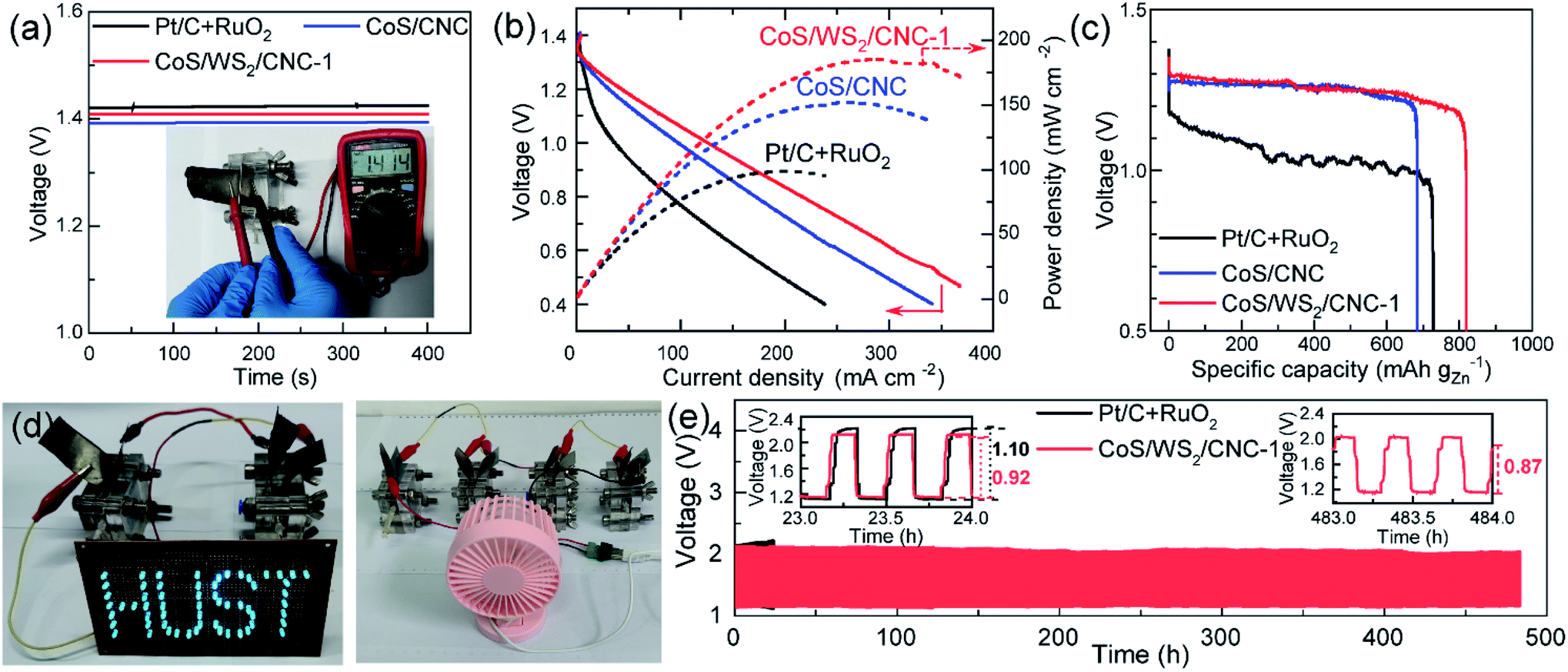 | ||
| Fig. 5 Electrochemical performances of Zn–air batteries. (a) Open circuit voltage versus time (inset: digital image of the open-circuit voltage of the CoS/WS2/CNC-1-based Zn–air battery measured by the voltmeter). (b) Discharge polarization and power density curves. (c) Discharging curves at 5 mA cm−2. (d) Digital image of a “HUST” display composed of 61 parallel-connected blue LEDs powered by two series-connected CoS/WS2/CNC-1-based Zn–air batteries and a 5 V electric fan launched by four series-connected ones. (e) Durability performance measured at 5 mA cm−2 with 20 min per cycle. | ||
Conclusions
In summary, we have reported a facile approach to preparing carbon nanocube-supported CoS/1T-WS2 heterostructures for oxygen electrocatalysis. This demonstrates that 1T-WS2 showed a strong electronic coupling effect with CoS for influencing the electronic structure and modulating the adsorption of oxygen intermediates, and high oxophilicity and a metallic character were beneficial for inhibiting the compositional change and accelerating the fast electron transfer during catalysis. As a result, the heterostructure catalyst exhibited an impressive ORR activity in terms of a positive half-wave potential of 0.868 V versus RHE and a high mass activity of 413.3 A gCo−1 at 0.80 V versus RHE, surpassing most of the state-of-the-art transition-metal sulfide catalysts reported to date. In particular, the activity was well preserved during the accelerated durability test regardless of whether being performed in the RDE or GDE setups. The OER activity was also incredibly enhanced compared to that without 1T-WS2, where an overpotential at 10 mA cm−2 shifted negatively by 10 mV and the turnover frequency was boosted by one order of magnitude. When it was explored as a cathode catalyst of aqueous Zn–air batteries, a peak power density of 183.5 mW cm−2 was achieved, along with a stable cycling performance over 480 h of operation. This research gives an insight into the rational design of highly active and durable first-row transition metal sulfide-based heterostructure electrocatalysts through interface engineering.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the National Natural Science Foundation of China (No. 21771069 and 21874051), the Science, Technology and Innovation Commission of Shenzhen Municipality (GJHZ2020731095001004), the Opening Fund of Hubei Key Laboratory of Material Chemistry and Service Failure (2021MCF02), and the HUST Academic Frontier Youth Team (2019QYTD11). The authors would like to thank the Analytical and Testing Center of HUST (Huazhong University of Science and Technology).References
- C. X. Zhao, J. N. Liu, J. Wang, D. Ren, B. Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- Y. Guo, T. Park, J. W. Yi, J. Henzie, J. Kim, Z. Wang, B. Jiang, Y. Bando, Y. Sugahara, J. Tang and Y. Yamauchi, Adv. Mater., 2019, 31, 1807134 CrossRef.
- Y. Liang, H. Lei, S. Wang, Z. Wang and W. Mai, Sci. China Mater., 2021, 64, 1868–1875 CrossRef CAS.
- Z. Li, Q. Wang, X. W. Bai, M. Y. Wang, Z. Z. Yang, Y. G. Du, G. E. Sterbinsky, D. J. Wu, Z. Z. Yang, H. J. Tian, F. P. Pan, M. Gu, Y. Y. Liu, Z. X. Feng and Y. Yang, Energy Environ. Sci., 2021, 14, 5035–5043 RSC.
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed.
- B. Yan, D. Krishnamurthy, C. H. Hendon, S. Deshpande, Y. Surendranath and V. Viswanathan, Joule, 2017, 1, 600–612 CrossRef CAS.
- S. Jin, ACS Energy Lett., 2017, 2, 1937–1938 CrossRef CAS.
- C. Hu, J. Liu, J. Wang, W. She, J. Xiao, J. Xi, Z. Bai and S. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 33124–33134 CrossRef CAS.
- X. Zhang, B. Li, M. Lan, S. Yang, Q. Xie, J. Xiao, F. Xiao and S. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 18683–18692 CrossRef CAS.
- M. Lan, C. Xie, B. Li, S. Yang, F. Xiao, S. Wang and J. Xiao, ACS Appl. Mater. Interfaces, 2022, 14, 11538–11546 CrossRef CAS PubMed.
- S. Ramakrishnan, J. Balamurugan, M. Vinothkannan, A. R. Kim, S. Sengodan and D. J. Yoo, Appl. Catal., B, 2020, 279, 119381 CrossRef CAS.
- H. Yang, B. Wang, H. Li, B. Ni, K. Wang, Q. Zhang and X. Wang, Adv. Energy Mater., 2018, 8, 1801839 CrossRef.
- X. Han, X. Wu, C. Zhong, Y. Deng, N. Zhao and W. Hu, Nano Energy, 2017, 31, 541–550 CrossRef CAS.
- X. Han, W. Zhang, X. Ma, C. Zhong, N. Zhao, W. Hu and Y. Deng, Adv. Mater., 2019, 31, 1808281 CrossRef PubMed.
- Y. Wang, W. Jin, C. Xuan, J. Wang, J. Li, Q. Yu, B. Li, C. Wang, W. Cai and J. Wang, J. Power Sources, 2021, 512, 230430 CrossRef CAS.
- X. Liu, Y. Li, Z. Cao, Z. Yin, T. Ma and S. Chen, J. Mater. Chem. A, 2022, 10, 1617–1641 RSC.
- L. N. Lu, Y. L. Luo, H. J. Liu, Y. X. Chen, K. Xiao and Z. Q. Liu, Chem. Eng. J., 2022, 427, 132041 CrossRef CAS.
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. B. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy, G. Eda and M. Chhowalla, Nat. Mater., 2013, 12, 850–855 CrossRef CAS PubMed.
- J. Yin, Y. Li, F. Lv, M. Lu, K. Sun, W. Wang, L. Wang, F. Cheng, Y. Li, P. Xi and S. Guo, Adv. Mater., 2017, 29, 1704681 CrossRef PubMed.
- D. C. Nguyen, D. T. Tran, T. L. L. Doan, D. H. Kim, N. H. Kim and J. H. Lee, Adv. Energy Mater., 2020, 10, 1903289 CrossRef CAS.
- J. Sun, H. Xue, N. Guo, T. Song, Y. R. Hao, J. Sun, J. Zhang and Q. Wang, Angew. Chem., Int. Ed., 2021, 60, 19435–19441 CrossRef CAS PubMed.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- C. C. Tran, Y. Han, M. Garcia-Perez and S. Kaliaguine, Catal. Sci. Technol., 2019, 9, 1387–1397 RSC.
- C. L. Qin, A. X. Fan, X. Zhang, S. Q. Wang, X. L. Yuan and X. P. Dai, J. Mater. Chem. A, 2019, 7, 27594–27602 RSC.
- Y. Wu, F. Li, W. Chen, Q. Xiang, Y. Ma, H. Zhu, P. Tao, C. Song, W. Shang, T. Deng and J. Wu, Adv. Mater., 2018, 30, 1803151 CrossRef PubMed.
- M. Kim, M. A. R. Anjum, M. Choi, H. Y. Jeong, S. H. Choi, N. Park and J. S. Lee, Adv. Funct. Mater., 2020, 30, 2002536 CrossRef CAS.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. Dong, S. Liu, X. Zhuang and X. Feng, Angew. Chem., Int. Ed., 2016, 55, 6702–6707 CrossRef CAS PubMed.
- T. An, Y. Wang, J. Tang, W. Wei, X. Cui, A. M. Alenizi, L. Zhang and G. Zheng, J. Mater. Chem. A, 2016, 4, 13439–13443 RSC.
- Y. Yang, H. Yao, Z. Yu, S. M. Islam, H. He, M. Yuan, Y. Yue, K. Xu, W. Hao, G. Sun, H. Li, S. Ma, P. Zapol and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 10417–10430 CrossRef CAS PubMed.
- M. Ma, J. Xu, H. Wang, X. Zhang, S. Hu, W. Zhou and H. Liu, Appl. Catal., B, 2021, 297, 120455 CrossRef CAS.
- X. Ji, M. Ma, R. Ge, X. Ren, H. Wang, J. Liu, Z. Liu, A. M. Asiri and X. Sun, Inorg. Chem., 2017, 56, 14743–14746 CrossRef CAS.
- W. Hong, E. Meza and C. W. Li, J. Mater. Chem. A, 2021, 9, 19865–19873 RSC.
- C. Tan, Z. Luo, A. Chaturvedi, Y. Cai, Y. Du, Y. Gong, Y. Huang, Z. Lai, X. Zhang, L. Zheng, X. Qi, M. H. Goh, J. Wang, S. Han, X. J. Wu, L. Gu, C. Kloc and H. Zhang, Adv. Mater., 2018, 30, 1705509 CrossRef.
- H. Hu, Bu Y. Guan and X. W. Lou, Chem, 2016, 1, 102–113 CAS.
- L. Wang, Z. Wang, L. Xie, L. Zhu and X. Cao, ACS Appl. Mater. Interfaces, 2019, 11, 16619–16628 CrossRef CAS PubMed.
- J. Zhao, W. Niu, L. Zhang, H. Cai, M. Han, Y. Yuan, S. Majeed, S. Anjum and G. Xu, Macromolecules, 2013, 46, 140–145 CrossRef CAS.
- S. K. Verma, P. Kar, D. J. Yang and A. Choudhury, Sens. Actuators, B, 2015, 219, 199–208 CrossRef CAS.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W. C. Cheong, Z. Chen, Y. Wang, Y. Li, Y. Liu, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 2610–2618 CrossRef CAS.
- H. Zhu, J. Zhang, R. Yanzhang, M. Du, Q. Wang, G. Gao, J. Wu, G. Wu, M. Zhang, B. Liu, J. Yao and X. Zhang, Adv. Mater., 2015, 27, 4752–4759 CrossRef CAS.
- M. S. Kim, M. H. Naveen, R. Khan and J. H. Bang, J. Mater. Chem. A, 2020, 8, 7647–7652 RSC.
- B. Qiu, Q. Zhu, M. Du, L. Fan, M. Xing and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2684–2688 CrossRef CAS PubMed.
- B. Mahler, V. Hoepfner, K. Liao and G. A. Ozin, J. Am. Chem. Soc., 2014, 136, 14121–14127 CrossRef CAS PubMed.
- J. Zhang, D. Han, S. Wang, X. Zhang, R. Yang, Y. Ji and X. Yu, Electrochem. Commun., 2019, 99, 75–80 CrossRef CAS.
- H. Yin, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2018, 6, 10932–10938 RSC.
- L. Benoist, D. Gonbeau, G. Pfister-Guillouzo, E. Schmidt, G. Meunier and A. Levasseur, Surf. Interface Anal., 1994, 22, 206–210 CrossRef CAS.
- T. Y. Chen, Y. H. Chang, C. L. Hsu, K. H. Wei, C. Y. Chiang and L. J. Li, Int. J. Hydrogen Energy, 2013, 38, 12302–12309 CrossRef CAS.
- J. Wang, J. Xu, X. Guo, T. Shen, C. Xuan, B. Tian, Z. Wen, Y. Zhu and D. Wang, Appl. Catal., B, 2021, 298, 120539 CrossRef CAS.
- C. Chen, X.-T. Wang, J.-H. Zhong, J. Liu, G. I. N. Waterhouse and Z.-Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 22043–22050 CrossRef CAS PubMed.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- P. Cai, J. Huang, J. Chen and Z. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS PubMed.
- H. Zhang, Z. Wang, C. Ma, Z. Zhou, L. Cao, X. Gong, C. Dong and J. Yang, ChemSusChem, 2021, 14, 1388–1395 CrossRef CAS PubMed.
- P. Zhang, X. Hou, J. Mi, L. Liu and M. Dong, Chem. Phys. Lett., 2015, 641, 112–116 CrossRef CAS.
- Z. Wang, J. Huang, L. Wang, Y. Liu, W. Liu, S. Zhao and Z. Q. Liu, Angew. Chem., Int. Ed., 2022, 61, e202114696 CAS.
- X. T. Wang, T. Ouyang, L. Wang, J. H. Zhong, T. Ma and Z. Q. Liu, Angew. Chem., Int. Ed., 2019, 58, 13291–13296 CrossRef CAS PubMed.
- L. Zhong and S. Li, ACS Catal., 2020, 10, 4313–4318 CrossRef CAS.
- C. Chen, H. Su, L. N. Lu, Y. S. Hong, Y. Chen, K. Xiao, T. Ouyang, Y. Qin and Z. Q. Liu, Chem. Eng. J., 2021, 408, 127814 CrossRef CAS.
- H. Rabiee, L. Ge, X. Zhang, S. Hu, M. Li and Z. Yuan, Energy Environ. Sci., 2021, 14, 1959–2008 RSC.
- J. Schröder, V. A. Mints, A. Bornet, E. Berner, M. Fathi Tovini, J. Quinson, G. K. H. Wiberg, F. Bizzotto, H. A. El-Sayed and M. Arenz, JACS Au, 2021, 1, 247–251 CrossRef PubMed.
- K. Ehelebe, J. Knöppel, M. Bierling, B. Mayerhöfer, T. Böhm, N. Kulyk, S. Thiele, K. J. J. Mayrhofer and S. Cherevko, Angew. Chem., Int. Ed., 2021, 60, 8882–8888 CrossRef CAS PubMed.
- F. Dionigi, J. Zhu, Z. Zeng, T. Merzdorf, H. Sarodnik, M. Gliech, L. Pan, W. X. Li, J. Greeley and P. Strasser, Angew. Chem., Int. Ed., 2021, 60, 14446–14457 CrossRef CAS PubMed.
- J. M. Hu, J. Q. Zhang and C. N. Cao, Int. J. Hydrogen Energy, 2004, 29, 791–797 CrossRef CAS.
- Z. Cai, I. Yamada and S. Yagi, ACS Appl. Mater. Interfaces, 2020, 12, 5847–5856 CrossRef CAS PubMed.
- H. F. Wang, C. Tang, B. Wang, B. Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- G. Fu, J. Wang, Y. Chen, Y. Liu, Y. Tang, J. B. Goodenough and J. M. Lee, Adv. Energy Mater., 2018, 8, 1802263 CrossRef.
- Z. Pan, H. Chen, J. Yang, Y. Ma, Q. Zhang, Z. Kou, X. Ding, Y. Pang, L. Zhang, Q. Gu, C. Yan and J. Wang, Adv. Sci., 2019, 6, 1900628 CrossRef PubMed.
- D. Zhou, Y. Jia, H. Yang, W. Xu, K. Sun, J. Zhang, S. Wang, Y. Kuang, B. Liu and X. Sun, J. Mater. Chem. A, 2018, 6, 21162–21166 RSC.
- L. Zhuang, H. Tao, F. Xu, C. Lian, H. Liu, K. Wang, J. Li, W. Zhou, Z. Xu, Z. Shao and Z. Zhu, J. Mater. Chem. A, 2021, 9, 18329–18337 RSC.
- W. Zhang, X. Zhao, Y. Zhao, J. Zhang, X. Li, L. Fang and L. Li, ACS Appl. Mater. Interfaces, 2020, 12, 10280–10290 CrossRef CAS PubMed.
- X. F. Lu, S. L. Zhang, E. Shangguan, P. Zhang, S. Gao and X. W. Lou, Adv. Sci., 2020, 7, 2001178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02157a |
| This journal is © The Royal Society of Chemistry 2022 |