
Solar thermal-activated photocatalysis for hydrogen production and aqueous triethanolamine polymerization†
Jinghui
Wang‡
a,
Ziping
Wang‡
ad,
Xia
Wang
a,
Peihe
Li
*a,
Danhui
Sun
a,
Limei
Duan
a,
Jie
Bai
 c,
Sarina
Sarina
b,
Huaiyong
Zhu
b and
Jinghai
Liu
*a
c,
Sarina
Sarina
b,
Huaiyong
Zhu
b and
Jinghai
Liu
*a
aInner Mongolia Key Laboratory of Carbon Nanomaterials, Inner Mongolia Engineering Research Center of Lithium-Sulfur Battery Energy Storage, Nano Innovation Institute (NII), College of Chemistry and Materials Science, Inner Mongolia Minzu University, Tongliao 028000, People' s Republic of China. E-mail: jhliu2008@sinano.ac.cn; phli2018@foxmail.com; Fax: +86-475-8313570; Tel: +86-475-8314342
bSchool of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology, Brisbane, QLD 4001, Australia
cChemical Engineering College, Inner Mongolia University of Technology, Huhhot 010051, People' s Republic of China
dNational & Local United Engineering Laboratory for Power Battery, Department of Chemistry, Northeast Normal University, Changchun 130024, People' s Republic of China
First published on 4th July 2022
Abstract
The photocatalytic process plays a vital role in the direct conversion and storage of renewable solar energy into green hydrogen (H2) fuel, a long-term and sustainable technology pathway with the potential for limiting the growth of global carbon emissions. However, the kinetics of H2 production and photogenerated hole reactions are sluggish, which limit the intrinsic photoelectrochemical attributes of semiconductor materials, thus lowering the conversion efficiency of solar energy. Herein, we report a heterogeneous solar thermal activated photocatalysis (STAP) strategy for H2 production and triethanolamine (TEOA) polymerization initiated by highly active photogenerated electron–hole pairs. Under simulated solar light irradiation, solar thermal activation elevated the reaction temperature up to 40.7 °C with a 76.7 mmol g−1 h−1 photocatalytic H2 evolution (PHE) rate, which was 6.31 times faster than that at 11.1 °C (12.16 mmol g−1 h−1), ascribed to the solar thermal energy promoting H2 desorption from the surface of platinum (Pt)-deposited graphitic carbon nitride (g-C3N4/Pt). The detailed DFT calculations reveal that the solar thermal energy contributes significantly to activating the H2 desorption kinetics by reducing the energy barrier (ΔGD) of H2 desorption from the Pt-carbon nitride (Pt-CN) active site and diminishing the bond-dissociation energy (kcal mol−1) of the Pt–H bond. Furthermore, the STAP-optimized g-C3N4-T/Pt improved the PHE rate up to 92.1 mmol g−1 h−1, which was close to the level of commercial TiO2 (P25) at 104.0 mmol g−1 h−1. Besides, we also found that STAP facilitates aqueous TEOA polymerization, thus favoring the high-efficiency utilization of excited-state holes toward mediating green synthesis.
 Jinghai Liu | Jinghai Liu received his PhD in Physical Chemistry from the Suzhou Institute of Nano-Tech and Nano-Bionics (SINANO), Chinese Academy of Sciences, in 2012. Currently, he is Professor at the Nano Innovation Institute (NII) and the College of Chemistry and Materials Science at the Inner Mongolia Minzu University (IMUN). His research interests include the precise synthesis of functional carbon/inorganic solid materials and devices at the nano/atomic scale for photocatalysis, electrochemical energy storage, and bioelectronics with an emphasis on interdisciplinary solid-state chemistry and nanoscience, and very impactful “Fundamental to Applied” research. He won the second prize of the Inner Mongolia Natural Science Award (2020). |
1 Introduction
Solar-driven hydrogen (H2) production from water globally attracts unprecedented attention in the area of renewable solar energy harvesting, together with the sustainable green H2 fuel production process to reduce carbon emission goals. Solar thermolysis,1–5 photovoltaic-powered electrolysis (PV-E),6–8 photoelectrochemical cells (PEC),9–11 and particulate photocatalysis12–14 are well-developed routes, where particulate photocatalysis presents scalable and cost-competitive advantages in technoeconomic analysis toward lowering the cost of hydrogen down to US$ 2.00–4.00 kg−1.15 However, the solar-to-hydrogen (STH) efficiency still remains low for photocatalytic H2 production. Also, advancing the H2 production rate and the effective exploitation of photogenerated hole reactions on the surface of particulate photocatalysts could be the main considerations in designing surface sites and new chemical processes.The long residence time (τ) of the hydrogen molecule on a solid surface site is thermodynamically dependent on the desorption energy (ΔHdes) and temperature (T), according to the relationship 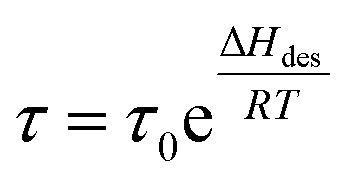 .16 The shorter the residence time, the faster the H2 production rate. The desorption energy is strongly dependent on the binding interaction between the active site and adsorbed H atom (Hads); thus, an appropriate adsorption and desorption kinetics is highly desired for designing the H2 evolution active site. The desorption of Hads for rapid H2 formation has been employed to boost photocatalytic H2 evolution over electron-enriched mediated active S sites with a weakening S–Hads bond.17 Except for the modulation of the electronic configuration of the active site, the temperature is another crucial factor that affects the desorption and residence time of H2 on the solid site. To shorten the residence time, this is associated with the system temperature elevation, which can be generated by solar thermal energy, especially the thermal effects after exposure to infrared (IR) radiation.18–20 Traditionally, a condensed medium (e.g., mixture of ethanol and water) is employed to control the system temperature from overheating under solar light irradiation, which would waste the converted thermal energy from IR light in the solar spectrum (up more than 50% (ref. 21 and 22)) in sustainable solar energy utilization and energy conservation. Therefore, we propose a heterogeneous solar thermal-activated photocatalysis (STAP) strategy for H2 production, combining the particulate semiconductor photocatalysis with solar thermal-promoted H2 desorption kinetics. As well demonstrated in the model of hydrogenated magnesium (MgH2), a higher temperature favors hydrogen desorption from the Mg site.23,24
.16 The shorter the residence time, the faster the H2 production rate. The desorption energy is strongly dependent on the binding interaction between the active site and adsorbed H atom (Hads); thus, an appropriate adsorption and desorption kinetics is highly desired for designing the H2 evolution active site. The desorption of Hads for rapid H2 formation has been employed to boost photocatalytic H2 evolution over electron-enriched mediated active S sites with a weakening S–Hads bond.17 Except for the modulation of the electronic configuration of the active site, the temperature is another crucial factor that affects the desorption and residence time of H2 on the solid site. To shorten the residence time, this is associated with the system temperature elevation, which can be generated by solar thermal energy, especially the thermal effects after exposure to infrared (IR) radiation.18–20 Traditionally, a condensed medium (e.g., mixture of ethanol and water) is employed to control the system temperature from overheating under solar light irradiation, which would waste the converted thermal energy from IR light in the solar spectrum (up more than 50% (ref. 21 and 22)) in sustainable solar energy utilization and energy conservation. Therefore, we propose a heterogeneous solar thermal-activated photocatalysis (STAP) strategy for H2 production, combining the particulate semiconductor photocatalysis with solar thermal-promoted H2 desorption kinetics. As well demonstrated in the model of hydrogenated magnesium (MgH2), a higher temperature favors hydrogen desorption from the Mg site.23,24
Graphite carbon nitride (g-C3N4) as a metal-free polymeric semiconductor photocatalyst has attracted broad research interests.25,26 A variety of g-C3N4 composites have been explored for photocatalytic H2 evolution (PHE), including RhPx/g-C3N4,27 In2O3-cube/g-C3N4,28 Ag-N2C2/CN,29 Au-TTA/g-C3N4-CdS,30 ZnIn2S4/g-C3N4,31 Co3S4-CN,32 TiO2/Ti3C2 MXene/Carbon Nitride,33 2D/2D Phosphorene/g-C3N4,34 amorphous carbon/g-C3N4,35 and 2D/2D Fe2O3/g-C3N4.36 The efficient composite route can effectively increase the H2 evolution rate and the quantum efficiency, where the developed TiO2/Ti3C2 MXene/g-C3N4 represents a supreme PHE rate at 15.29 mmol g−1 h−1.33 However, for these g-C3N4-based particulates' photocatalytic H2 production, photoexcited electrons involving the water (proton) reduction half-reaction is particularly focused on, with usually an additive sacrificial donor (e.g., triethanolamine, TEOA). Less attention has been paid to explore the interfacial photogenerated holes in mediating the oxidation reaction. Coupling the photocatalytic H2 production and holes-mediated transformation organic synthesis in particulate photocatalysis system represents a new trend, where the oxidation of alcohols, dehydrocoupling of thiols to disulfides, and oxidative cross-coupling have been tentatively investigated.37–39 We also realized synchronous photocatalytic H2 evolution and anaerobic oxidation of alcohols to ketones in acetonitrile (MeCN) by Pt/g-C3N4-mediated hole oxidation.40 However, the progress of photocatalytic holes-mediated organic transformation and polymerization synthesis in water still faces challenges.
With these considerations, we reveal an approach of heterogeneous solar thermal-activated photocatalysis (STAP) to realize photocatalytic H2 evolution and aqueous polymerization in particulate photocatalysis under solar light irradiation. With Pt/g-C3N4 and Pt/TiO2 (P25) in water and TEOA solution as a model particulate photocatalytic system, we demonstrate that STAP promotes the PHE rate and TEOA polymerization. Six types of monochromatic radiations at 380 nm, 420 nm, 450 nm, 500 nm, 600 nm, and 800 nm were used to identify the photocatalysis and solar thermal activation in STAP, where the photocatalysis is dependent on the light absorption of semiconductors in the UV-Vis spectrum range, while solar thermal activation is causing by the heating effects of IR at 800 nm. The Gibbs energy (ΔGA) of the H atom adsorbed and the Gibbs energy (ΔGD) of the H2 molecule desorbed from the Pt sites at variable temperatures were then calculated to understand the STAP accelerating desorption kinetics of H2 molecules during H2 production. The morphology and electronic structure of g-C3N4/Pt were further investigated to deeply understand the relationship of the structure-determining performances of the Pt-carbon nitride (Pt-CN) active site. To demonstrate the general feasibility of STAP H2 production, g-C3N4 obtained from five different synthetic routes and commercial TiO2 (P25) were carefully examined. The STAP-optimized g-C3N4-T/Pt gives a system temperature up to 41.1 °C with a high PHE to 92.1 mmol g−1 h−1, mostly close to the that of P25. Then, UV-Vis absorption spectroscopy, organic elementary analysis, FT-IR spectroscopy, and high-resolution mass spectrometry (MS) were used to determine the aqueous TEOA polymerization and the required conditions for STAP aqueous TEOA polymerization.
2 Experimental
2.1 Materials and methods
All reagents were analytical grade and used exactly as received. Titanium dioxide (P25, 99.0%), urea (CO(NH2)2, AR, ≥99.0%), chloroplatinic acid (H2PtCl6·6H2O, AR), glacial acetic acid (CH3COOH, AR, ≥99.5%), and nitric acid (HNO3, AR, 65–68%) were provided by Sinopharm Chemical Reagent Co., Ltd, China. Triethanolamine (C6H15NO3, AR, 99%+) was purchased from Shanghai Adamas Reagent Co., Ltd, China.All series of g-C3N4 materials were synthesized by a self-supporting pyrolysis process, according to our previously reported method. The products were rinsed with nitric acid (0.1 mol L−1) and deionized (DI) water, collected by filtration, and dried at 80 °C.
2.2 Characterization
X-ray diffraction (XRD) patterns were collected on a SmartLab 9 kW (Rigaku, Cu Kα λ = 1.5406 Å). UV-vis diffuse reflectance spectra (UV-vis DRS) were recorded on an Agilent Cary 300 (BaSO4 as a standard). FTIR transmission spectra were obtained by a Nicolet 6700 Fourier-transform infrared (IR) spectrometer using KBr pellets. The XPS profiles and chemical state analysis were performed by an ESCALAB Xi+ X-ray photoelectron spectrometer using Al Kα X-ray as the excitation source. TEM, HAADF-scanning transmission electron microscopy (STEM) images, and elemental mappings were measured by a JEM-F200 transmission electron microscope (JEOL, 200 KV). The N2 adsorption/desorption isotherms were obtained using a QUADRASORB SI automated surface area and pore size analyzer (Quantachrome). The specific surface area (SSA) was calculated using the Brunauer–Emmett–Teller (BET) method, and the PSD was analyzed using the NL-DFT method. On a Zahner-Zennium-PP211 with CIMPS (controllable intensity modulation photoelectrochemical system), Mott–Schottky measurements were performed using a three-electrode setup with Hg/HgCl2 as the reference electrode, Pt sheet as the counter electrode, and Na2SO4 (0.2 M) aqueous solution as the electrolyte. An AQE focus orbitrap high-resolution mass spectrometer was used to determine the molecular weight. NMR spectra were collected at 500 MHz (1H) and 125 MHz (13C) on a Bruker 500 spectrometer using D2O as the solvent and TMS as the internal standard.2.3 Photocatalytic hydrogen evolution
Photocatalytic hydrogen evolution reaction (HER) was conducted in a top irradiation type Pyrex glass vessel reaction cell connected to a commercial online system (LabSolar-III, Perfectlight Co.). Variable amounts of the photocatalyst were dispersed in a solvent with 54 mL DI water and 6 mL triethanolamine (used as sacrificial reagent). After the system was degassed for 1 h under vacuum conditions, a simulated solar light (300 W Xe lamp with an AM 1.5 filter) irradiation was started. Photocatalytic HER was carried out for 2 h. An online gas chromatograph (GC7900, TianMei, Shanghai) with a TCD detector was used to detect H2 evolution.3 Computational methods
All theoretical calculations were performed to get a deep understanding of the electronic structure and molecular orbital of ground state geometry at the DFT level within the Gaussian 09 program package. The geometry optimization of g-C3N4 and Pt@g-C3N4 clusters were carried out at the Becke's three parameters (B3) nonlocal exchange and Lee–Yang–Parr (LYP) nonlocal correlation functionals (B3LYP) using 6-311++g (d, p) basis set for C, N, H, and LANL2DZ for Pt atoms with the temperature increasing from 18 °C to 36 °C. Molecular electrostatic potential surfaces as well as HOMOs and LUMOs clouds of systems were mapped using the computer software GaussView 05. The atomic charge in molecules was performed using the NBO 3.1 program. The free energy can be defined as follows| ΔGH* = ΔEH* + ΔEZPE − TΔS |
4 Results and discussion
The concept of heterogeneous solar thermal activated photocatalysis (STAP) is elucidated in Scheme 1, where traditionally the high energetic photons in the ultraviolet-visible (UV-Vis) spectrum of solar light can excite a semiconductor photocatalyst to generate electrons and holes, which then kinetically separate and transfer to participate into the surface catalysis reactions or combine competitively. Furthermore, the low-energetic photons in the infrared light (IR) spectrum would vibrate the polar water molecules resonance, producing thermal energy, which would quickly heat the system and elevate the temperature. The solar thermal activation, that is IR-heating effects, converted the thermal energy, which might accelerate the desorption kinetics of the hydrogen molecules from catalytically active Pt sites, while also activating the polymerization of triethanolamine (TEOA) molecules in water during the photocatalytic process.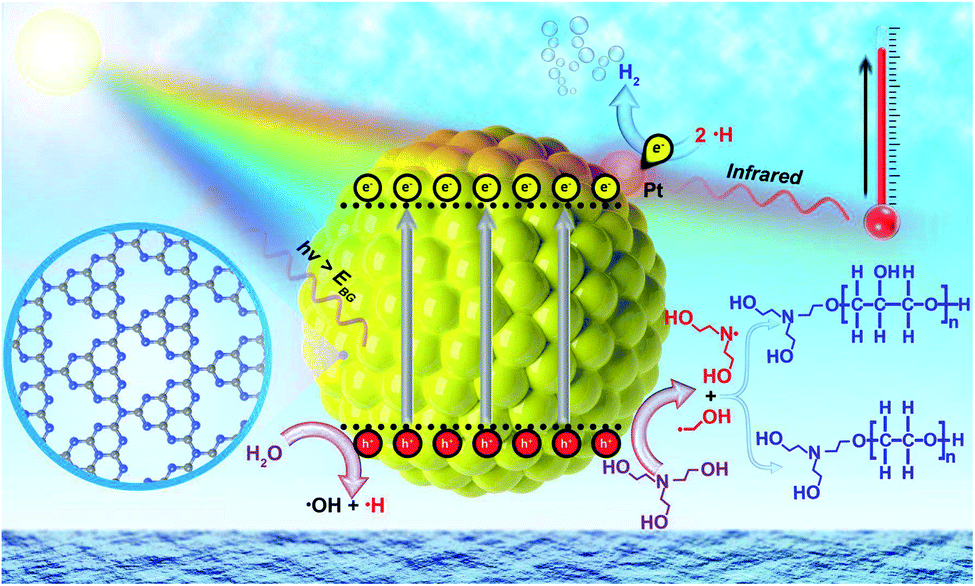 | ||
| Scheme 1 Schematic illustration of heterogeneous solar thermal-activated photocatalysis (STAP) for hydrogen (H2) production and aqueous triethanolamine (TEOA) polymerization. | ||
The photocatalytic hydrogen evolution (PHE) and solar thermal activation under solar light irradiation were demonstrated using monochromatic light and platinum (Pt)-deposited graphitic carbon nitride (g-C3N4/Pt) as a model photocatalyst. We firstly observed H2 evolution only for monochromatic light with wavelengths shorter than 500 nm, when the PHE rates of 2.478 mmol g−1 h−1 at 380 nm and 4.844 mmol g−1 h−1 at 420 nm changed into undetectable ones at 600 nm and 800 nm, respectively (Fig. 1a). This explicitly confirms that H2 evolution is a photocatalytic process under the control of light absorption of the g-C3N4 semiconductor (Fig. S2†). Meanwhile, we examined the elevation in the system temperature to present the solar thermal phenomenon. Under 800 nm monochromatic light, it sharply rises to 35.7 °C (308.85 K), indicating the noteworthy heating effects of IR in comparison with the other monochromatic lights in the UV-Vis spectrum range. Interestingly, we also discovered that the rate of PHE and temperature elevation are both dependent on the quantity of the photocatalyst in the reaction system, with both exhibiting a similar declining trend, turning into smaller ones as the amounts increases but not a linear relationship (Fig. 1b).
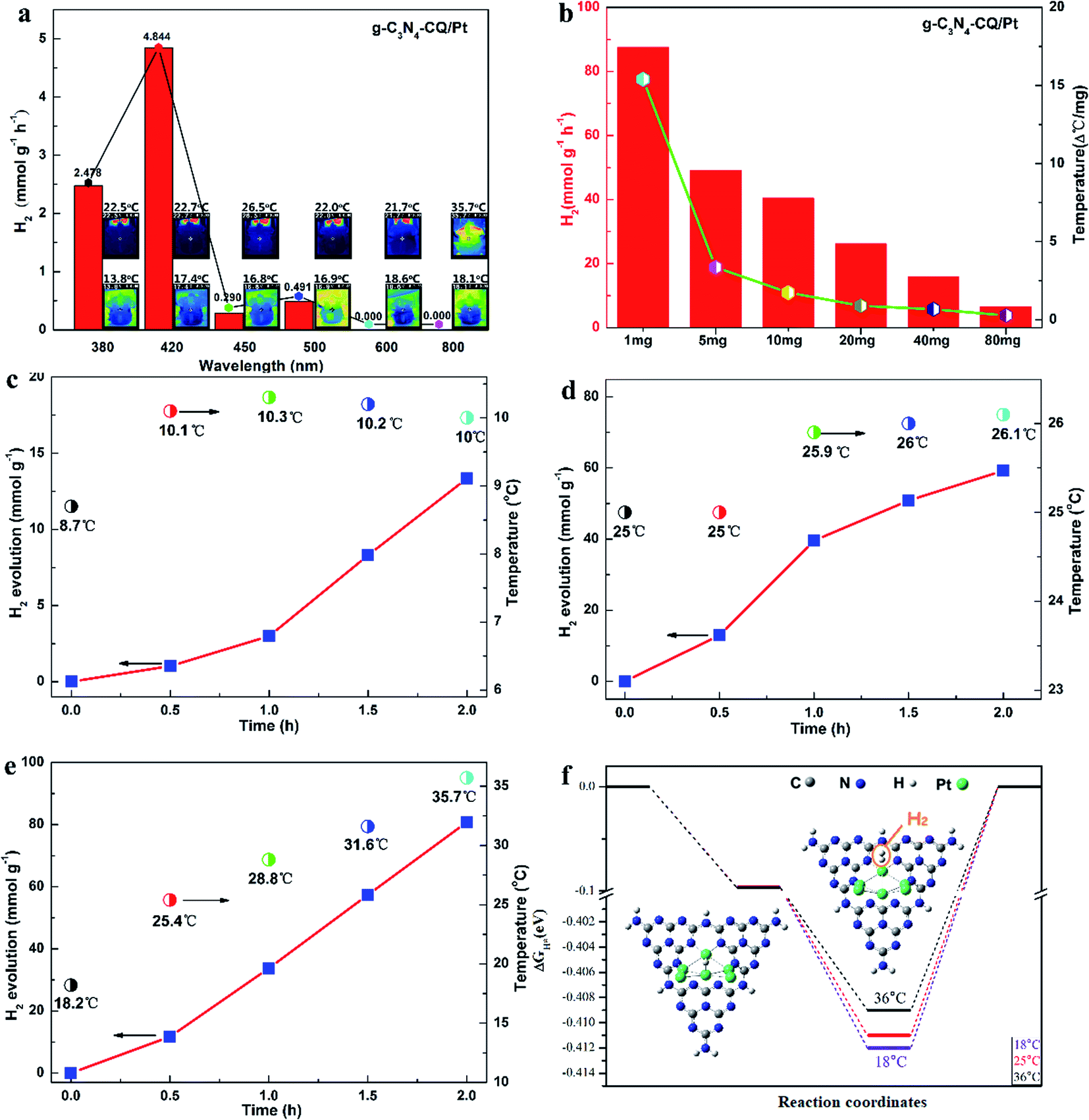 | ||
| Fig. 1 STAP H2 evolution. (a) Relationships between the PHE rate and heating effects of typical monochromatic light in the solar spectrum. Photocatalyst loading: 10 mg. (b) Dependence of the PHE rate and system temperature elevation on the photocatalyst loading. (c) PHE rate for the system temperature controlled at 10 °C ± 1 °C. (d) PHE rate for the system temperature controlled at 25 °C ± 1 °C. (e) PHE rate for the solar thermal system with variable increasing temperatures. (f) Calculated Gibbs energy (ΔGA) of H-atom adsorption and Gibbs energy (ΔGD) of H2 molecule desorption. Inset: structural model for the H atom and H2 molecule adsorbed on Pt sites. | ||
The solar thermal-activated PHE has further been illustrated by manipulating the temperature conditions. We utilized a constant temperature water/triethanolamine (TEOA) mixture by externally circulating the Pyrex glass vessel. When the reaction system is controlled at 10 ± 1 °C, the PHE rate is only 6.5 mmol g−1 h−1 (Fig. 1c) after solar light irradiation for 2 h. At a higher temperature of 25 ± 1 °C, the PHE rate increases to 30.0 mmol g−1 h−1, a 4.6-fold increase (Fig. 1d). For the solar thermal-activated temperature elevation, the initial temperature is 18.2 °C, and gradually goes up over 25.4 °C after half an hour until 35.7 °C as the irradiation time is prolonged to 2 h. Synchronously, the PHE rate increases rapidly and reaches 40.4 mmol g−1 h−1 at 35.7 °C (Fig. 1e), which is 6.2 times higher than that at 10 ± 1 °C. From the comparison of the PHE kinetics, we also observed the intense dependence of H2 evolution on the temperature elevation, where the 57.4 mmol g−1 at 31.6 °C for STAP is obviously higher than the one of 50.8 mmol g−1 with the temperature controlled at 26.0 °C under the same solar light irradiation conditions after 1.5 h. The H2 evolution increase of 47.0 mmol g−1 for STAP with the temperature elevated from 28.8 °C to 35.7 °C is 2.4 times higher than that for a constant temperature of 26.0 °C under the same solar light irradiation for 1 h. These evidences sufficiently verify the phenomenon that photocatalytic H2 evolution kinetics can be activated by solar thermal energy.
In order to give further insights into the mechanism for this STAP H2 evolution kinetics, we calculated the Gibbs energy (ΔGA) of the H atom adsorbed at platinum (Pt) sites (H-Pt), and the Gibbs energy (ΔGD) of the H2 molecule desorbed from these Pt sites (H2-Pt) at variable solar thermal temperatures, where the Pt cluster-inserted carbon nitride (heptazine units) works as a structural model (Fig. 1f, inset). As the system temperature increases from 25.0 °C to 36.0 °C, for the H–Pt adsorption state (initial step), ΔGA shows a subtle downward shift of 0.3 meV. In comparison, ΔGD gives an upward shift of 3 meV for the H2–Pt adsorption state (final step), which is 10 times larger than that of ΔGA (Fig. 1f). These results indicate that H2 evolution from Pt sites is a slow chemical adsorption process with thermal activation energy (Ea), and the large upward ΔGD suppresses the desorption activation energy (Ed) of H2 molecules departing from the Pt sites. Thus, increasing system temperature is more favorable for H2 molecule desorption from the surface of the Pt cluster. This provides sufficient evidence to deeply elaborate the solar thermal-activated photocatalytic H2 evolution by accelerating the desorption kinetics of H2 molecules during solar thermal activation, thus elevating the system temperature.
To further understand the relationships of the structures determining the performance of solar thermal-activated photocatalysis (STAP) for H2 production, we investigated the morphology and electronic structure of Pt/g-C3N4. The scanning transmission electron microscopy (STEM) image shows a general crumpled two-dimensional (2D) morphology with highly curved edges (Fig. 2a), which is mainly composed of carbon (C) and nitrogen (N) elements along with a little amount of oxygen (O) (Fig. 2d–f). The XRD patterns at 27.4° and 13.0° (Fig. S1†), and the intensive FTIR bands at 1637.9, 1575.3, 1459.7, and 1406.8 cm−1 (Fig. S6†) are the typical microstructural features of graphitic carbon nitride (g-C3N4) with graphitic phase-like (002) layered stacking and heptazine molecular structural unit in (100) plane. The high-resolution XPS spectrum further confirms the surface's chemical structure, which is mainly constructed by the heptazine-type carbon (C) at 288.1 eV and nitrogen (N) at 398.5 eV, and the graphitic C at 284.6 eV and N at 400. 8 eV (Fig. S9†). The platinum (Pt) nanoparticles (NPs) with a size of ∼2 nm are highly dispersed on the surface (Fig. 2b and c). Two oxidation states (Pt4+/Pt2+) were determined for these Pt NPs with binding energies of Pt 4f5/2 at 76.2 eV and Pt 4f7/2 at 72.6 eV for Pt4+, and Pt 4f5/2 at 74.2 eV and Pt 4f7/2 at 71.0 eV for Pt2+ (Fig. S10†).
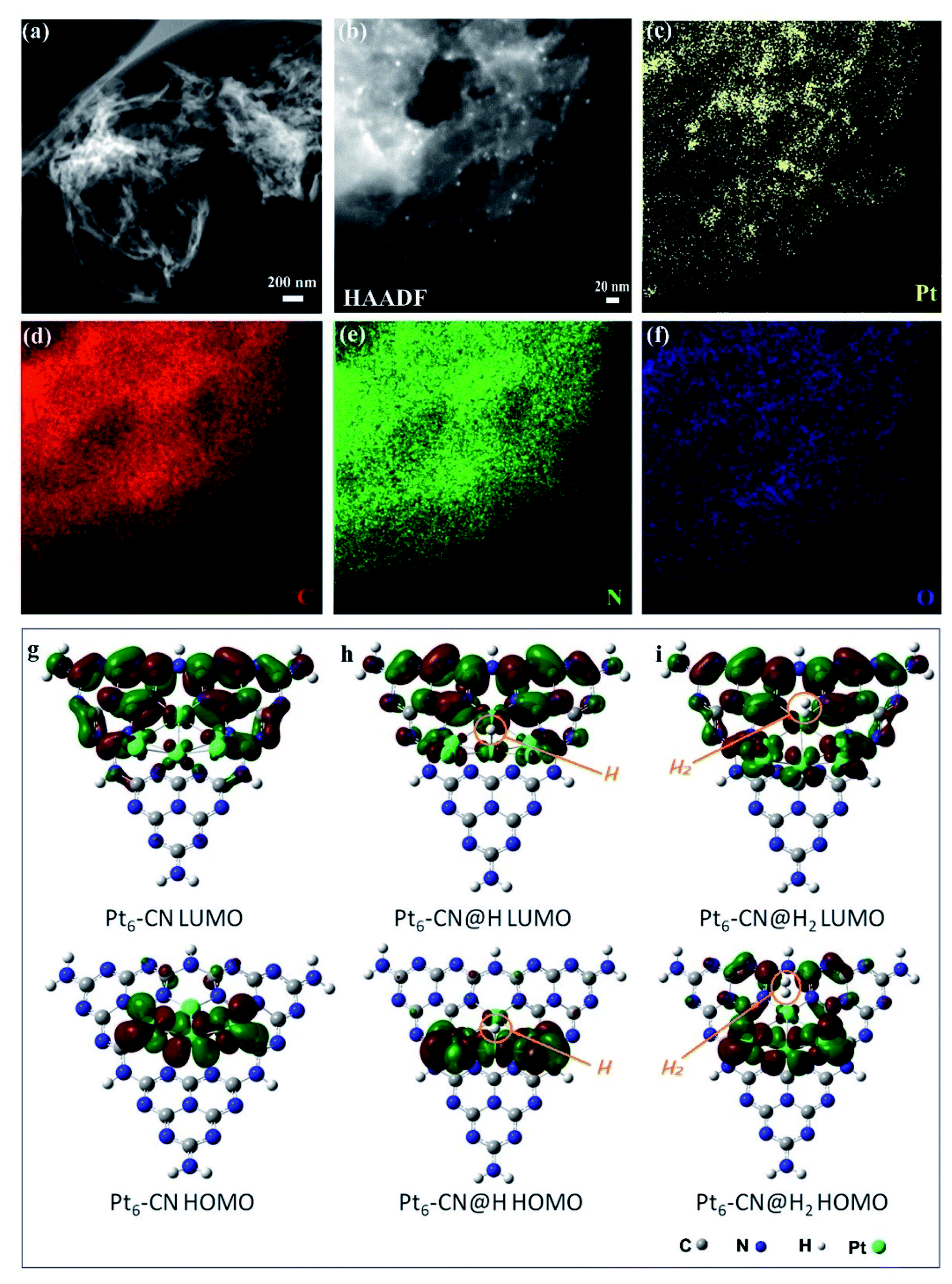 | ||
| Fig. 2 Morphology and electronic structures of platinum-deposited carbon nitride (Pt/g-C3N4). (a) STEM image with a scale bar of 200 nm. (b) HAADF-STEM image at large magnification. Elementary mappings for Pt (c), C (d), N (e), and O (f). (g–i) Frontier molecular orbital (HOMO–LUMO) of N-bonded Pt6 cluster site for H2 evolution. | ||
The electronic structure and molecular orbital (MO) of the ground state geometry were further calculated with the geometry-optimized Pt6@g-C3N4 cluster (Pt6-CN) model. We observed a stable wrinkled configuration for the g-C3N4 cluster with an in-built electrostatic field, where a positive potential zone localizes at the heptazine units and a negative one appears along with the vacancies and the edges, originating from the dipole moments and polarization of C–N bonds and point defects (Fig. S24a and b†). The Pt6 cluster prefers to anchor at the more negative potential vacancy coordinated with heptazine-N atoms to form a stable configuration. Interestingly, we find that the Pt6 cluster has a significant influence on the spatial distribution of the in-built electrostatic field, where the negative potential zone extends and mainly localizes in the region of the Pt6 cluster (Fig. S24c†). Electrostatically, a negative potential accumulated at the top Pt6 cluster facilitates the adsorption of the positively charged H atom, which gives fascinating insights into Pt as a catalytic active site for H2 evolution.
Subsequently, we investigated the frontier molecular orbital (HOMO and LUMO) of Pt6-CN to deeply understand the catalytic active site for STAP, from the perspective of electron density, charge distribution, and bond dissociation energy. The HOMO is mainly composed of the d-orbital of Pt atoms, while the LUMO is derived from the delocalized antibonding π-MO (π*) by the combination of p-orbitals of C and N atoms in heptazine and the small part of d-orbitals of the coordinated Pt atoms (Fig. 2g and S22†). As indicated by frontier molecular orbital (FMO) theory, the 5d electrons of the Pt atom occupying the HOMO orbital participate in the H2 evolution reaction by combining with the electrophilic H to form the Pt–H bond. Next, the HOMO orbital electron density at the Pt–H site (Pt6-CN@H) indicates the subsequent reaction with another electrophilic H to from the Pt–H–H bond (Fig. 2h and S23†). Finally, the H2 molecule departs from the Pt site by complete desorption, evidenced by the fact that no HOMO orbital around the H2 molecule was observed at the Pt–H–H site (Pt6-CN@H2, Fig. 2i and S23†). Furthermore, the orbital density of HOMO and LUMO at both the intermediate states (Pt6-CN@H and Pt6-CN@H2) presents no reduction, indicating the good catalytic activity.
To obtain a detailed insight into the desorption process, we examined the charge density of the atoms at the active site and the bond dissociation energy of the Pt–H bond with the formula ΔEBDE (Pt–H) = E(Pt–CN) + E(H) − E(Pt–CN@H). The natural bond orbital (NBO) analysis shows that the charge at the N and Pt atoms of the Pt6-CN site is redistributed after the H atom is adsorbed, with more negative charge accumulated (0.268e gain) at the Pt atom bonded to H (Fig. S23†). After H2 desorption, the charge almost withdraws to the initial state only with a total of 0.03e loss at the valence electron orbitals (6s and 5d). Simultaneously, the calculated bond dissociation energy changes slightly, giving an 86 cal mol−1 drop from 57.887 kcal mol−1 at 18.0 °C as the temperature rises to 36.0 °C, which indicates the positive effects of solar thermal activation to the hydrogen desorption kinetics. These results show a clear understanding of STAP H2 production by affecting the electrostatic potential, charge density, and bond dissociation energy of the H adsorbed intermediate states at the Pt–CN catalytic site to facilitate the H2 desorption kinetics as the system temperature rises.
To further demonstrate the general feasibility of STAP H2 production, we explored the correlation of H2 evolution yield with system temperature elevation at a variable time under the same simulated solar light irradiation for six different photocatalysts, including five carbon nitrides (g-C3N4-CQ/Pt, g-C3N4-HG/Pt, g-C3N4-OM/Pt, g-C3N4-H1/Pt, and g-C3N4-T/Pt) and one titanium dioxide (TiO2/Pt). The color in the color scale bar represents a corresponding system temperature. Interestingly, we observed a similar phenomenon of STAP H2 evolution kinetics, as revealed by g-C3N4-CQ/Pt, as the system temperature elevation for all these photocatalysts was examined. For example, the solar thermal-activated temperature elevation up to 40.7 °C for g-C3N4-HG, the PHE rate increased to 76.7 mmol g−1 h−1, which is 2.27 times higher than the one at 26.4 °C and 6.31 times higher than that at 11.1 °C (Fig. 3b). Also, we find the intense dependence of system temperature elevation on the catalysts under the same irradiation conditions, where g-C3N4-HG/Pt and g-C3N4-H1/Pt can raise it up to about 40.7 °C relative to the temperature at ∼34.9 °C for g-C3N4-OM/Pt (Fig. 3b and c). Correspondingly, the PHE rate of 78.82 mmol g−1 h−1 for g-C3N4-H1/Pt is 1.78 times higher than the one for g-C3N4-OM/Pt (Fig. 3d).
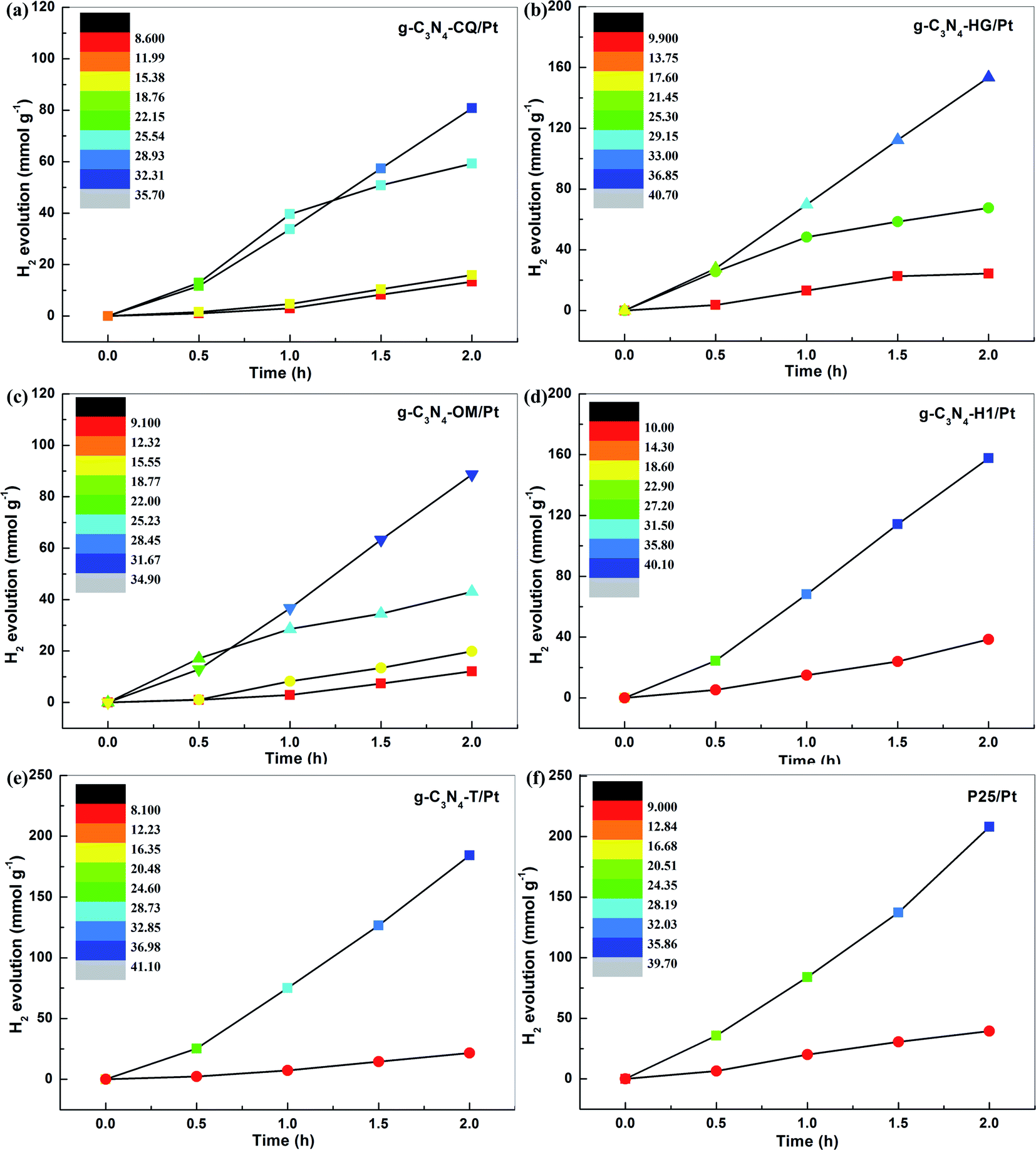 | ||
| Fig. 3 The general phenomenon for STAP H2 evolution kinetics. The H2 evolution yield correlated with system temperature elevation at a variable time under simulated solar light irradiation for the photocatalyst of (a) g-C3N4-CQ/Pt, (b) g-C3N4-HG/Pt, (c) g-C3N4-OM/Pt, (d) g-C3N4-H1/Pt, (e) g-C3N4-T/Pt, (f) P25 (TiO2)/Pt. Inset: color scale bar representing system temperature elevation. Photocatalyst loading, 10 mg. | ||
We then selected an inorganic photocatalyst, typical commercial TiO2 (Degussa-P25), to examine the feasibility of STAP H2 production. Fortunately, the same phenomenon was also observed for TiO2/Pt, where solar thermal activation raises the system temperature up to 39.7 °C, giving a PHE rate of 104.0 mmol g−1 h−1, which is 5.26 times higher than the one at 10.9 °C under simulated solar light irradiation for 2 h (Fig. 2f). Under this direction, we screened g-C3N4 with the intention of exploring its application potential as an efficient photocatalyst for H2 production with regard to commercial P25. The STAP-optimized g-C3N4-T/Pt gives a system temperature elevated to 41.1 °C with a high PHE to 92.1 mmol g−1 h−1, which is very close to the one of P25, manifesting that the g-C3N4 material could be an extraordinary alternative metal-free photocatalyst for green solar H2 production in the future.
Besides, we also found that STAP facilitates aqueous triethanolamine (TEOA) polymerization. Traditionally, TEOA works as a hole scavenger during heterogeneous photocatalytic H2 production. For STAP, surprisingly, TEOA underwent a polymerization reaction in water. The direct evidence is the color, where the polymeric product visually changes into reddish brown from colorless TEOA after STAP H2 production under simulated solar light irradiation for 2 h (Fig. 4a, inset). In the experimental process of controlling the temperature at 10 ± 1 °C, no color change could be observed in 2 h. We could only observe the occurrence of a brown color in the upper solution of the reaction system after placing it in the dark naturally for two or three years, indicating the sluggish kinetics of TEOA polymerization initiated by photogenerated hole-mediating reaction in the dark. STAP accelerates the aqueous TEOA polymerization kinetics. UV-Vis absorption spectroscopy further reveals that these polymeric products of TEOA with six photocatalysts give a similar absorption peak at about 300 nm (Fig. 4a). The organic elementary analysis gives an atomic ratio of C132N10H224O108 for the polymeric product, which is distinct from the formula of C6H15NO3 for the precursor TEOA molecule (Table S1†).
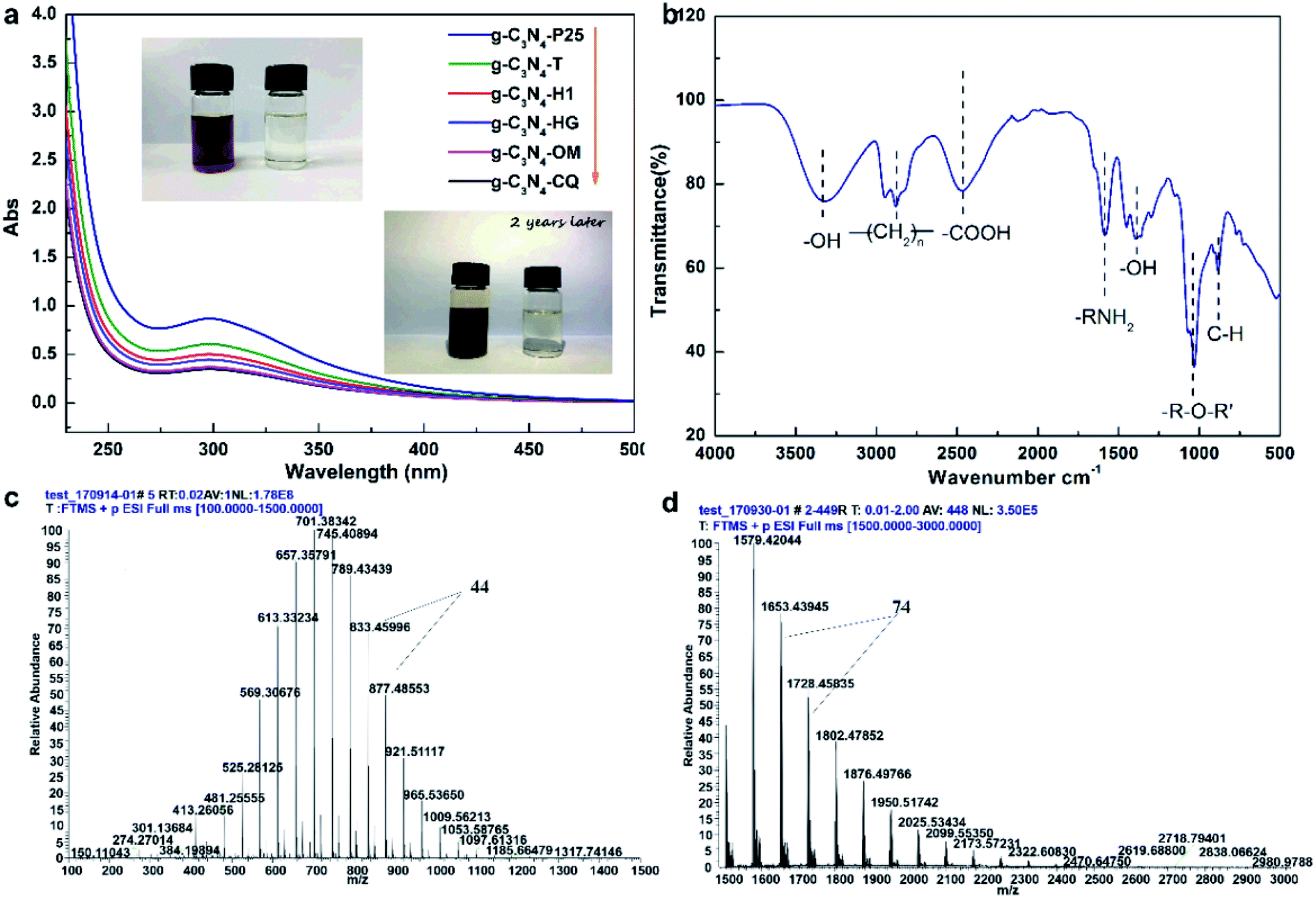 | ||
| Fig. 4 Aqueous triethanolamine (TEOA) polymerization by STAP. (a) UV-Vis absorption spectrum. Inset: the photograph of colorless TEOA (left) and brown polymeric products in water (right). (b) FTIR spectrum. (c) Typical mass spectrum for small molecular weight. (d) Typical mass spectrum for large molecular weight. | ||
The structure and aqueous polymerization process were also tentatively explored. FT-IR spectroscopy discloses the functional groups on polymeric C132N10H224O108 with a typical vibration of O–H stretching (3324.9 cm−1), C–H stretching (2944.4–2827.9 cm−1), C–C stretching (1402.1 cm−1), tertiary N–C stretching (1363.3 cm−1), C–O stretching (1299.0 cm−1), C–OH stretching (1143.6–1066.5 cm−1), and C–N bending (1028.2 cm−1), unlike the ones for TEOA. High-resolution mass spectrometry (MS) was further employed to probe the mass and related degree of polymerization. The results show that the molecular weight has a broad distribution from 481 to 2980, a typical polydisperse characteristic of molecular weight distribution for a macromolecule. The molecular fraction mainly distributes between 569 and 877 with a repeating molecular weight unit of 44 (Fig. 4c), and from 1579 to 1950 with 74 (Fig. 4d). To further represent the aqueous polymerization process of chain initiation, propagation, and termination clearly, we proposed free radical triggering condensation polymerization mediated by STAP hole reactions. The initiation reactions start from the chain decomposition of aqueous TEOA and H2O to produce ·NC4H10O2 and ·C2H4OH, and ·H and ·OH, by hole-mediated radical formation. The chain propagation steps include the combination of ·OH and ·C2H4OH, and subsequent condensation polymerization to generate oxygen-1,2-ethanediyl (–CH2CH2O–)n chain and 2-hydroxyl-oxygen-1,3-ethanediyl (–CH2CHOHCH2O–)n chain as the repeating units with molecular weight of 44 and 74, respectively. The principal termination reaction involves the radical reactions between ·OH and TEOA together with two main polymeric chains having oxygen-2-(diethanolamino)ethyl, HOCH2CH2–N(–CH2CH2OH)–CH2CH2O, and H as the capping agents (Fig. S26†).
To further probe the required conditions for STAP aqueous TEOA polymerization, we examined the four key factors including photocatalyst, illumination, H2 evolution, and system temperature. With g-C3N4-CQ/Pt and TiO2/Pt as the model photocatalysts together with g-C3N4-CQ and H2PtCl6 as the control, we found that the solar thermal-activated temperature elevation along with H2 evolution are concurrently required to realize TEOA polymerization (Table 1). Under the conditions of illumination and higher temperature of 50 °C attained by solar thermal activation, hydrogen is evolved and TEOA polymerization occurs. In contrast, under controlled temperature at 15 °C and illumination, only H2 can be detected without TEOA polymerization. No illumination occurred under controlled temperature at 50 °C, and neither H2 nor TEOA polymerization was produced. For g-C3N4 without Pt loading, no H2 evolution and TEOA polymerization can be detected even at a higher controlled temperature of 50 °C, indicating the significant roles of Pt (Table S2†) in H2 production and mediating TEOA polymerization. However, for the catalytic system with only H2PtCl6 at a higher controlled temperature of 50 °C under illumination, no H2 evolution occurs but TEOA polymerization does, confirming the significant contribution of photocatalysis in TEOA polymerization.
| Photocatalyst | Illuminationb | H2 evolution | Temperature (°C) | TEOA polymerizatione |
|---|---|---|---|---|
| a g-C3N4-CQ. b Illumination under simulated solar light. c Solar thermal-activated temperature elevation. d Controlled temperature. e Reddish brown color in 2 h. | ||||
| g-C3N4/Pta | Yes | Yes | 50c | Yes |
| g-C3N4/Pta | Yes | Yes | 15d | No |
| g-C3N4/Pta | No | No | 50d | No |
| g-C3N4a | Yes | No | 15d | No |
| g-C3N4a | Yes | No | 50d | No |
| TiO2(P25)/Pt | Yes | Yes | 50 c | Yes |
| H2PtCl6·6H2O | Yes | No | 50 d | No |
5 Conclusion
In summary, we present solar thermal-activated photocatalysis (STAP) as an efficient sustainable technology for green hydrogen (H2) fuel production and synthetic chemistry. The solar thermal-optimized g-C3N4/Pt photocatalyst gives system temperature elevation with a high H2 production efficiency close to that of commercial P25 (TiO2), and triggers aqueous triethanolamine (TEOA) polymerization. STAP activated the platinum-carbon nitride (Pt–CN) solid site to facilitate H2 desorption kinetics and to accelerate hole-mediated aqueous TEOA polymerization kinetics. This work promotes photocatalysis as a potential future technology for global sustainable development goals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank funding support from the National Natural Science Foundation of China (21961024 and 21961025), Inner Mongolia Natural Science Foundation (2018JQ05). Supported by Incentive Funding from Nano Innovation Institute (NII) of Inner Mongolia University for Nationalities (IMUN). Inner Mongolia Autonomous Region Science & Technology Planning Project for Applied Technology Research and Development (2019GG261). Inner Mongolia Autonomous Region Funding Project for Science & Technology Achievement Transformation (CGZH2018156). Scientific Research Project of Inner Mongolia University for Nationalities (NMDYB19041), and Open Project from the Inner Mongolia Key Laboratory of Carbon Nanomaterials (MDK2019012). We gratefully acknowledge financial support from Australian Research Council (DE 190101450).References
- S. E. Hosseini and M. A. Wahid, Int. J. Energy Res., 2020, 44, 4110–4131 CrossRef.
- Z. P. Chen, Q. Q. Jiang, F. Cheng, J. H. Tong, M. Yang, Z. X. Jiang and C. Li, J. Mater. Chem. A, 2019, 7, 6099–6112 RSC.
- C. N. R. Rao and S. Dey, Proc. Natl. Acad. Sci. U.S.A., 2017, 114, 13385–13393 CrossRef CAS PubMed.
- X. Qian, J. G. He, E. Mastronardo, B. Baldassarri, C. Wolverton and S. M. Haile, Chem. Mater., 2020, 32(21), 9335–9346 CrossRef CAS.
- Z. J. L. Bare, R. J. Morelock and C. B. Musgrave, Adv. Funct. Mater., 2022, 32, 2200201 CrossRef CAS.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- X. L. Yi, L. Z. Song, S. X. Ouyang, N. Wang, H. Y. Chen, J. B. Wang, J. Lv and J. H. Ye, Small, 2021, 17, 2102222 CrossRef CAS PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef PubMed.
- J. W. Ager, M. R. Shaner, K. A. Walczak, I. D. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811–2824 RSC.
- Y. H. Chiu, T. H. Lai, M. Y. Kuo, P. Y. Hsieh and Y. J. Hsu, APL Mater., 2019, 7, 080901 CrossRef.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260–2288 CrossRef CAS.
- A. Grimm, W. A. de Jong and G. J. Kramer, Int. J. Hydrogen Energy, 2020, 45, 22545–22555 CrossRef CAS.
- G. A. Somorjai, Chemistry in Two Dimensions: Surfaces, Cornell University Press, 1981, ch. 1, p. 29 Search PubMed.
- D. D. Gao, J. C. Xu, L. X. Wang, B. C. Zhu, H. G. Yu and J. G. Yu, Adv. Mater., 2021, 2108475 Search PubMed.
- J. C. Zhu, W. W. Shao, X. D. Li, X. C. Jiao, J. F. Zhu, Y. F. Sun and Y. Xie, J. Am. Chem. Soc., 2021, 143, 18233–18241 CrossRef CAS PubMed.
- J. Jing, S. Chen and Q. Lu, Adv. Biosyst., 2019, 3, 1800334–1800341 CrossRef.
- S. H. Du, X. N. Bian, Y. X. Zhao, R. Shi and T. R. Zhang, Chem. Res. Chin. Univ., 2022, 38, 723–734 CrossRef CAS.
- S. T. Tang, W. T. Qiu, X. W. Xu, S. X. Xiao, Y. X. Tong, X. W. Wang and S. H. Yang, Adv. Funct. Mater., 2022, 2110284 CrossRef CAS.
- (a) C. Song, Z. Wang, Z. Yin, D. Xiao and D. Ma, Chem. Catal., 2022, 2, 52–83 CrossRef; (b) S. H. Guo, X. H. Li, J. Li and B. Q. Wei, Nat. Commun., 2021, 12, 1343–1345 CrossRef CAS PubMed.
- E. Evard, I. Gabis and V. A. Yartys, Int. J. Hydrogen Energy, 2010, 35, 9060–9069 CrossRef CAS.
- I. P. Jain, C. Lal and A. Jain, Int. J. Hydrogen Energy, 2010, 35, 5133–5144 CrossRef CAS.
- Y. P. Xing, X. K. Wang, S. H. Hao, X. L. Zhang, X. Wang, W. X. Ma, G. Zhao and X. J. Xu, Chin. Chem. Lett., 2021, 32, 13–20 CrossRef CAS.
- Z. H. Chen, B. Chong, N. Wells, G. D. Yang and L. Z. Wang, Chin. Chem. Lett., 2022, 33, 2579–2584 CrossRef CAS.
- H. Dong, M. Xiao, S. Yu, H. Wu, Y. Wang, J. Sun, G. Chen and C. Li, ACS Catal., 2019, 10, 458–462 CrossRef.
- W. Wang, X. Bai, Q. Ci, L. Du, X. Ren and D. L. Phillips, Adv. Funct. Mater., 2021, 31, 2103978 CrossRef CAS.
- X. H. Jiang, L. S. Zhang, H. Y. Liu, D. S. Wu, F. Y. Wu, L. Tian, L. L. Liu, J. P. Zou, S. L. Luo and B. B. Chen, Angew. Chem., Int. Ed., 2020, 59, 23112–23116 CrossRef CAS PubMed.
- J. Fang, Y. Chen, W. Wang, L. Fang, C. Lu, C. Zhu, J. Kou, Y. Ni and Z. Xu, Appl. Catal. B Environ., 2019, 258, 117762–117772 CrossRef CAS.
- Z. Gao, K. Chen, L. Wang, B. Bai, H. Liu and Q. Wang, Appl. Catal. B Environ., 2020, 268, 118462 CrossRef CAS.
- J. Zhou, J. Zhang, J. Zhao, H. Wang and R. Liu, J. Mater. Chem. A, 2021, 9, 16522–16531 RSC.
- H. Zeng, Z. Li, G. Li, X. Cui, M. Jin, T. Xie, L. Liu, M. Jiang, X. Zhong, Y. Zhang, H. Zhang, K. Ba, Z. Yan, Y. Wang, S. Song, K. Huang and S. Feng, Adv. Energy Mater., 2021, 12, 2102765 CrossRef.
- J. Ran, W. Guo, H. Wang, B. Zhu, J. Yu and S. Z. Qiao, Adv. Mater., 2018, 30, 1800128 CrossRef PubMed.
- Q. Xu, B. Cheng, J. Yu and G. Liu, Carbon, 2017, 118, 241–249 CrossRef CAS.
- Q. Xu, B. Zhu, C. Jiang, B. Cheng and J. Yu, Sol. RRL, 2018, 2, 1800006–1800016 CrossRef.
- M. Y. Qi, M. Conte, M. Anpo, Z. R. Tang and Y. J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed.
- S. Li, E. W. Shuler, D. Willinger, H. T. Nguyen, S. Kim, H. C. Kang, J. J. Lee, W. Zheng, C. G. Yoo, B. D. Sherman and G. Leem, ACS Appl. Mater. Interfaces, 2022, 14, 22799–22809 CrossRef CAS PubMed.
- H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef CAS PubMed.
- D. H. Sun, P. H. Li, X. Wang, Y. Y. Wang, J. H. Wang, Y. Wang, Y. Lu, L. M. Duan, S. Sarina, H. Y. Zhu and J. H. Liu, Chem. Commun., 2020, 56, 11847–11850 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02066a |
| ‡ J. H. W. and Z. P. W. contribute equally to the work. |
| This journal is © The Royal Society of Chemistry 2022 |